Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 19
Epidemiological Features and Pathogenic Mechanisms of Steatocystoma Multiplex: A Systematic Review
Authors Sun J ![]() , Gu J
, Gu J ![]() , Wang Y, Liu Z, Ma C
, Wang Y, Liu Z, Ma C
Received 19 May 2026
Accepted for publication 8 July 2026
Published 14 July 2026 Volume 2026:19 625870
DOI https://doi.org/10.2147/CCID.S625870
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monica K. Li
Jiachen Sun, Jingyan Gu, Yuan Wang, Zilian Liu, Chuan Ma
Department of Dermatology, Peking University Third Hospital, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chuan Ma; Zilian Liu, Email [email protected]; [email protected]
Background: Steatocystoma multiplex (SM) is a rare hereditary skin disorder arising from the pilosebaceous unit, clinically characterized by multiple asymptomatic sebum-containing cysts predominantly distributed on the chest, axillae, and upper arms. However, the epidemiological features and pathogenic mechanisms of SM remain poorly characterized.
Methods: A systematic search of PubMed, Embase, and CNKI was conducted for studies published between January 2003 and December 2023. Case reports and observational studies reporting individual-level clinical data on confirmed SM patients were eligible. Demographic, clinical, and genetic data were extracted and analyzed using appropriate statistical tests.
Results: A total of 145 cases from 82 publications were included. SM showed no clear geographical or ethnic predilection. The median age at onset was 23 years and the median age at diagnosis was 31 years, reflecting a diagnostic delay of approximately 8 years. Familial cases (26.2%) had a significantly earlier age at onset (13.5 vs. 30.0 years, P < 0.001) and diagnosis (22.0 vs. 35.0 years, P < 0.001), and higher rates of infected lesions and nail dystrophy. Hidradenitis suppurativa was more prevalent in females (12.5% vs. 1.5%, P = 0.019), and sex-based differences in lesion distribution were observed. KRT17 was the most frequently mutated gene, with Arg94 as the predominant mutation site. KRT17 mutations are thought to impair cytoskeletal integrity within the pilosebaceous unit, while hormonal factors - particularly androgen-driven sebaceous gland activation during puberty - are implicated as additional modulators of disease onset and lesion distribution.
Conclusion: This review characterizes the epidemiological and clinical features of SM, demonstrating meaningful differences between familial and sporadic cases and between sexes. Genotype–phenotype discordance and KRT17-negative cases suggest that modifier genes and hormonal factors contribute to phenotypic determination. The substantial diagnostic delay highlights the need for improved clinical awareness, and mutation-targeted therapeutic strategies warrant further investigation.
Keywords: steatocystoma multiplex, systematic review, epidemiological features, pathogenic mechanisms
Introduction
Steatocystoma multiplex (SM) is a rare hereditary skin disorder arising from the pilosebaceous unit.1–3 Clinically, the cysts typically range from 2 to 20 mm in diameter, presenting as soft, yellowish, non-tender nodules with a smooth surface that may occasionally discharge an oily or creamy fluid upon puncture. Lesions most commonly involve the chest, axillae, upper arms, and trunk, though facial, scrotal, and acral involvement has also been reported. The condition can manifest across a wide range of ages, from infancy to late adulthood, and although benign in nature, exerts a considerable impact on patients’ appearance and psychosocial well-being. Differential diagnosis of SM includes several conditions with overlapping clinical features, such as epidermoid and pilar cysts, lipomas, eruptive vellus hair cysts, and hidradenitis suppurativa, and distinction relies primarily on the characteristic sebaceous cyst content and histopathological findings. However, clinical differentiation of these entities can be challenging, and ultrasonography has emerged as a valuable non-invasive adjunct tool, enabling characterization of cyst morphology, internal echogenicity, and vascularity patterns that aid in distinguishing SM from mimicking conditions.4 Although most cases are sporadic, SM may follow an autosomal dominant inheritance pattern with variable expressivity, and nonsynonymous mutations in the KRT17 gene have been identified as the principal genetic basis of familial disease.5,6
To date, the epidemiological features of SM, including its temporal and geographical distribution, remain poorly delineated. Likewise, the clinical characteristics and patterns of cutaneous involvement have yet to be systematically defined, particularly with respect to lesion distribution preferences, gender-related differences, and the distinct presentations between familial and sporadic cases. The molecular mechanisms underlying cystogenesis are likewise not fully elucidated.
Therefore, based on a systematic appraisal of the published literature, this review aims to: (1) characterize the epidemiological features and clinical patterns of SM, with emphasis on lesion distribution preferences, gender-related differences, and the distinctions between familial and sporadic presentations; (2) synthesize current evidence on the pathogenic mechanisms underlying SM.
Methods
Search Strategy
This systematic review was retrospectively registered in PROSPERO (Registration ID: CRD420261444736). A comprehensive literature search was conducted across three electronic databases (PubMed, Embase, and CNKI), to identify relevant studies published between January 2003 and December 2023. This 20-year search window was defined in accordance with the project initiation date of January 2024. The search strategy employed the following terms, combined using Boolean operators: “multiple steatocystoma”, “steatocystoma multiplex”, and “steatocystomatosis”. The final search was completed on January 10, 2024. To minimize linguistic bias, non-English publications were translated using Google Translate and included where eligibility criteria were met.
Eligibility Criteria
Studies were considered eligible for inclusion if they fulfilled the following criteria: (1) case reports, case series, or observational studies reporting individual-level clinical data on patients with SM; (2) patients with a confirmed diagnosis of SM established on the basis of clinical presentation and/or histopathological examination; and (3) studies providing, at minimum, basic demographic information (age and sex) together with clinical characteristics. Studies were excluded if they met any of the following criteria: (1) review articles, editorials, letters to the editor, or conference abstracts lacking original case-level data; (2) reports with insufficient clinical information precluding reliable data extraction; or (3) duplicate publications describing the same patient, in which case only the most comprehensive report was retained.
Study Selection and Data Extraction
Two investigators independently screened the titles and abstracts of all retrieved records, followed by full-text review of potentially eligible studies. Any discrepancies between reviewers were resolved through discussion and consensus, with arbitration by a third investigator when necessary. Data extraction was performed using a standardized, pre-piloted data extraction form, capturing the following variables where available: demographic characteristics, age at onset, age at diagnosis, family history, anatomical distribution of lesions, and genetic findings. After application of the eligibility criteria, the final dataset comprised 145 SM cases reported across 82 publications.
Statistical Analysis
All statistical analyses were performed using IBM SPSS Statistics (version 27.0; IBM, USA). The normality of continuous variables was assessed using the Kolmogorov–Smirnov test. Normally distributed variables were expressed as mean ± standard deviation and compared using the independent-samples t-test for two-group comparisons. Non-normally distributed variables were expressed as median (interquartile range, IQR: Q1–Q3) and compared using the Mann–Whitney U-test. Categorical variables were summarized as frequencies and percentages, and compared using the Pearson χ2-test or Fisher’s exact test. A two-tailed P value < 0.05 was considered statistically significant.
Results
Demographic and Epidemiological Characteristics of SM
Temporal and Geographical Distribution
To characterize the global epidemiological profile of SM, we analyzed 145 cases documented in the database over a 20-year period. The annual frequency of reported cases remained relatively stable throughout the study period, with the exception of 2012, when a single Italian dermatological cohort contributed 32 cases,7 rendering Italy the country with the highest number of reported SM cases over the two decades (Figure 1a). Among Asian countries, China, South Korea, and Japan followed with 20, 13, and 12 cases, respectively (Figure 1b). Overall, SM does not appear to exhibit a geographical or ethnic predilection, suggesting that genetic rather than environmental factors may be the primary driver of disease occurrence.
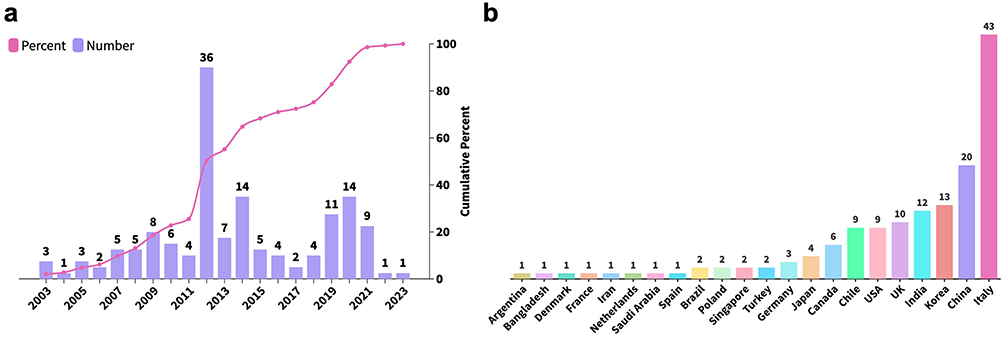 |
Figure 1 Temporal and geographic distribution of 145 SM Cases. (a) Temporal distribution curve for 145 SM patients, with bar graphs denoting the annual number of reported cases and an overlaid line graph depicting the cumulative percentage of SM cases over time. (b) Geographic distribution of SM patients. |
General Demographic Features
Detailed demographic information for all included cases is presented in Table S1. The age at onset spanned a wide range, from 1 to 81.5 years, with a median of 23 years. The age at diagnosis was similarly broad, ranging from 1 to 82 years, with a median of 31 years, suggesting a median diagnostic delay of approximately 8 years. Infected lesions were identified in 11 cases (7.6%), and nail dystrophy was documented in 8 cases (5.5%). With respect to comorbidities, hidradenitis suppurativa (HS) was the most frequently reported, affecting 10 patients, potentially reflecting shared pathological involvement of the folliculosebaceous unit.
Sex-Based Differences
Of the 145 cases, 65 were male and 72 were female (Table 1 and Table S1). Males demonstrated a marginally earlier median age at onset (20.0 years, IQR 14.5–35.5; Figure 2a) and age at diagnosis (28.0 years, IQR 22.0–43.0; Figure 2b), compared to females (26.0 years, IQR 17.5–40.5, and 37.0 years, IQR 25.0–49.8, respectively). However, neither difference reached statistical significance (P = 0.092 and P = 0.061, respectively). No significant sex-based differences were observed with respect to family history, infection, or nail dystrophy (P = 0.751, 0.443, and 0.721, respectively). HS was significantly more prevalent in female patients (9/72, 12.5%) than in male patients (1/65, 1.5%, P = 0.019). The overall anatomical distribution of lesions was broadly comparable between sexes (Figure 2c), with the notable exceptions of axillary involvement, which was more frequent in females (18.25% vs 7.62% in males), and whole-body involvement, which was more prevalent in males (6.67% vs 0.73% in females).
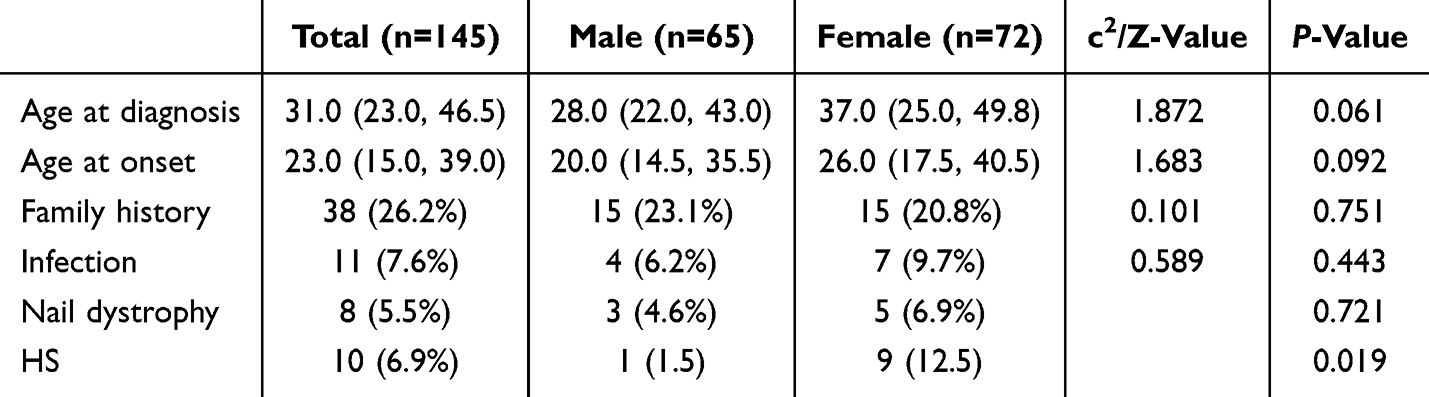 |
Table 1 Demographic Data and Gender Differences of 145 SM Patients |
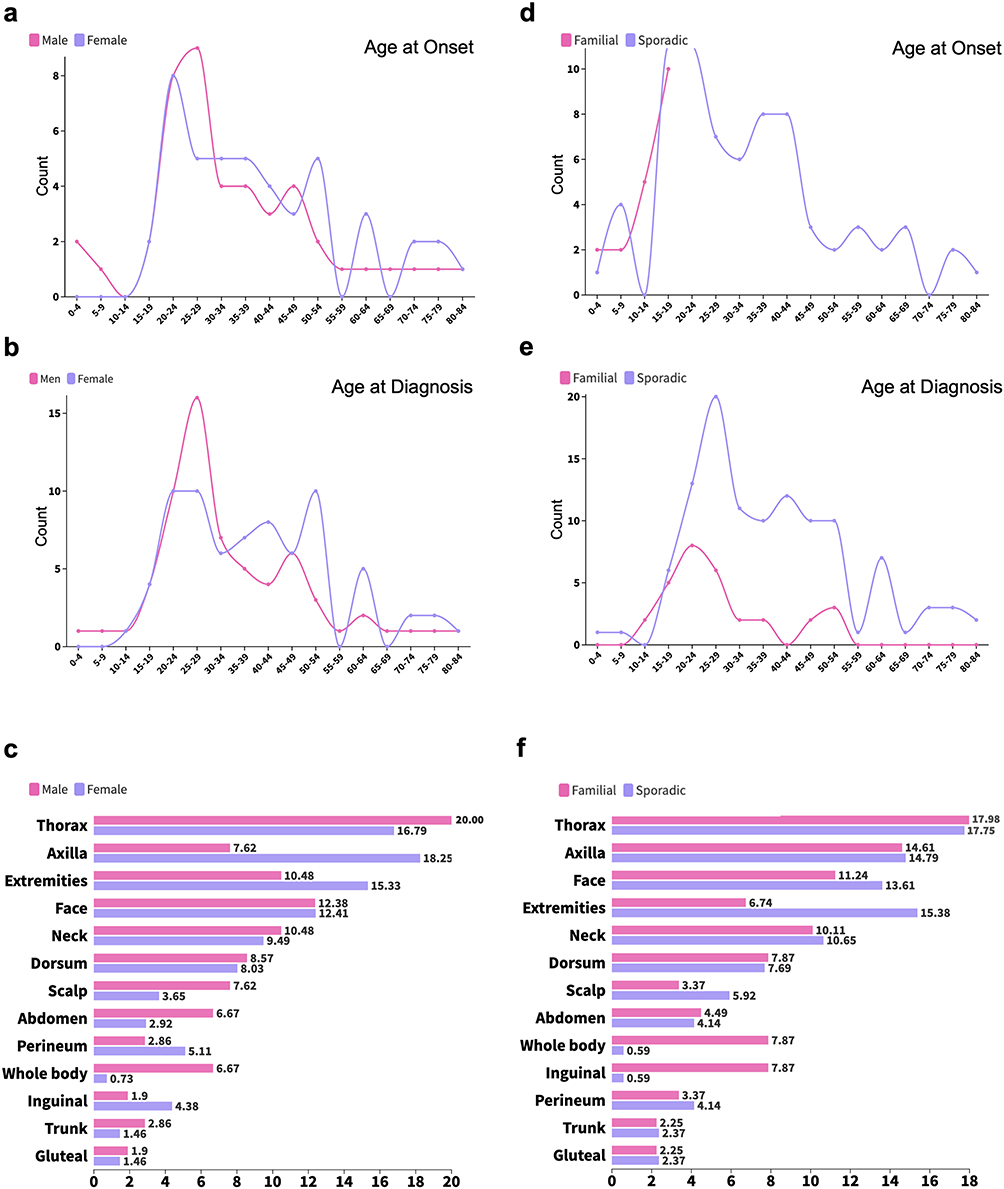 |
Figure 2 Clinical characteristics of SM patients stratified by sex and inheritance pattern. (a–c) illustrate sex-based differences in age at onset (a), age at diagnosis (b), and lesion distribution (c). (d–f) compare familial and sporadic SM patients with respect to age at onset (d), age at diagnosis (e), and lesion distribution patterns (f). |
Familial Versus Sporadic Presentations
Familial occurrence was identified in 38 cases (26.2%, Table 2). Consistent with the autosomal dominant inheritance pattern of SM, familial cases exhibited a significantly earlier median age at onset (13.5 years, IQR 7.5–15.8; Figure 2d) and age at diagnosis (22.0 years, IQR 17.0–30.5; Figure 2e) compared to sporadic cases (30.0 years, IQR 19.0–41.0, P < 0.001; and 35.0 years, IQR 26.0–49.0, P < 0.001, respectively). Sex distribution did not differ significantly between familial and sporadic cases (P = 0.751). Familial cases demonstrated a markedly higher incidence of infected lesions (26.3% vs 0.9%, P < 0.001) and nail dystrophy (13.2% vs 2.8%, P = 0.029). No significant difference in the prevalence of HS was observed between familial and sporadic cases (5.3% vs 7.5%, P = 1.000). With respect to anatomical distribution, familial cases showed greater inguinal and whole-body involvement (both 7.87% vs 0.59% in sporadic cases), whereas extremity involvement was less frequent in familial cases (6.74% vs 15.38%; Figure 2f).
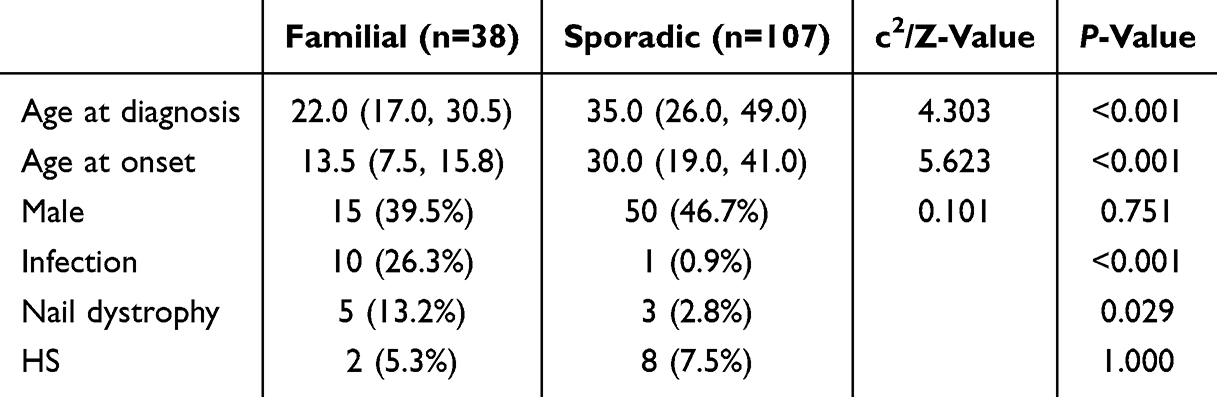 |
Table 2 Familial and Sporadic Differences Among 145 SM Patients |
Genetic Mutation Analysis in SM
Among 145 SM patients, 21 patients reported genetic mutations (Table S1). Genetic mutation data were not explicitly documented for the remaining patients, further limiting our understanding of the pathogenic mechanisms underlying SM. KRT17, a well-established pathogenic gene of SM, exhibited the highest mutation frequency, with 13 distinct mutation sites (Figure 3) detected in 19 patients. The most frequently mutated site is Arg94, with 4 patients exhibiting the p.Arg94Cys mutation,8–11 2 with p.Arg94His,12,13 and 1 with p.Arg94Gly.14 Three patients with KRT17 mutations also presented with pachyonychia congenita type 2 (PC-2), with mutations respectively located at p.Met88Thr,15 p.Asn92Ser,16 and p.Leu99Pro,17 all within the Coil 1A region of KRT17. These patients exhibited no discernible pattern in the distribution of their SM lesions. However, other mutations in the Coil 1A region of KRT17, including p.Asn92Thr,18 p.Arg94His,12,13 p.Arg94Cys,11 and p.Arg94Gly,14 have not been reported to be associated with PC-2. Nail dystrophy in SM patients is generally considered to be associated with PC-2, yet we identified a Japanese patient with nail dystrophy who did not have a KRT17 mutation but instead had a PKD1 mutation (c.9855_9856insAC), leading to autosomal dominant polycystic kidney disease.19 This patient also exhibited diffuse whole-body lesion distribution with infection. Additionally, a patient with neurofibromatosis type 1 (NF1), carrying the NF1 mutation c.730G>A, presented with symptoms of SM.20
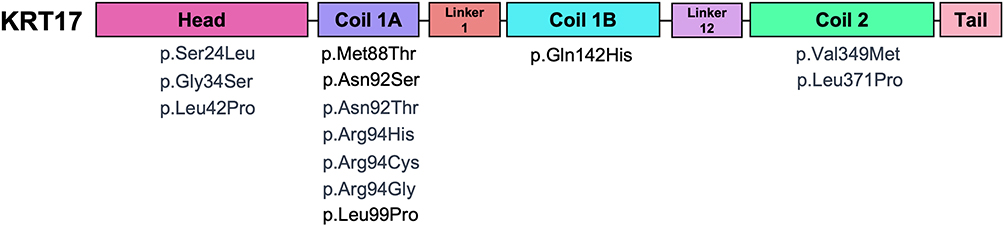 |
Figure 3 The spatial distribution of KRT17 mutations mapped onto corresponding functional domains. |
Pathogenesis
Cellular and Histopathological Origin
SM is fundamentally a disorder of the pilosebaceous unit, with cysts believed to originate from an abnormal lining of the sebaceous duct.21 Histologically, the cyst wall is composed of stratified squamous epithelium arranged in characteristic folded layers, with flattened sebaceous lobules embedded within the wall - a defining pathological feature that distinguishes SM from other cutaneous cysts, including epidermal inclusion cysts and trichilemmal cysts. The luminal contents consist of sebaceous material, further corroborating the sebaceous ductal origin of these lesions.22
The initial pathogenic event is thought to involve keratinous plug formation within the sebaceous duct, resulting in progressive sebum accumulation and cystic dilatation.23 In parallel, pluripotent ectodermal cells that retain embryonic capacity to form sebaceous structures may contribute to sustained cyst wall proliferation. The near-universal onset of SM during or around puberty is consistent with the marked augmentation of sebaceous gland activity driven by pubertal hormonal changes, a relationship further elaborated in Hormonal Influences and Sex-Based Pathogenic Differences.
Genetic Basis: KRT17 Mutations and Protein Dysfunction
KRT17 mutations represent the most frequently identified genetic alterations in SM. The KRT17 protein, in common with other members of the keratin family, is characterized by a central rod domain flanked by non-helical head (N-terminal) and tail (C-terminal) domains.24 The rod domain is organized into four helical coil segments (1A, 1B, 2A, and 2B) separated by non-helical linker regions, a hierarchical architecture that is indispensable for the formation of coiled-coil dimers and their subsequent assembly into intermediate filaments.25 Mutations within the coiled-coil domains may disrupt this assembly process, thereby compromising cytoskeletal integrity and impairing the structural homeostasis of the pilosebaceous unit.26,27 Notably, all three KRT17 mutations identified in the present cohort as associated with PC-2 were localized to the coil 1A region - a segment particularly critical for maintaining protein stability and function - suggesting that perturbations in this domain may confer susceptibility to both SM and the broader PC-2 phenotypic spectrum.28,29
Beyond its structural role, KRT17 functions as a multifunctional cytoskeletal regulator involved in keratinocyte proliferation and differentiation, modulation of inflammatory responses, and hair follicle cycling.28,30,31 Furthermore, KRT17 has been demonstrated to inhibit dermal fibroblast migration through an integrin α11-mediated signaling pathway, implicating it in the broader regulation of the cutaneous microenvironment.32 The considerable diversity of pathogenic mutation sites across the KRT17 gene poses a significant challenge to the development of targeted small-molecule inhibitors, as each variant may necessitate a distinct therapeutic approach.27
Familial vs Sporadic SM: Genotype–Phenotype Implications
The same KRT17 mutation may give rise to markedly divergent clinical phenotypes across individuals and families, manifesting as isolated SM, PC-2, or an overlapping phenotype encompassing features of both conditions. This genotype–phenotype discordance implies that KRT17 mutation alone is insufficient to fully determine the clinical outcome; modifier genes, hormonal milieu, and environmental factors are likely to act in concert to shape the final phenotype.33 The existence of sporadic cases, in which no identifiable KRT17 mutation is detected, further underscores the pathogenic heterogeneity of SM and suggests that additional, as yet uncharacterized genetic or epigenetic mechanisms may be operative.
Prior studies have not systematically quantified the demographic and clinical differences between familial and sporadic SM. In the present study, we addressed this gap through statistical analysis, demonstrating that familial SM is associated with a significantly earlier age at onset and age at diagnosis compared to sporadic cases. Moreover, familial SM exhibited distinct anatomical site preferences, with relatively less extremity involvement and a higher proportion of inguinal lesions compared to sporadic SM. The predilection for the inguinal region in familial cases may reflect the selective vulnerability of the apocrine gland-rich microenvironment in this anatomical territory to the functional consequences of KRT17 mutations, though the precise mechanistic basis for this site-specific distribution warrants further investigation.
Hormonal Influences and Sex-Based Pathogenic Differences
Hormonal factors, particularly the elevation of androgens during puberty, have been proposed to promote the onset and progression of SM, a hypothesis consistent with the well-established pubertal timing of disease emergence.2 Androgens are known to upregulate sebaceous gland activity and modulate the biology of androgen-sensitive apocrine glands; their surge during puberty may therefore lower the threshold for cyst formation in genetically predisposed individuals.34
Sex-specific differences in lesion distribution observed in the present cohort provide further indirect evidence for a hormonal contribution to SM pathogenesis. The axilla - an anatomical region richly endowed with androgen-sensitive apocrine glands - was the most frequently affected site among female patients (18.25% vs 7.62% in males), suggesting that androgen-driven apocrine gland activation may preferentially influence lesion localization in this sex. Conversely, whole-body involvement was more prevalent in males (6.67% vs 0.73% in females), a pattern that may reflect the broader distribution of androgen-sensitive sebaceous units in male skin. Furthermore, although the difference did not reach statistical significance, males demonstrated a marginally earlier median age at onset (20.0 years) compared to females (26.0 years), a trend that may be attributable to the earlier and more pronounced androgenic stimulation of sebaceous glands in males during puberty. Taken together, these observations suggest that hormonal factors - operating in a sex-specific manner - interact with the underlying genetic background, most notably KRT17 mutations, to modulate the timing and anatomical distribution of SM. However, the precise molecular interplay between sex hormone signaling and keratin dysfunction remains incompletely understood and represents an important avenue for future investigation.
Conclusion
This systematic review provides the most comprehensive epidemiological and clinical characterization of SM to date, revealing significant demographic distinctions between familial and sporadic presentations, sex-based differences in lesion distribution, and a substantial median diagnostic delay of approximately 8 years. At the pathogenic level, while KRT17 mutations remain the central genetic determinant, marked genotype–phenotype discordance and the existence of KRT17-negative cases implicate additional modifier genes, hormonal signaling, and epigenetic factors as co-determinants of phenotypic expression. The inherent limitations of retrospective case-based evidence call for prospective multicenter studies to further delineate SM’s natural history. Ultimately, the persistent challenge of disease recurrence and the absence of a definitive cure underscore the urgent need for mutation-targeted strategies - including in situ gene editing directed at pathogenic KRT17 variants — as the most promising path toward durable disease resolution.
Data Sharing Statement
The data that support the findings of this study are available from the Prof. Chuan Ma ([email protected]) upon reasonable request.
Consent for Publication
This manuscript has not been published and is not under consideration for publication elsewhere. The details of the manuscript can be published and all the authors providing consent have been shown the article contents to be published.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Beijing Natural Science Foundation Youth Program (Grant No. 7264350).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Shamloul G, Khachemoune A. An updated review of the sebaceous gland and its role in health and diseases Part 2: pathophysiological clinical disorders of sebaceous glands. Dermatol Ther. 2021;34(2):e14862. doi:10.1111/dth.14862
2. Shamloul G, Khachemoune A. An updated review of the sebaceous gland and its role in health and diseases Part 1: embryology, evolution, structure, and function of sebaceous glands. Dermatol Ther. 2021;34(1):e14695. doi:10.1111/dth.14695
3. Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139(3):475–9. doi:10.1046/j.1365-2133.1998.02413.x
4. Zussino M, Nazzaro G, Moltrasio C, Marzano AV. Coexistence of steatocystoma multiplex and hidradenitis suppurativa: assessment of this unique association by means of ultrasonography and Color Doppler. Skin Res Technol. 2019;25(6):877–880. doi:10.1111/srt.12751
5. Koprulu M, Naeem M, Nalbant G, et al. KERATIN 17-related recessive atypical pachyonychia congenita with variable hair and tooth anomalies. Eur J Hum Genet. 2022;30(11):1292–1296. doi:10.1038/s41431-022-01128-4
6. Ahmed G, Prabha N, Ganguly S. Familial Steatocystoma Multiplex Generalisita Suppuritiva: oral Rifampicin and Clindamycin Combination Worth a Trial. Indian J Dermatol. 2021;66(5):553–555. doi:10.4103/ijd.IJD_117_20
7. Marzano AV, Tavecchio S, Balice Y, Polloni I, Veraldi S. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53(3):198–201. doi:10.1111/j.1440-0960.2011.00851.x
8. Ren X, Huang D, Wei W, et al. Clinical Investigation and Molecular Pathogenesis Study of Multiple Steatocystoma in a Family. Chin Jf Drugs Clin Pract. 2013;13(6):3.
9. Liu Q, Wu W, Lu J, Wang P, Qiao F. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12(4):5072–5076. doi:10.3892/mmr.2015.4063
10. Wang J, Li J, Li X, et al. A recurrent mutation in the KRT17 gene responsible for severe steatocystoma multiplex in a large Chinese family. Clin Exp Dermatol. 2018;43(2):205–208. doi:10.1111/ced.13311
11. Antal AS, Kulichova D, Redler S, Betz RC, Ruzicka T. Steatocystoma multiplex: keratin 17 - the key player? Br J Dermatol. 2012;167(6):1395–1397. doi:10.1111/j.1365-2133.2012.11073.x
12. Jianfeng W, Yong C, Fusheng Z, et al. Study on KRT17 Gene Mutation in a Family with Multiple Steatocystoma. Chin J Leprosy Skin Dis. 2008;24(12):3.
13. Wang JF, Lu WS, Sun LD, et al. Novel missense mutation of keratin in Chinese family with steatocystoma multiplex. J Eur Acad Dermatol Venereol. 2009;23(6):723–724. doi:10.1111/j.1468-3083.2009.03180.x
14. Dongjie Z, Cheng Z, Meili H, Xiaolei M, Zhang J. A novel mutation (p. Arg94Gly) of keratin 17 in a Chinese family with steatocystoma multiplex. Eur J Dermatol. 2011;21(1):142–144. doi:10.1684/ejd.2011.1207
15. Oh SW, Kim MY, Lee JS, Kim SC. Keratin 17 mutation in pachyonychia congenita type 2 patient with early onset steatocystoma multiplex and Hutchinson-like tooth deformity. J Dermatol. 2006;33(3):161–164. doi:10.1111/j.1346-8138.2006.00037.x
16. Ofaiche J, Duchatelet S, Fraitag S, Nassif A, Nougue J, Hovnanian A. Familial pachyonychia congenita with steatocystoma multiplex and multiple abscesses of the scalp due to the p.Asn92Ser mutation in keratin 17. Br J Dermatol. 2014;171(6):1565–1567. doi:10.1111/bjd.13123
17. Kanda M, Natsuga K, Nishie W, et al. Morphological and genetic analysis of steatocystoma multiplex in an Asian family with pachyonychia congenita type 2 harbouring a KRT17 missense mutation. Br J Dermatol. 2009;160(2):465–468. doi:10.1111/j.1365-2133.2008.08983.x
18. Zhang B, Sun L, Fu X, Yu G, Liu H, Zhang F. Mutation analysis of the KRT17 gene in steatocystoma multiplex and a brief literature review. Clin Exp Dermatol. 2020;45(1):132–134. doi:10.1111/ced.14030
19. Yoneda K, Nakai K, Demitsu T, Kubota Y. Polycystic kidney disease with steatocystoma multiplex: evidences for a disruptive effect of mutated polycystin-1 on keratin 17 polymerisation. Acta Derm Venereol. 2015;95(3):353–354. doi:10.2340/00015555-1934
20. Zhang Y, Fang M, Ding X, Tang L, Zhang X. Familial neurofibromatosis type 1 has diverse manifestations in skin and is associated with steatocystoma multiplex. Clin Exp Dermatol. 2021;46(6):1166–1169. doi:10.1111/ced.14618
21. Carroll L, Ieremia E. The histopathological spectrum of sebaceous tumours: key features, diagnostic criteria and differential diagnosis. Diagnostic Histopathol. 2025;31(2):98–106. doi:10.1016/j.mpdhp.2024.11.006
22. Kamra HT, Gadgil PA, Ovhal AG, Narkhede RR. Steatocystoma multiplex-a rare genetic disorder: a case report and review of the literature. J Clin Diagn Res. 2013;7(1):166–168. doi:10.7860/JCDR/2012/4691.2698
23. Sharma A, Agrawal S, Dhurat R, Shukla D, Vishwanath T. An Unusual Case of Facial Steatocystoma Multiplex: a Clinicopathologic and Dermoscopic Report. Dermatopathology. 2018;5(2):58–63. doi:10.1159/000488584
24. Jacob JT, Nair RR, Poll BG, et al. Keratin 17 regulates nuclear morphology and chromatin organization. J Cell Sci. 2020;133(20):jcs254094. doi:10.1242/jcs.254094
25. Sato H, Koide T, Sagai T, et al. The genomic organization of type I keratin genes in mice. Genomics. 1999;56(3):303–309. doi:10.1006/geno.1998.5721
26. Zieman AG, Coulombe PA. Pathophysiology of pachyonychia congenita-associated palmoplantar keratoderma: new insights into skin epithelial homeostasis and avenues for treatment. Br J Dermatol. 2020;182(3):564–573. doi:10.1111/bjd.18033
27. Ghazawi FM, Hassani-Ardakani K, Henriques L, Jafarian F. Identification of a novel substitution mutation (R103C) in the rod domain of the keratin 17 gene associated with pachyonychia congenita type 2. Int J Dermatol. 2019;58(2):233–236. doi:10.1111/ijd.14082
28. Ho M, Thompson B, Fisk JN, et al. Update of the keratin gene family: evolution, tissue-specific expression patterns, and relevance to clinical disorders. Hum Genomics. 2022;16(1):1. doi:10.1186/s40246-021-00374-9
29. Wang H, Zhu C, Dong L, et al. The variant c.274A>G (p.Asn92Asp) in KRT17 in a patient with pachyonychia congenita and a novel clinical feature of acne inversa. Front Genet. 2024;15:1365581. doi:10.3389/fgene.2024.1365581
30. Yang L, Zhang S, Wang G. Keratin 17 in disease pathogenesis: from cancer to dermatoses. J Pathol. 2019;247(2):158–165. doi:10.1002/path.5178
31. Natsuga K, Shinkuma S, Kanda M, et al. Possible modifier effects of keratin 17 gene mutation on keratitis-ichthyosis-deafness syndrome. Br J Dermatol. 2012;166(4):903–905. doi:10.1111/j.1365-2133.2011.10696.x
32. Zhou P, Li Y, Zhang S, et al. KRT17 From Keratinocytes With High Glucose Stimulation Inhibit Dermal Fibroblasts Migration Through Integrin alpha11. J Endocr Soc. 2024;8(2):bvad176. doi:10.1210/jendso/bvad176
33. Fimbres DCP, Wolfe SA, Kelley CE. Proliferation of steatocystomas in 2 transgender men. JAAD Case Rep. 2022;26:70–72. doi:10.1016/j.jdcr.2022.06.020
34. Del Rosso JQ, Kircik L. The cutaneous effects of androgens and androgen-mediated sebum production and their pathophysiologic and therapeutic importance in acne vulgaris. J Dermatol Treat. 2024;35(1):2298878. doi:10.1080/09546634.2023.2298878
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.