Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Epidemiological support for genetic variability at hypothalamic–pituitary–adrenal axis and serotonergic system as risk factors for major depression
Authors Ching-López A, Cervilla J, Rivera M, Molina E, McKenney K, Ruiz-Perez I, Rodríguez-Barranco M ![]() , Gutiérrez B
, Gutiérrez B
Received 12 June 2015
Accepted for publication 30 July 2015
Published 22 October 2015 Volume 2015:11 Pages 2743—2754
DOI https://doi.org/10.2147/NDT.S90369
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Roger Pinder
Ana Ching-López,1 Jorge Cervilla,1–3 Margarita Rivera,1–3 Esther Molina,4 Kathryn McKenney,2 Isabel Ruiz-Perez,3,5,6 Miguel Rodríguez-Barranco,5 Blanca Gutiérrez1,2
1Department of Psychiatry, Institute of Neurosciences, School of Medicine, University of Granada, Granada, 2CIBER en Salud Mental (CIBERSAM), University of Granada, Granada, 3Instituto de Investigación Biosanitaria Ibs. Granada, Granada, 4Department of Nursing, University of Seville, Seville, 5Andalusian School of Public Health, Granada, 6CIBER de Epidemiología y Salud Pública (CIBERESP), Granada, Spain
Background: Major depressive disorder (MDD) is a serious, and common psychiatric disorder worldwide. By the year 2020, MDD will be the second cause of disability in the world. The Granad∑p study is the first, to the best of our knowledge, epidemiological study of mental disorders carried out in Andalusia (South Spain), being one of its main objectives to identify genetic and environmental risk factors for MDD and other major psychiatric disorders. In this study, we focused on the possible association of 91 candidate single nucleotide polymorphisms (SNPs) with MDD.
Methods: A total of 711 community-based individuals participated in the Granad∑p study. All individuals were extensively assessed for clinical, psychological, sociodemographic, life style, and other environmental variables. A biological sample was also collected for subsequent genetic analyses in 91 candidate SNPs for MDD. DSM-IV diagnosis of MDD was used as the outcome variable. Logistic regression analysis assuming an additive genetic model was performed to test the association between MDD and the genetic data. The experiment-wide significance threshold adjusted with the SNP spectral decomposition method provided a maximum P-value (8×10-3) required to identify an association. Haplotype analyses were also performed.
Results: One SNP (rs623580) located in the tryptophan hydroxylase 1 gene (TPH1; chromosome 11), one intergenic variant (rs9526236) upstream of the 5-hydroxytryptamine receptor 2A gene (HTR2A; chromosome 13), and five polymorphisms (rs17689966, rs173365, rs7209436, rs110402, and rs242924) located in the corticotropin-releasing hormone receptor 1 gene (CRHR1; chromosome 17), all showed suggestive trends for association with MDD (P<0.05). Within CRHR1 gene, the TATGA haplotype combination was found to increase significantly the risk for MDD with an odds ratio =1.68 (95% CI: 1.16–2.42, P=0.006).
Conclusion: Although limited, perhaps due to insufficient sample size power, our results seem to support the notion that the hypothalamic–pituitary–adrenal and serotonergic systems are likely to be involved in the genetic susceptibility for MDD. Future studies, including larger samples, should be addressed for further validation and replication of the present findings.
Keywords: genetic association analysis, major depression, HPA axis, serotonergic system
Introduction
Major depression is the most common psychiatric disorder, with an estimated lifetime prevalence that varies from 8% to 12% across studies.1,2 It is a serious mental illness characterized by persistent sadness and anhedonia, as well as alterations in vegetative, psychomotor, and cognitive functions. It can deeply impair the person’s ability to function or cope with daily life, being a leading cause of disability worldwide.3
Genetic factors clearly play a substantial role in the etiology of major depression, as evidenced by family and twin studies that report heritability estimates ranging from 17% to 75%, with an average of 37%.4–6 Since major depression is responsible for a significant burden on our health systems,3 the identification of underlying genetic factors has recently been recognized to be among the greatest challenges that researchers are facing.7 A better understanding of the genetic bases of major depression could provide us new insights into the etiology of such illness and lead to improved diagnosis, treatments, and prevention strategies.
Although several genome-wide association studies (GWAS)8 have been published in the last few years, none of them has robustly identified a locus that exceeds genome-wide significance for major depressive disorder (MDD) or genetically related traits.8 The lack of conclusive results could be explained, at least, in part, by an underlying genetic vulnerability caused by the joint effect of multiple loci of small effect, difficult to detect from GWAS approaches.
In contrast to such results, some promising findings have been reported from classical genetic association studies. A recent review by Flint and Kendler8 shows that from almost 200 candidate genes for MDD tested in different studies, only a few provided some robust results. This review also includes a meta-analysis focused on 26 of those candidate genes for MDD, of which seven yielded a significant P-value (SLC6A4, APOE, DRD4, GNB3, HTR1A, MTHFR, and SLC6A3).
Such suggestive results come from a neurobiologically informed strategy of genetic search (based on the existing knowledge of the underlying neural substrates of the disorder) which can be particularly informative in unraveling the genetic architecture of MDD. With this approach in the context of an epidemiological study on mental disorders with a representative sample of the province of Granada, southern Spain (the GranadΣp study), we designed a custom single nucleotide polymorphism (SNP) array containing 91 SNPs of relevance for MDD. The SNPs were chosen taking into account key candidate genes previously described in the literature, novel genes from putatively important pathways, and top hits from published GWAS on MDD. Our main aim was to evaluate the possible association of these 91 candidate SNPs with MDD.
Materials and methods
Study context and design
The GranadΣp study is a cross-sectional epidemiological study based on a relatively large sample representative of the adult population living in the province of Granada (southern Spain). This study was designed to serve as a self-contained pilot study for a larger cross-sectional study using a sample of the entire Andalusia region (Plan Integral de Salud Mental en Andalucía epidemiological mental health study [PISMA-ep study]9), and both used the same assessment methods. Both studies were cross-sectional surveys aiming to establish the prevalence of major psychiatric disorders in Andalusia and to identify genetic and environmental risk factors for such conditions.
The recruitment of the sample took place between October 2011 and December 2012 involving the province of Granada (n=711). All individuals in this study were randomly selected from the Andalusian Health Service users database, which is estimated to cover 98% of the population. Participants were assessed for clinical, psychological, sociodemographic, life style, and other environmental variables. Individuals who also agreed to participate in the genetic analysis gave specific informed consent and provided a biological sample. Participants were mostly Spanish of Spanish ancestry; although Spanish nationality was not an inclusion criterion and people coming from other European countries with European ancestry could also be included in the study.
The GranadΣp study was approved by the Research Ethic Committee of the University of Granada. The common methodology of both the GranadΣp and PISMA-ep studies has been described in more detail elsewhere.9
The sample
Sampling was performed in a two-stage cluster sampling with stratification of the primary sampling units depending on whether they were rural or urban. Rurality was determined according to the rurality of Andalusian municipalities index.10 The median of the rurality index for all Andalusian municipalities was taken into account to make the distinctions between rural and urban municipalities.
In the first stage, municipalities were sampled by probability proportional to size cluster sampling. Clusters were selected with proportional allocation within strata. The total number of selected municipalities represented approximately 15% of the total of municipalities in the province of Granada. In the second stage, the individuals that made up the sample were selected using a simple random sampling procedure.
Eight hundred and nine noninstitutionalized community-based participants living in the province of Granada agreed to take part in the survey. All interviews were performed between October 2011 and September 2012. To be able to estimate a 2% mental disorder prevalence with ±0.8% accuracy at a 95% confidence interval (CI) the sample was estimated at 1,176 participants.
All living participants between the ages of 18 and 80 were selected for participation in the study, divided into four age groups. Exclusion criteria were: being outside the age range, having lived in Granada province for less than a year, not being able to communicate fluently in Spanish, being too ill to be able to complete the interview, having a diagnosis of dementia or mental retardation, living in an institution (hospital, prison, etc), having moved or not living normally at the address we had been given by the users database, and erroneous data from the users database (eg, an incomplete address). Excluded participants were replaced with other individual matched for age, sex, and location.
Characterization of major depressive disorder
The Mini-International Neuropsychiatric Interview (MINI)11 was used to ascertain psychiatric diagnoses in our sample. Interviews were conducted by professionally trained psychologists and took place either in the participant’s local primary health care center or in their homes.
The psychiatric interview section was composed of the MINI interview that generates Axis I DSM-IV and ICD-10 diagnoses for 16 mental disorders. This interview consists of one or two screening questions asked to all participants at the beginning of each of the diagnostic sections. Any participants responding positively to the screening questions are then asked the complete set of that section’s questions leading to ascertainment of that section’s specific diagnosis. Prevalence of MDD was calculated using the information from the MINI interview, where diagnoses of mental disorders were determined by their answers about symptoms. A DSM-IV compatible diagnosis of MDD, generated from the MINI interview, was used as the outcome variable in this study.
Genetic analysis
SNP selection
Ninety-one SNPs were selected for analysis in this study. These SNPs were chosen among the most replicated in previous genetic association studies on depression and among those described as suggestive or clearly associated with MDD in previous GWAS published between 2010 and 2011.12–18 We mined the public databases for general information about the genes and polymorphic variants of interest, including some haplotype tagging and potentially functional SNPs, as well as markers with minor allele frequencies >0.1. This information was then combined and compared with the list of SNPs available from Illumina, Inc. (San Diego, CA, USA).
Genotyping
A biological sample was obtained from each participant using the Oragene DNA saliva collection kit (OG-500; DNA Genotek Inc, Ottawa, ON, Canada). DNA extraction was performed by using standard procedures. DNA concentration was measured by absorbance using the Infinite® M200 PRO multimode reader (Tecan US Inc, Research Triangle Park, NC, USA).
Genotyping was performed using TaqMan® OpenArray™ Genotyping System (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions. Raw data were analyzed with the TaqMan® Genotyper v1.2 software (Thermo Fisher Scientific).
Stringent quality control criteria were applied to both SNP and individual data. SNPs were excluded if they had a missing genotype rate higher than 1% or showed departure from the Hardy–Weinberg equilibrium (P<0.01). Individuals with genotypic data showing a missing rate >10% or those with a non-European ancestry were also excluded.
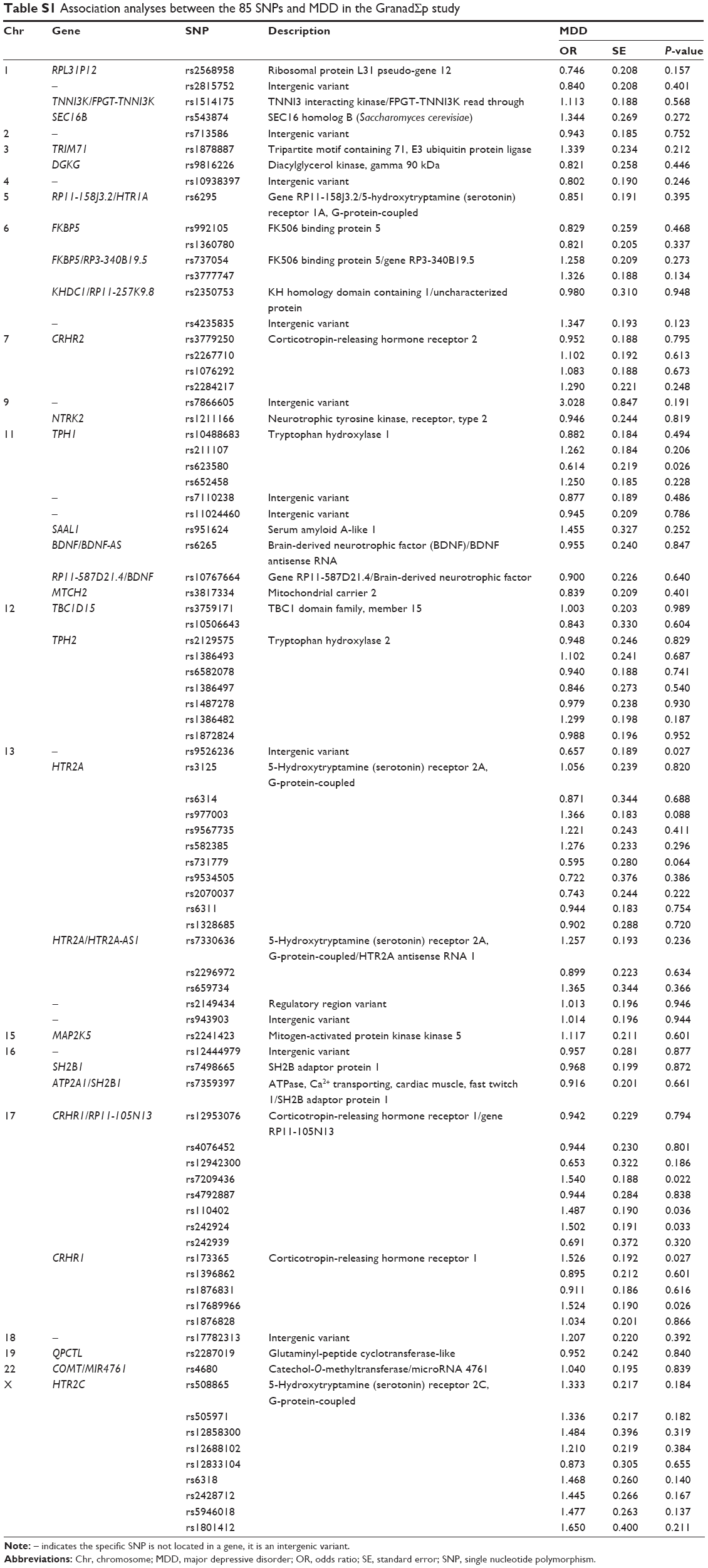
After quality control procedures, 85 SNPs and 567 individuals were finally included in the analyses. Detailed information about those SNPs, such as location, function, or possible allelic variants, is given in Table S1.
Statistical analysis
The Hardy–Weinberg equilibrium was checked in the entire sample, and both in depressed individuals and controls, by using Plink (http://pngu.mgh.harvard.edu/~purcell/plink/).19
The same statistical package was also used to test the association between MDD and the SNP data assuming an additive genetic model.
The number of effective-independent tests performed was calculated with the single nucleotide polymorphism spectral decomposition (SNPSpD) method. This method corrects for nonindependence of SNPs in linkage disequilibrium (LD) with each other.20 Under this method, the effective number of independent marker loci was 61, and the experiment-wide significance threshold required to keep type I error rate at 5% was 8×10−3.
We further estimated the haplotype frequencies with the standard expectation–maximization algorithm and performed simple tests of association based on the distribution of probabilistically inferred set of haplotypes in depressed individuals and controls.
Haplotype-specific odds ratio (OR) and 95% CI were calculated. Statistical significance was set at P<0.05 for haplotype association analysis.
Statistical power was calculated by using the software QUANTO v.1.2.4 (http://biostats.usc.edu/Quanto.html). QUANTO allowed us to calculate both the effect size we could detect (OR ranging from 2 to 5) and the power we had (ranging from 50% to 80% with a P<8×10−3) taking into account: i) our sample size (65 cases and 505 controls); ii) the prevalence of the illness (15%); iii) the frequency of the risk alleles (ranging from 0.10 to 0.50), and iv) an additive genetic model.
Results
The sample
Out of 1,176 community-based individuals who were approached and invited to take part in this study, 711 participants (305 men and 406 women; mean age ± SD: 49.7±16.7) provided written informed consent and donated a biological sample. The final study sample included 567 individuals (Figure 1), 67 (11.8%) participants fulfilled criteria for MDD (25 men, 42 women; mean age ± SD: 51.25±14.64), and 500 were controls (230 men, 270 women; mean age ± SD: 49.34±17.05). The control group did not have diagnosis of any Axis I-DSM-IV psychiatric disorder. Sociodemographic and clinical characteristics of the sample are summarized in Table 1. In brief, 312 (55%) were women; mean age was 49.6 years (SD=16.8) ranging from 19 to 83; more than half the participants lived in urban areas; most were educated at least to secondary school level, were in some sort of stable relationship, and were unemployed (Table 1).
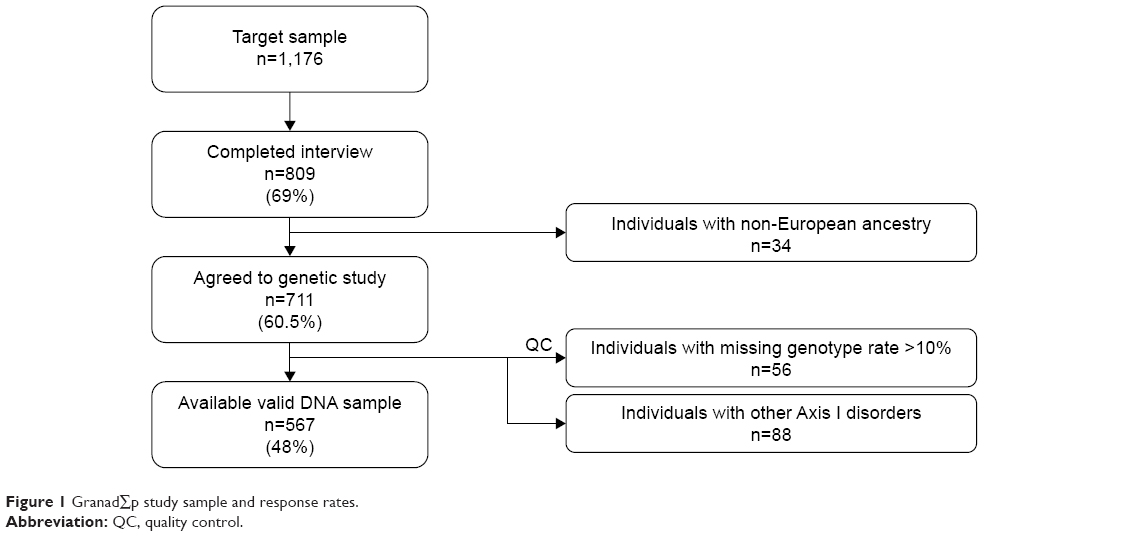 | Figure 1 GranadΣp study sample and response rates. |
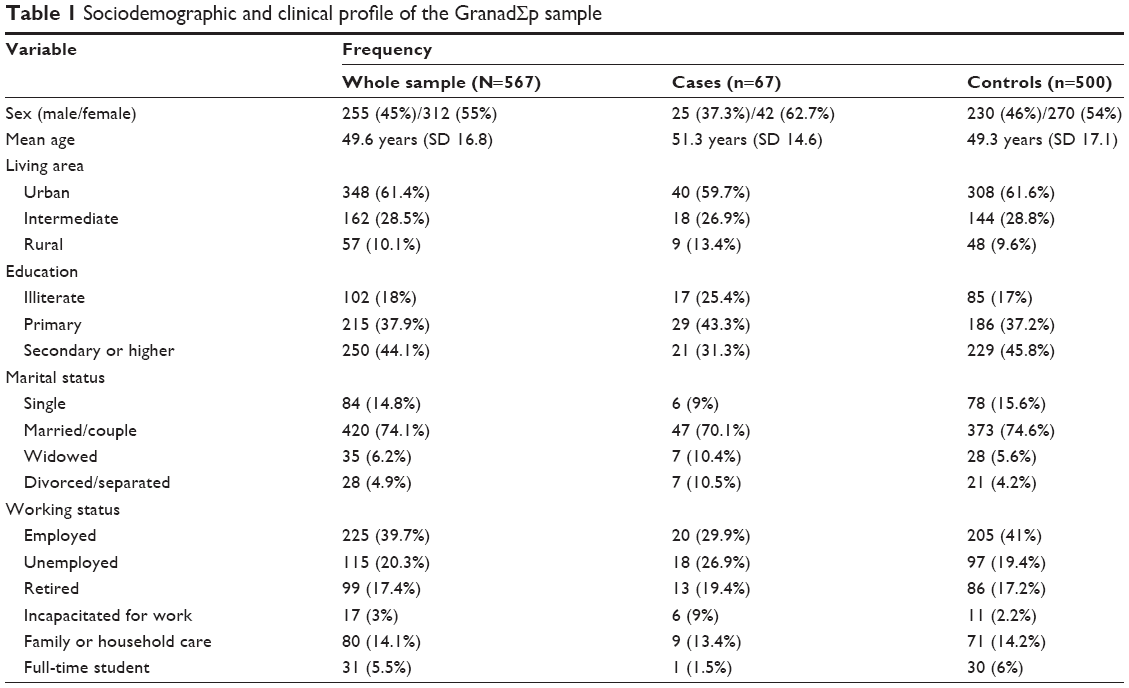 | Table 1 Sociodemographic and clinical profile of the GranadΣp sample |
Participants who agreed to take part in the genetic study did not vary significantly from those who refused to give a genetic sample in terms of sex (female 57.1% vs 54.7%, χ2=0.14, P=0.71), mean age (49.72 vs 52.09 years, Student’s t=0.57, P=0.45), living area (χ2=3.87, P=0.14), education (χ2=7.64, P=0.27), marital status (χ2=2.33, P=0.68), working status (χ2=4.10, P=0.54), or prevalence of DSM-IV MDD (10.5% vs 9.4%, χ2=0.09, P=0.77).
Genetic association analysis
Genotype frequencies were found to be in Hardy–Weinberg equilibrium in the whole sample, and both in cases and controls (P>0.05 in all cases).
None of the 85 SNPs were significantly associated with MDD after multiple testing correction (P>8×10−3 in all comparisons). However, one SNP (rs623580) located in the tryptophan hydroxylase 1 gene (TPH1; chromosome 11), one intergenic variant (rs9526236) between the esterase D gene (ESD) and the 5-hydroxytryptamine receptor 2A gene (HTR2A; chromosome 13), and five polymorphisms (rs17689966, rs173365, rs7209436, rs110402, and rs242924) located in the corticotropin-releasing hormone receptor 1 gene (CRHR1; chromosome 17), all showed suggestive trends for association with MDD, reaching traditionally conventional levels of statistical significance (P<0.05).
Detailed association results for these seven SNPs are shown in Table 2. Figure 2 and Table S1 show the association results of the 85 SNPs included in the analyses.
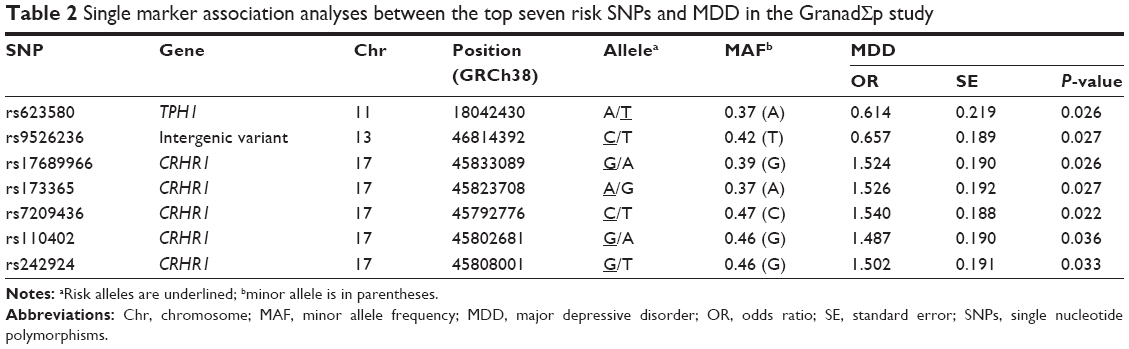 | Table 2 Single marker association analyses between the top seven risk SNPs and MDD in the GranadΣp study |
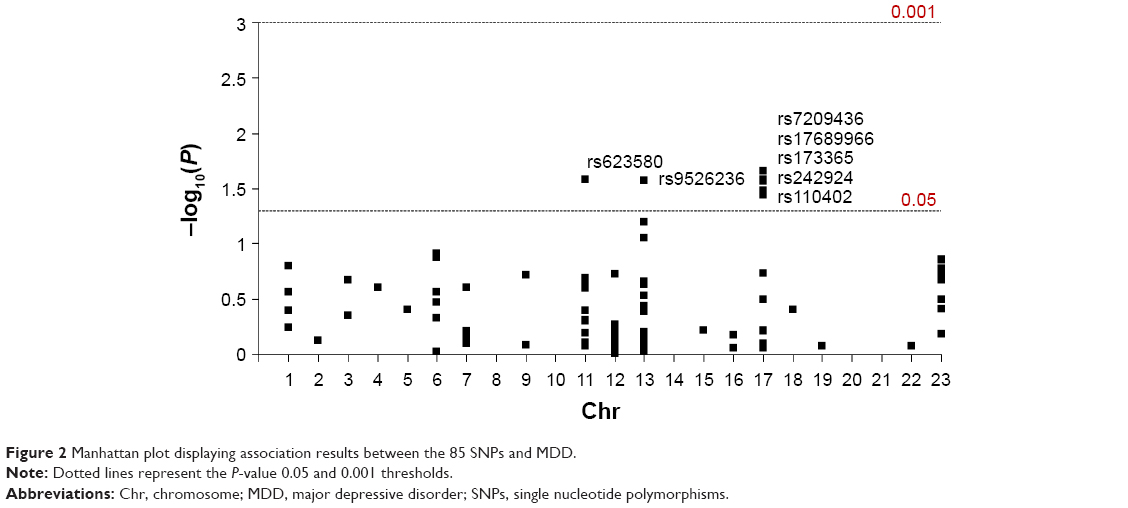 | Figure 2 Manhattan plot displaying association results between the 85 SNPs and MDD. |
Haplotype analysis
A five-marker haplotype analysis was performed including the five CRHR1 SNPs univariantly associated with MDD. Table 3 shows haplotype frequencies both in cases and controls. Among all the possible haplotype combinations, three of them accounted for more than 96% of the total variability observed both in depressive cases and controls. However, these haplotypes were not equally distributed in both groups (χ2=8.70, df=3, P=0.034). Particularly, the haplotype TATGA (rs7209436–rs110402–rs242924–rs173365–rs17689966) was found to be over-represented in MDD cases (42%) when compared with controls (30%) (OR=1.68; 95% CI: 1.16–2.43; P=0.005) (Table 3).
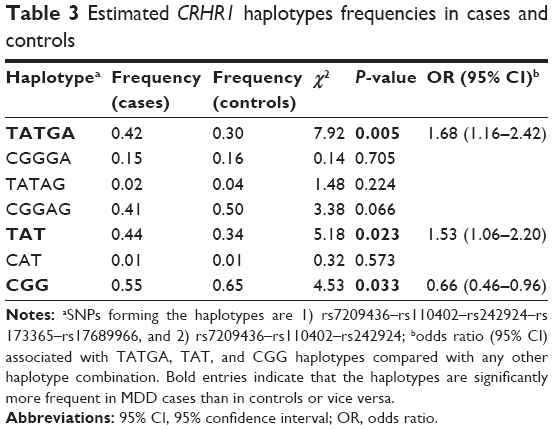 | Table 3 Estimated CRHR1 haplotypes frequencies in cases and controls |
A three-marker haplotype analysis was also performed including only the three SNPs that constitute the known CRHR1 TAT haplotype (which involves the following SNPs: rs7209436–rs110402–rs242924; Table 3). Such a haplotype combination (inserted in the five-SNPs haplotype described) has previously been described as a risk haplotype for depression in other studies.21–24
In our sample, the haplotype TAT was also more frequent in MDD cases (44%) than in controls (34%) (OR=1.53; 95% CI: 1.06–2.20; P=0.023), whereas the haplotype CGG was significantly more frequent in controls (65%) than in MDD cases (44%) (OR=0.66; 95% CI: 0.46–0.96; P=0.028).
Discussion
Despite significant advances in the search of the genetic basis of MDD, knowledge of the specific genes involved, genetic mechanisms, and physiological intermediates underlying the origin of such illness remains still very limited. Candidate gene studies, mostly focused on genes involved in neurotransmitter circuits or in reactions to stress, have yielded some suggestive results, although not conclusive enough. One of the reasons could be the polygenic nature of the illness and the hypothetically large number of loci involved, each of small effect. Moreover, as reported in Maier et al,25 MDD seems to be less homogeneous across populations than other psychiatric disorders such as schizophrenia and bipolar disorder. Replication studies in independent samples are absolutely necessary to test the robustness of such association reports.
In this study, we aimed to replicate previously reported associations between candidate genes and MDD26–29 in a sample of the general adult population of Andalusia (South of Spain). Although none of the associations tested were statistically significant after multiple testing correction (P>8×10−3), seven SNPs in three candidate genes (TPH1, HTR2A, and CRHR1) showed trends toward association (P<0.05), and one haplotype combination at CRHR1 was found to increase significantly the risk for MDD in our sample.
Association of two key serotonergic pathway genes (TPH1 and HTR2A) with MDD
TPH1 and HTR2A are key genes in the serotonergic neurotransmission. Disturbances in the serotonin (5-hydroxytryptamine, 5-HT) system constitute the neurobiological abnormality most extensively studied and consistently associated with MDD.30,31 The neurotransmitter serotonin modulates various functions related to homeostasis and responses to the environment, which in turn are linked to MDD. In addition, most antidepressants have a direct or indirect influence on serotonergic activity.32,33 Several lines of evidence suggest that abnormalities in the functioning of the serotonergic system are present in psychiatric conditions such as depression, schizophrenia, and obsessive compulsive disorders, as well as suicide and aggression.34
TPH1 gene encodes a tryptophan hydroxylase (TPH) isoform, a rate-limiting enzyme involved in the synthesis of neurotransmitter serotonin.32,35 Although TPH gene sequence variants and multiple psychiatric disorders have been associated over time, most mutations are found in noncoding regions of the gene, and limited information about their functional consequences is available. The administration of tryptophan and subsequent stimulation of serotonin production has an antidepressant effect, whereas the inhibition of TPH may precipitate depression.34 In 2002, Kim et al found that TPH expression is upregulated by chronic treatment with selective serotonin reuptake inhibitors, which provide an additional link between the antidepressant effect and TPH activity.36
The rs623580 (3804T/A) is an upstream genetic variant located in a regulatory region within the 5′-UTR of the TPH1 gene at chromosome 11.37 Previous studies involving this polymorphism have reported the negative results with affective disorders37,38 and suicide-related behavior.39 However, Kwak et al40 in a GWAS of 8,842 individuals found that this polymorphism was associated with body mass index, a measure of obesity many times related to MDD.41–44
There is a large amount of data implicating the serotonin system in the pathophysiology of affective disorders, but much of the attention is given specifically to genes coding for serotonin receptors and transporters.32,26 Moreover, almost every compound ever synthesized in order to inhibit serotonin reuptake has been proved to be a clinically effective antidepressant.45 The HTR2A is particularly relevant in the field of biological psychiatry due to its role as an important target for psychotropic drugs and its altered expression in several neuropsychiatric disorders such as MDD and schizophrenia.26,46,47 HTR2A gene in chromosome 13 is implicated in the regulation of serotonergic neurotransmission48 and the hypothalamic–pituitary–adrenal (HPA) axis.49,50 HTR2A has been extensively studied in genetic association studies of many psychiatric conditions, but the results are inconclusive and do not allow us to draw any definite conclusion about the potential implication of the HTR2A gene in MDD.51
In our study, the rs9526236 polymorphism (HTR2A) showed a trend for association with MDD. Although it has not been investigated in previous studies, its location upstream of the promoter region of HTR2A gene and its potential functionality makes it a good candidate variant to be further investigated in future studies given that both MDD and some antidepressants effects are linked to functionality of 5HT2A receptors.
Association between CRHR1 and MDD
CRHR1 encodes a G-protein-coupled receptor that binds neuropeptides of the corticotropin-releasing hormone (CRH) family that are major regulators of the HPA pathway.52 The encoded protein is essential for the activation of signal transduction pathways that regulate diverse physiological processes including stress, reproduction, immune response, and obesity.53
In response to stressful events, this receptor modifies the extent and duration of the response mediating the action of CRH on the pituitary gland to secrete corticotropin into the bloodstream. Corticotropin stimulates the production of cortisol in the adrenal cortex.24 According to the hypothalamic–pituitary–cortisol hypothesis of depression, abnormalities in the cortisol response to stress may underlie depression.45
In this study, we have found five SNPs (rs7209436, rs110402, rs242924, rs173365, and rs17689966) within CRHR1 gene suggestive of being associated with MDD. The haplotype analysis (including these five CRHR1 markers) revealed significant differences in the distribution of haplotype combinations between cases and controls. Particularly, TATGA haplotype (rs7209436–rs110402–rs242924–rs173365–rs17689966) was found in our sample to be significantly more frequent in cases with MDD than in controls (42% vs 30%, P=0.006).
Several SNPs at CRHR1 locus have been associated with the origin of MDD in previous studies.54,55 Some authors suggest that variation at CRHR1 could modulate reactivity to stress, and that altered CRHR1 function would be associated with stress-related psychopathology, particularly anxiety, and depressive disorders.56
CRHR1 haplotypes have also been associated with MDD,54,55 as well as with MDD mediated by childhood trauma21,24 and clinical response to antidepressant treatment.54,57 Our results are in agreement with these previous reports and, although our power is limited by our sample size, our data are in congruence with the pivotal neurobiological role that CRHR1 seems to play in the regulation of emotions. No doubt, CRHR1 is a highly suggestive candidate gene for MDD which would deserve further attention in future analysis.
Taking all together, our results are in agreement with previous neurochemical findings reporting disturbances at both HPA and serotonergic systems as highly likely implicated in MDD. Although a full comprehension of the nature of the relationships existing between both (HPA and serotonergic) systems is yet needed, it is well known that the HPA axis functioning can be modulated by various neuronal signaling molecules, including serotonin58 and, conversely, serotonergic activity can be modulated by HPA axis (ie, CNS concentrations of serotonin can decrease after a stimulation of the tryptophan metabolism induced by glucocorticoids).59 Since such relationship exists, we could think that TPH1, HTR2A, and CRHR1 genes work synergistically and they jointly contribute to confer a genetic risk profile for MDD which may differ according to the specific allele combinations carried by each subject. This hypothesis has not been tested in our study as our sample has a limited size but, in future studies, it would be desirable to analyze the possible joint effect of TPH1, HTR2A, and CRHR1 genes on MDD.
Strengths and limitations
One of the main strengths of our study is its design, based on a candidate gene approach. Candidate gene studies are relatively low-cost and quick to perform, are focused on previous knowledge related to the genes and mechanisms underlying the etiology of the disease, and involve a relatively small number of statistical tests in comparison with traditional GWAS, so that the significance threshold can be less stringent.60 Another strength of our study is that the sample is representative of the general population and considerably well characterized for clinical, psychological, sociodemographic, life style, and environmental variables. No potential problems of population stratification are expected as the sample comes from a homogeneous ethnic background, being all individuals Caucasian of Spanish ancestry. The main limitation of our study is its sample size which has a limited power to detect small and even moderate effects, as the power calculation performed in the statistics section reveals. A reduced number of candidate SNPs have also been investigated for this study. However, we prioritized theory-driven replication of previous associations between key genes and MDD, other genes from putatively important pathways (genes already known or even novel genes identified in recent GWAS) might not have been included in the analysis.
Conclusion
In conclusion, our results support that the HPA and serotonergic neurotransmission variability are both likely to be involved in the genetic susceptibility for MDD. This is in agreement with neurochemical findings which report disturbances at both HPA and serotonergic systems as highly likely implicated in MDD, although a full comprehension of the nature of such relationships is yet needed. Future studies including larger samples will be needed for further validation and replication of the present findings.
Acknowledgments
This work was mostly funded by an Andalusian Health System Health Council grant (PI0322/2009) and partially by Astra-Zeneca in agreement with CIBERSAM. It was also supported by a PhD grant from the Spanish Ministry of Education (AP2010-3563), and by the Andalusian Council of Innovation (CTS-6682).
Author contributions
All authors have been involved in drafting the manuscript or revising it critically for important intellectual content, and have approved the final version of the manuscript to be published. Ana Ching-López performed the genotyping work, analyzed data and wrote the initial draft of the manuscript. Jorge Cervilla originally conceived the study, acted as principal investigator, and director of the study as well as helped in training the interviewers, analyzed data, and writing the manuscript. Margarita Rivera participated in the analysis of genetic data and writing the manuscript. Esther Molina coordinated the collection of biological samples, extracted DNA, and prepared work samples as well as supervised genotyping and analyzed part of the genetic data. Kathryn McKenney participated in the study design, interviewers training, data collection, and data management. Isabel Ruiz actively participated in the study design, data collection, and in the final data analysis. Miguel Rodríguez-Barranco actively participated in the study design, data collection, and in the final data analysis. Blanca Gutiérrez conceived, coordinated, and supervised the genetic GranadΣp study as well as actively participated in analyzing data.
Disclosure
The authors report no conflicts of interest in this work.
References
Demyttenaere K, Bruffaerts R, Posada-Villa J, et al. Prevalence, severity, and unmet need for treatment of mental disorders in the World Health Organization World Mental Health Surveys. JAMA. 2004;291(21):2581–2590. | ||
Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095–3105. | ||
World Health Organization: The Global Burden of Disease: 2004 Update. Geneva: WHO Press; 2008:146. | ||
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157(10):1552–1562. | ||
Kendler KS, Gatz M, Gardner CO, Pedersen NL. A Swedish national twin study of lifetime major depression. Am J Psychiatry. 2006;163(1):109–114. | ||
Kendler KS, Gatz M, Gardner CO, Pedersen NL. Personality and major depression: a Swedish longitudinal, population-based twin study. Arch Gen Psychiatry. 2006;63(10):1113–1120. | ||
Collins PY, Patel V, Joestl SS, et al. Grand challenges in global mental health. Nature. 2011;475(7354):27–30. | ||
Flint J, Kendler KS. The genetics of major depression. Neuron. 2014;81(3):484–503. | ||
Cervilla J, Ruiz I, Rodríguez-Barranco M, et al. Study protocol and methods in the Andalusian epidemiological mental health study (PISMA-ep study). Rev Psiquiatr Salud Ment. 2015. in Press. | ||
Ocaña-Riola R, Sánchez-Cantalejo C. Rurality index for small areas in Spain. Soc Indic Res. 2005;73(2):247–266. | ||
Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(Suppl. 20):22–33;quiz 34–57. | ||
Sullivan PF, de Geus EJ, Willemsen G, et al. Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol Psychiatry. 2009;14:359–375. | ||
Lewis CM, Ng MY, Butler AW, et al. Genome-wide association study of major recurrent depression in the UK population. Am J Psychiatry. 2010;167:949–957. | ||
Muglia P, Tozzi F, Galwey NW, et al. Genome-wide association study of recurrent major depressive disorder in two European case-control cohorts. Mol Psychiatry. 2010;15:589–601. | ||
Rietschel M, Mattheisen M, Frank J, et al. Genome-wide association-, replication-, and neuroimaging study implicates HOMER1 in the etiology of major depression. Biol Psychiatry. 2010;68:578–585. | ||
Kohli MA, Lucae S, Saemann PG, et al. The neuronal transporter gene SLC6A15 confers risk to major depression. Neuron. 2011;70: 252–265. | ||
Shi J, Potash JB, Knowles JA, et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol Psychiatry. 2011;16:193–201. | ||
Shyn SI, Shi J, Kraft JB, et al. Novel loci for major depression identified by genome-wide association study of sequenced treatment alternatives to relieve depression and meta-analysis of three studies. Mol Psychiatry. 2011;16:202–215. | ||
Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. | ||
Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74(4):765–769. | ||
Bradley RG, Binder EB, Epstein MP, et al. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch Gen Psychiatry. 2008;65(2):190–200. | ||
Kranzler HR, Feinn R, Nelson EC, et al. A CRHR1 haplotype moderates the effect of adverse childhood experiences on lifetime risk of major depressive episode in African-American women. Am J Med Genet B Neuropsychiatr Genet. 2011;156b(8):960–968. | ||
Nugent NR, Tyrka AR, Carpenter LL, Price LH. Gene–environment interactions: early life stress and risk for depressive and anxiety disorders. Psychopharmacology. 2011;214(1):175–196. | ||
Polanczyk G, Caspi A, Williams B, et al. Protective effect of CRHR1 gene variants on the development of adult depression following childhood maltreatment: replication and extension. Arch Gen Psychiatry. 2009;66(9):978–985. | ||
Maier R, Moser G, Chen G, Ripke S, Cross-disorder working group of the Psychiatric Genomics Consortium. Joint analysis of psychiatric disorder increases accuracy risk prediction for schizophrenia, bipolar disorder, and major depressive disorder. Am J Hum Genet. 2015;96:283–294. | ||
Anguelova M, Benkelfat C, Turecki G. A systematic review of association studies investigating genes coding for serotonin receptors and the serotonin transporter: I. Affective disorders. Mol Psychiatry. 2003;8(6):574–591. | ||
López-León S, Janssens AC, González-Zuloeta Ladd AM, et al. Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatry. 2008;13:772–785. | ||
Kishi T, Tsunoka T, Ikeda M, et al. Serotonin 1A receptor gene and major depressive disorder: an association study and meta-analysis. Hum Genet. 2009;54:629–633. | ||
Fukuo Y, Kishi T, Yoshimura R, et al. Serotonin 6 receptor gene and mood disorders: case–control study and meta-analysis. Neurosci Res. 2010;67:250–255. | ||
Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 2008;31(9): 464–468. | ||
Lopez JF, Chalmers DT, Little KY, Watson SJ. A.E. Bennett Research Award. Regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol Psychiatry. 1998;43(8):547–573. | ||
Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry. 2010;15(5):473–500. | ||
Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med. 2009;60:355–366. | ||
Wang L, Erlandsen H, Haavik J, Knappskog PM, Stevens RC. Three-dimensional structure of human tryptophan hydroxylase and its implications for the biosynthesis of the neurotransmitters serotonin and melatonin. Biochemistry. 2002;41(42):12569–12574. | ||
Andre K, Kampman O, Viikki M, et al. TPH1 A218C polymorphism and temperament in major depression. BMC Psychiatry. 2013;13:118. | ||
Kim SW, Park SY, Hwang O. Up-regulation of tryptophan hydroxylase expression and serotonin synthesis by sertraline. Mol Pharmacol. 2002;61(4):778–785. | ||
Lai TJ, Wu CY, Tsai HW, Lin YM, Sun HS. Polymorphism screening and haplotype analysis of the tryptophan hydroxylase gene (TPH1) and association with bipolar affective disorder in Taiwan. BMC Med Genet. 2005;6:14. | ||
Shi J, Badner JA, Hattori E, et al. Neurotransmission and bipolar disorder: a systematic family-based association study. Am J Med Genet B Neuropsychiatr Genet. 2008;147b(7):1270–1277. | ||
Brent D, Melhem N, Ferrell R, et al. Association of FKBP5 polymorphisms with suicidal events in the Treatment of Resistant Depression in Adolescents (TORDIA) study. Am J Psychiatry. 2010;167(2):190–197. | ||
Kwak SH, Park BL, Kim H, et al. Association of variations in TPH1 and HTR2B with gestational weight gain and measures of obesity. Obesity. 2012;20(1):233–238. | ||
Opel N, Redlich R, Grotegerd D, et al. Obesity and major depression: body-mass index (BMI) is associated with a severe course of disease and specific neurostructural alterations. Psychoneuroendocrinology. 2015;51:219–226. | ||
Luppino FS, de Wit LM, Bouvy PF, et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry. 2010;67(3):220–229. | ||
Hung CF, Rivera M, Craddock N, et al. Relationship between obesity and the risk of clinically significant depression: Mendelian randomisation study. Br J Psychiatry. 2014;205(1):24–28. | ||
de Wit L, Luppino F, van Straten A, Penninx B, Zitman F, Cuijpers P. Depression and obesity: a meta-analysis of community-based studies. Psychiatry Res. 2010;178(2):230–235. | ||
Belmaker RH, Agam G. Major depressive disorder. N Engl J Med. 2008;358:55–68. | ||
Norton N, Owen MJ. HTR2A: association and expression studies in neuropsychiatric genetics. Ann Med. 2005;37(2):121–129. | ||
Bray NJ, Buckland PR, Hall H, Owen MJ, O’Donovan MC. The serotonin-2A receptor gene locus does not contain common polymorphism affecting mRNA levels in adult brain. Mol Psychiatry. 2004;9(1):109–114. | ||
Falkenberg VR, Gurbaxani BM, Unger ER, Rajeevan MS. Functional genomics of serotonin receptor 2A (HTR2A): interaction of polymorphism, methylation, expression and disease association. Neuromol Med. 2011;13(1):66–76. | ||
Lanfumey L, Mongeau R, Cohen-Salmon C, Hamon M. Corticosteroid–serotonin interactions in the neurobiological mechanisms of stress-related disorders. Neurosci Biobehav Rev. 2008;32(6):1174–1184. | ||
Porter RJ, Gallagher P, Watson S, Young AH. Corticosteroid–serotonin interactions in depression: a review of the human evidence. Psychopharmacology. 2004;173(1–2):1–17. | ||
Zhao X, Sun L, Sun YH, et al. Association of HTR2A T102C and A-1438G polymorphisms with susceptibility to major depressive disorder: a meta-analysis. Neurol Sci. 2014;35(12):1857–1866. | ||
Schatzberg AF, Keller J, Tennakoon L, et al. HPA axis genetic variation, cortisol and psychosis in major depression. Mol Psychiatry. 2014;19(2):220–227. | ||
Binder EB, Nemeroff CB. The CRF system, stress, depression and anxiety – insights from human genetic studies. Mol Psychiatry. 2010;15:574–588. | ||
Liu Z, Zhu F, Wang G, et al. Association study of corticotropin-releasing hormone receptor1 gene polymorphisms and antidepressant response in major depressive disorders. Neurosci Lett. 2007;414(2):155–158. | ||
Ishitobi Y, Nakayama S, Yamaguchi K, et al. Association of CRHR1 and CRHR2 with major depressive disorder and panic disorder in a Japanese population. Am J Med Genet B Neuropsychiatr Genet. 2012;159b(4):429–436. | ||
Rogers J, Raveendran M, Fawcett GL, et al. CRHR1 genotypes, neural circuits and the diathesis for anxiety and depression. Mol Psychiatry. 2013;18(6):700–707. | ||
Licinio J, O’Kirwan F, Irizarry K, et al. Association of a corticotropin-releasing hormone receptor 1 haplotype and antidepressant treatment response in Mexican-Americans. Mol Psychiatry. 2004;9(12):1075–1082. | ||
Leonard BE, Myint A. The psychoneuroimmunology of depression. Hum Psychopharmacol. 2009;24(3):165–175. | ||
Green AR, Sourkes TL, Young SN. Liver and brain tryptophan metabolism following hydrocortisone administration to rats and gerbils. Br J Pharmacol. 1975;53(2):287–292. | ||
Patnala R, Clements J, Batra J. Candidate gene association studies: a comprehensive guide to useful in silico tools. BMC Genet. 2013;14:39. |
Supplementary material
 | Table S1 Association analyses between the 85 SNPs and MDD in the GranadΣp study |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.