Back to Journals » International Journal of Nanomedicine » Volume 14
Enzyme-responsive mesoporous silica nanoparticles for tumor cells and mitochondria multistage-targeted drug delivery
Authors Naz S ![]() , Wang M
, Wang M ![]() , Han Y
, Han Y ![]() , Hu B, Teng L, Zhou J
, Hu B, Teng L, Zhou J ![]() , Zhang H
, Zhang H ![]() , Chen J
, Chen J
Received 19 January 2019
Accepted for publication 12 March 2019
Published 10 April 2019 Volume 2019:14 Pages 2533—2542
DOI https://doi.org/10.2147/IJN.S202210
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Safia Naz,1,* Mingyu Wang,1,* Yuning Han,1 Bin Hu,1 Liping Teng,2 Juan Zhou,1 Huijie Zhang,1 Jinghua Chen1
1Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, School of Pharmaceutical Sciences, Jiangnan University, Wuxi, People’s Republic of China; 2Wuxi School of Medicine, Jiangnan University, Wuxi, People’s Republic of China
*These authors contributed equally to this work
Background: Drug delivery systems (DDS) capable of targeting both cell and organelle levels are highly desirable for effective cancer therapy. In this study, we developed a novel enzyme-responsive, multistage-targeted anticancer DDS based on mesoporous silica nanoparticles (MSNs), which possessed both CD44-targeting and mitochondrial-targeting properties.
Materials and methods: Triphenylphosphine (TPP), a mitochondria-targeting compound, was grafted onto the surface of MSNs firstly. Then, Doxorubicin (Dox) was encapsulated into the pore of MSNs, followed by capping with tumor-targeting molecules hyaluronic acid (HA) through electrostatic interactions to form the final product consist of Dox loaded, TPP attached, HA capped mesoporous silica nanoparticles (MSN-DPH).
Results: Our results suggested that MSN-DPH was preferentially taken up by cancer cells via CD44 receptor-mediated endocytosis. Moreover, MSN-DPH mainly accumulated in mitochondria owing to the mitochondrial-targeting ability of TPP. Degradation of HA by overexpressed HAase facilitated the release of Dox in cancer cells. Thus, MSN-DPH efficiently killed the cancer cells while exhibited much lower cytotoxicity to normal cells.
Conclusion: This study demonstrates a promising multistage-targeted DDS for cancer chemotherapy.
Keywords: mesoporous silica nanoparticles, drug delivery, enzyme-responsive, multistage-targeting, cancer therapy
Introduction
Nowadays, cancer remains a major threat to human life.1–3 Chemotherapy is one of the most effective options among current clinical cancer therapies. However, most of the traditional chemotherapeutic drugs cause severe side effects since they cannot distinguish between cancer and normal cells. Their therapeutic efficiency was also limited due to the rapid clearance during circulation and premature drug release.4,5 Development of novel drug delivery systems (DDSs) capable of minimizing adverse effects while improving therapeutic efficacy is crucial for successful cancer therapy.
Nano-sized DDSs possessed the well-known enhanced permeability and retention effect and was effective in reducing the drawbacks of traditional chemotherapeutic drugs.6 So far, various types of nanocarriers have been developed.4,7–12 Among these nanocarriers, mesoporous silica nanoparticles (MSNs) have attracted enormous interests.13,14 MSNs possessed unique features including high specific surface area, tunable particle and pore size, good biocompatibility and easy surface functionalization.13,15–18 These features offer promise to develop MSN-based “on-command” DDSs. To date, various MSN-based DDSs using different capping agents have been developed.13,15,16 These smart “gatekeepers” could protect the loaded drugs during circulation in blood and respond to certain stimulus to release the drug.19–23 Furthermore, targeting moieties could be anchored on the surface of MSNs to generate active targeted DDSs.24–27 These DDSs could target tumor cells overexpressing the corresponding receptors and efficiently deliver anticancer drugs into the cells. Among the existing capping and targeting molecules, hyaluronic acid (HA) constitutes advantageous properties and attracts increasing attentions. HA is a naturally occurring polysaccharide.28 As the main component of extracellular matrix, HA possesses favorable biocompatibility, non-immunogenicity and biodegradability.15,28,29 HA specifically interact with CD44 receptor, which is overexpressed in many cancer cells. Thus, HA functions as multifunctional agent with both capping and targeting ability. Furthermore, HA could be readily degraded by hyaluronidase (HAase) in acidic microenvironment of tumor tissues while stable in normal tissues due to the neutral pH.30,31 HA-capped MSN has shown great progress in targeted cancer therapy.15,16,28 Although promising, most of these DDSs could only target tumor cells. Drawbacks such as multidrug resistance is inevitable, which has become a major barrier to cancer therapy. Development of multistage-targeted DDSs capable of altering the intracellular localization of drugs and delivering them to specific subcellular organelle is highly desired.
Recently, mitochondria have emerged as a hot target in cancer therapy due to their central roles in regulating cell metabolism and apoptosis. Delivery of anticancer drugs to mitochondria may bypass the classical resistance pathways while maintaining or improving their therapeutic efficacy.32,33 In addition, induction of mitochondria-mediated apoptosis could also enhance the efficiency of cancer therapy.34 Triphenylphosphine (TPP) is one of the most promising mitochondria-targeting ligands, which can be driven into mitochondria by mitochondrial membrane potential.35–37 Battogtokh et al developed a mitochondria-targeted anticancer DDS by conjugating docetaxel (DTX) with TPP and further loading the TPP–DTX conjugate into folate-cholesteryl albumin (FA-chol-BSA) nanoparticles. In vitro and in vivo results revealed that the as-prepared TPP–DTX@FA-chol-BSA NPs primarily accumulated in the mitochondria and exhibited better antitumor effect.1
In this study, we developed an enzyme-responsive multistage-targeted anticancer DDS based on MSNs. As illustrated in Scheme 1, MSNs were grafted with TPP for mitochondrial-targeting and further modified with HA. HA capping could not only improve the CD44 receptor-mediated cancer cell targeting, but also block the pores as a “gatekeeper” to protect the Dox pre-leakage. The as-prepared MSN-DPH (consist of Dox loaded, TPP attached, HA capped mesoporous silica nanoparticles) was fully characterized and their HAase-responsive drug release was studied. Meanwhile, the CD44 receptor-mediated intracellular uptake as well as the mitochondrial-targeting ability of the MSN-DPH were also investigated. Finally, the in vitro antitumor effect of MSN-DPH was examined in detail.
 | Scheme 1 Schematic illustration of the preparation and application of MSN-DPH (consist of Dox loaded, TPP attached, HA capped mesoporous silica nanoparticles) for multistage-targeted delivery of Dox. Abbreviations: Dox, Doxorubicin; HA, hyaluronic acid; MSN, mesoporous silica nanoparticle; TPP, triphenylphosphine. |
Materials and methods
Materials
HA (MW: 30,000) was purchased from Freda Biopharm Co. (Shandong, People’s Republic of China). (3-aminopropyl) triethoxysilane (APTS) was obtained from Macklin (Shanghai, People’s Republic of China). N-hydroxysuccini-mide (NHS) was purchased from BBI Life Sciences (Shanghai, People’s Republic of China). Triethanolamine (TEA), tetraethyl orthosilicate (TEOS), hexadecyl trimethyl ammonium chloride (CTAC), methanol and ethanol were bought from Sinopharm Biotech (Shanghai, People’s Republic of China). 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) was purchased from Aladdin (Shanghai, People’s Republic of China). Doxorubicin hydrochloride (Dox·HCl) was purchased from Yuancheng (Wuhan, People’s Republic of China). HAase was obtained from Macklin (Shanghai, People’s Republic of China). African green monkey SV40-transformed kidney fibroblast cells (Cos-7) and human gastric carcinoma cells (MGC-803) cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). Dulbecco’s modified Eagle’s medium (DMEM), RPMI 1640 medium, FBS and penicillin-streptomycin solution were purchased from Gibco (MA, USA).
Characterizations
Transmission electron microscopy images were obtained using a JEM-2100 (JEOL, Japan) transmission electron microscopy operating at an acceleration voltage of 200 kV. Nitrogen adsorption-desorption isotherms were recorded using a Quadrsorb SI analyzer (Quantachrome, USA) at −196°C under continuous adsorption conditions. Fourier transform infrared spectra (FT-IR) were measured on a NicoletTM iSTM 10 spectrophotometer (Thermo Fisher Scientific, USA) at 4 cm−1 resolution with 32 scans. Dynamic light scattering and zeta potential were measured using a ZEN 3600 instrument (Malvern, UK). UV absorbance spectra were recorded using a UV-2550 spectrophotometer (Shimadzu, Japan). Confocal scanning laser microscopy images were taken on a TCS SP8 microscope (Leica, Germany). Flow cytometry analysis was carried out using a CytoFLEX flow cytometer (Beckman Coulter, USA).
Synthesis and ammonization of MSNs (MSN-NH2)
CTAC (0.5 g) and appropriate amounts of TEA were dissolved completely in distilled water (20 mL) at 95°C and stirred for 1 hr. Then, TEOS (1.5 mL) was added manually using a syringe at a speed of approximately 1 mL/min. After continuous stirring for 1 hr, the as-synthesized MSNs were collected by centrifugation at 12,000 rpm for 30 mins and then washed with ethanol to remove the residual reactants. To remove surfactants completely, the as-synthesized MSNs were dispersed under ultrasound for 20 mins and refluxed in a mixed solution of ethanol (12 mL) and hydrochloric acid (36–38%, 1.5 mL) at 60°C overnight. Then, the solution was centrifuged and washed several times with ethanol. The same operation was repeated twice. The final product was dried under vacuum.
To obtain amino-functionalized MSNs, MSNs (1 g) were refluxed with anhydrous toluene (80 mL) containing APTS (0.75 mL) for 20 hrs and washed by ethanol for several times. After drying under vacuum overnight, MSN-NH2 was obtained.
Synthesis of TPP propionic acid and preparation of MSN-TPP
TPP (8.5 g, 32.5 mmol) was added to a solution of 3-bromopropionic acid (5 g, 32.5 mmol) in acetonitrile (13 mL). The mixture was stirred at 80°C for 24 hrs and then concentrated in vacuum. The residue was taken up with a minimal amount of chloroform and the product was precipitated from diethyl ether. The white precipitate was filtered and recrystallized from acetonitrile to obtain TPP propionic acid as colorless solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 2.9–3.0 (m, 2H, CH2CO), 3.6–3.7 (m, 2H, Ph3PCH2), 7.6–7.7 (m, 15H, ArH). 13C NMR (100 MHz, CDCl3) δ (ppm): 18.7 (d, Ph3PCH2), 27.9 (s, CH2CO), 117.4 (d, 3C, ArCH (ispo)), 131.2 (d, 6C, ArCH), 133.5 (d, 6C, ArCH), 135.2 (d, 3C, ArCH), 171.0 (d, CO).
TPP propionic acid (22 mg) was dissolved in distilled water (10 mL); then, EDC (10 mg) and NHS (5 mg) were added. The pH value of the solution was adjusted to 4–6, and the reaction was stirred for 30 mins to active carboxylate groups. After that, MSN-NH2 (50 mg) was added to complete esterification reaction. Nanoparticles were collected by centrifugation and washed by ethanol for several times. The product (MSN-TPP) was dried by vacuum drying.
Preparation of dox loaded MSNs (MSN-DPH)
>Dox (15 mg) was dissolved in distilled water (30 mL); then, MSN-TPP (150 mg) was added and dispersed by ultrasonic bath. After 12 hrs of stirring, additional Dox (15 mg) was added and kept for 12 hrs to ensure the maximum loading. Thereafter, HA (150 mg) was added to package the particle. Finally, the solution was centrifuged and the precipitation was washed by PBS for four times. The concentration of Dox was measured by UV−vis spectrophotometer and calculated according to the standard curve. The loading capacity was calculated from the concentration of the unloaded Dox in the supernatant. The product (MSN-DPH) was dried by vacuum drying.
Release of dox from MSN-dox and MSN-DPH
MSN-Dox (2 mg) and MSNs-DPH (2 mg) were dispersed in 2 mL of PBS (0.01 M, pH 7.4), respectively, and then sealed in dialysis bags (cutoff 3500 KDa). The bags containing MSN-Dox were immersed in PBS (20 mL, n=3). The bags containing MSNs-DPH were immersed in PBS (20 mL, n= 3) with and without HAase, respectively. Then, the bags were incubated on shaking table at 37°C. At each time intervals, PBS (3 mL) was taken out and analyzed by UV–vis spectrophotometer. Simultaneously, 3 mL of fresh PBS was added for supplementary. The concentration of Dox was determined by comparing the absorbance at 480 nm with the standard curve.
Cellular uptake and intracellular localization of MSN-DPH
MGC-803 cells were seeded on a 20 mm cell culture dishes at a density of 4×104 cells per dish and cultured for 24 hrs. The cells were then treated with MSN-DPH for 6, 12 and 24 hrs, respectively. The cells were then stained with Mitolite Green and DAPI, followed by washing with PBS and immobilized with 4% paraformaldehyde. Finally, the fixed cells were washed with PBS and then observed by confocal microscopy.
For cytometry assay, Cos-7 and MGC-803 cells were seeded on 12-well plate at a density of 4×104 cells per well and cultured for 24 hrs. Then, MSN-Dox and MSN-DPH (20 µg/mL) were added into the well and incubated continuously for 6, 12 and 24 hrs, respectively. Thereafter, the cells were digested by trypsin, collected by centrifugation at 1,000 rpm and resuspended in 1% paraformaldehyde solution. The intracellular fluorescence was analyzed using a CytoFLEX flow cytometer (Beckman Coulter).
In vitro cytotoxicity of MSN-DPH
MSN and MSN-DPH were dispersed in DMEM and RPMI 1640 medium at a concentration of 1 mg/mL. Cos-7 and MGC-803 cells were maintained in DMEM medium and 1,640 medium supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C in a humidified incubator (5% CO2). Cells were grown in 96-well plate overnight at a density of 5,000 cells per well (100 μL). MSN and MSN-DPH at the indicated concentrations (from 5 to 25 μg/mL) were added into the well. After 48 hrs of incubation, the medium was removed and 100 μL of MTT solution was added. After 4 hrs, 100 μL of DMSO was added into the well. Finally, the absorbance at 570 nm was measured by a Multiskan MK3 microplate reader to determine the cell viabilities (Thermo Scientific, USA).
Statistical analysis
Statistical analysis was performed using Student’s t-test. Data are presented as mean±SD.
Results and discussion
Preparation and characterization of MSN-DPH
MSNs with controlled particle size were synthesized under alkaline condition using cationic surfactants as the structure-directing agent.38 As shown in Figure 1A, the as-synthesized MSNs presented regular round shape with obvious pore channels, which had a size of about 50 nm. The MSNs were further conjugated with TPP. Then, Dox was loaded, followed by capping with HA, which yielded MSN-DPH. MSN-DPH still maintained spherical shape whereas a slight increase of particle size was observed, which was due to the introduction of HA (Figure 1B). This result was further confirmed by dynamic light scattering analysis. MSN possessed a hydrodynamic diameter of approximately 70 nm, which was increased to 110 nm after functionalization with TPP and HA (Figure 1C and D).
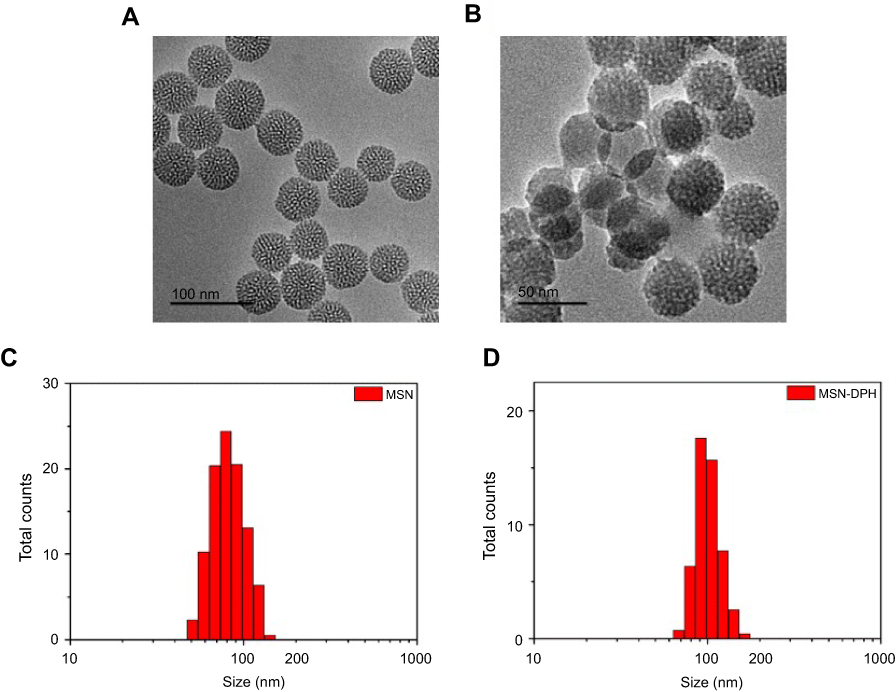 | Figure 1 Characterizations of the MSNs and MSN-DPH (consist of Dox loaded, TPP attached, HA capped mesoporous silica nanoparticles). Transmission electron microscopy images of (A) MSN and (B) MSN-DPH. Hydrodynamic size distribution of MSN (C) and MSN-DPH (D).Abbreviation: MSN, mesoporous silica nanoparticle. |
FT-IR spectra verified the successful synthesis of MSN-DPH. MSNs exhibited the typical Si-O vibration at 1,100 cm−1 (Figure 2A).39 No additional peaks at 2,800 cm−1 corresponding to the C-H vibration of CTAC could be observed, indicating the good purity of MSNs. MSN-NH2 revealed a new band of amino groups (–NH2) at around 1,530 cm−1, indicating the functionalization of amino groups onto MSN.39,40 The conjugation of TPP to MSNs was evidenced by the appearance of signals at 3,700–3,500 cm−1 and 1,690–1,630 cm−1, which were ascribed to N-H stretch and C=O stretch of amide group. An absorption band at around 1,640 cm−1 was clearly observed after HA capping, which was assigned to amide C=O stretching of the carboxyl groups in HA.28 The zeta potential of MSN changed from negative (-26 mV) to positive (+7 mV) after TPP conjugation (Figure 2B). After Dox loading and HA capping, the MSN-DPN exhibited almost neutral surface charge. Neutral nanoparticles were considered to possess longer circulation time in blood.41 Therefore, neutral MSN-DPH may have prolonged blood circulation time by means of avoiding been eliminated by immune system. Furthermore, the Brunauer–Emmett–Teller and Barret–Joyner–Halenda analyses were carried out to verify the mesoporous structure of nanoparticles. MSNs exhibited a typical mesoporous N2 adsorption–desorption isotherm with a pore diameter of about 3 nm (Figure 2C and D). In contrast, MSN-DPH no longer maintained the mesoporous structure. Moreover, a significant decrease in surface area (from 553 to 441 m2/g) and pore volume (from 0.37 to 0.1 cm3/g) was observed after Dox loading and HA capping. These results were due to the loading of Dox in pores of MSNs and the coverage of HA on the surface. MSN-DPH exhibited a high Dox loading capacity of about 110 mg Dox per 1 g of particles, which was determined by UV–vis analysis. The stability of MSN-DPH in physiological solutions was further studied. As shown in Figure 2E, no significant change was observed regarding the hydrodynamic sizes of MSN-DPH in PBS and medium after statically placed for 4 days. This result suggested that MSN-DPH was stable as carrier.
 | Figure 2 Characterizations of the MSN-DPH (consist of Dox loaded, TPP attached, HA capped mesoporous silica nanoparticles). (A) FT-IR spectra of MSN, MSN-NH2 and MSN-TPP. (B) Zeta potentials of MSN, MSN-NH2, MSN-TPP and MSN-DPH. Nitrogen adsorption–desorption isotherms (C) and corresponding pore size distribution (D) of MSN and MSN-DPH. (E) Stability of MSN-DPH in terms of hydrodynamic size in PBS and medium.Abbreviations: MSN, mesoporous silica nanoparticle; TPP, triphenylphosphine. |
HAase-responsive Dox release from MSN-DPH
Release of Dox from MSN-DPH and MSN-Dox in PBS (pH 7.4) was further determined by UV–vis analysis. As shown in Figure 3, a much faster release of Dox from MSN-Dox was observed, which reached to 70% in 72 hrs. In contrast, Dox was released from MSN-DPH in a slower and sustained manner due to the HA capping. This property might minimize Dox premature release from MSN-DPH during blood circulation and reduce the side effects to normal tissues. Furthermore, release of Dox from MSN-DPH was obviously accelerated after the addition of HAase. HAase hydrolyzed the outer HA layer of MSN-DPH and thus facilitated the release of Dox. Therefore, MSN-DPH may achieve an enhanced antitumor effect due to the accelerated Dox release in tumor cells triggered by HAase.
 | Figure 3 Cumulative release of Dox from MSN-Dox, MSN-DPH (consist of Dox loaded, TPP attached, HA capped mesoporous silica nanoparticles) and MSN-DPH with the presence of HAase versus time. Data were presented as mean±SD (n=3).Abbreviations: HAase, hyaluronidase; Dox, Doxorubicin; MSN, mesoporous silica nanoparticle. |
Multistage targeting capability of MSN-DPH
To investigate the targeting capability of MSN-DPH at cell level, MSN-Dox and MSN-DPH were incubated with MGC-803 cells, followed by measuring intracellular fluorescence through flow cytometer. As shown in Figure 4A and B, MSN-Dox and MSN-DPH could be gradually internalized by MGC-803 cells and the intensity of intracellular Dox fluorescence increased as the incubation time went by. Compared with MSN-Dox, MSN-DPH presented more cellular uptake, which was primarily attributed to the CD44-mediated endocytosis. Therefore, MSN-DPH might result in a high chance of drug accumulation in tumor tissues, which eventually leads to enhanced antitumor effect.
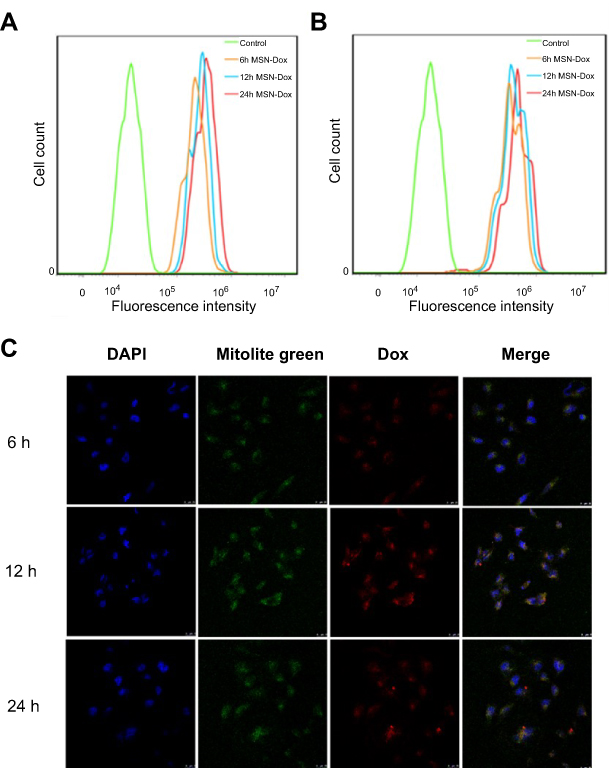 | Figure 4 Cellular uptake and intracellular localization of MSN-DPH (consist of Dox loaded, TPP attached, HA capped mesoporous silica nanoparticles). Flow cytometry profiles of MGC-803 cells incubated with MSN-Dox (A) and MSN-DPH (B) for various time. (C) Confocal microscopy images of MGC-803 cells incubated with MSN-DPH for various time. The nuclei of the cells were stained with DAPI (blue). Cell mitochondria was stained by Mitolite Green. Scale bars denote 25 μm. Abbreviations: Dox, Doxorubicin; MSN, mesoporous silica nanoparticle. |
The intracellular localization and mitochondrial-targeting capability of MSN-DPH were further carried out using multi-staining assay and observed by confocal fluorescence microscopy (Figure 4C). Cell mitochondria was stained by Mitolite Green, which was a mitochondria-specific fluorescent probe. In the initial 6 hrs of incubation, MSN-DPH was taken up by the cells and only a small amount of MSN-DPH could be observed in mitochondria, indicated by moderately overlapped Dox fluorescence and Mitolite Green. Although the efficiency was low at the beginning, until 12 hrs of incubation, obvious red fluorescence appeared in mitochondria and yellow spots arose in the merged images. These observations demonstrated that MSN-DPH had been successfully internalized by MGC-803 cells and the released Dox tended to accumulate in mitochondria. After 24 hrs of incubation, the red fluorescence not only overlapped completely with the green fluorescence but also gradually covered the blue fluorescence of DAPI, indicating the completed disassembling of HA layer and Dox release. Furthermore, no significant change of Mitolite Green fluorescence was observed with the increase of incubation time. This result suggested that the enhanced co-localization of MSN-DPH fluorescence with Mitolite Green fluorescence was not due to the increase of Mitolite Green fluorescence. This result confirmed again the excellent mitochondria-targeting capability of MSN-DPH. Taken together, MSN-DPH possessed a multistage targeting ability for tumor cells and mitochondria.
In vitro antitumor effect of MSN-DPH
Next, cytotoxicity assays were performed with two cell lines including normal cell Cos-7 and stomach cancer cell MGC-803. The cells were treated with MSN and MSN-DPH with different concentrations for 48 hrs, and then the cell viabilities were recorded. No significant cytotoxicity was observed when incubating MSN with Cos-7 and MGC-803 cells. All of the cell viabilities exceed 80% at the concentrations below 25 μg/mL. This result was in accordance with our previous report, which suggested the good biocompatibility of MSNs.42 In addition, the viabilities of Cos-7 cells exposure to MSN-DPH with different concentrations were still higher than 70% (Figure 5A). As normal cell lines, Cos-7 cells lack CD44 receptor on their surface and possess lower intracellular HAase levels.43,44 Therefore, only small amount of Dox could enter the Cos-7 cells and induce cell death. However, MSN-DPH exhibited much higher dose-dependent cytotoxicity to MGC-803 cells. When increasing the concentrations of MSN-DPH, the viabilities of MGC-803 cells were significantly decreased to 34% (Figure 5B). Enhanced antitumor effect of MSN-DPH was mainly contributed to the following reasons: 1) enhanced cellular uptake of Dox in MGC-083 cells via CD44 receptor-mediated endocytosis; 2) accelerated Dox release in MGC-803 cells triggered by overexpressed HAase; and 3) induction of mitochondria-mediated apoptosis by Dox accumulated in mitochondria. To further investigate the lethality of MSN-DPH toward MGC-803 cell, the live/dead viability/cytotoxicity assay was performed using confocal fluorescence microscopy. Live cells were viewed with green fluorescence, while dead cells were represented in red. As shown in Figure 6, few sporadic red fluorescence appeared in cells cultured with MSN, whereas almost half of the cells exhibited red fluorescence when treated with MSN-DPH for 24 hrs. These findings further verified that MSN-DPH possessed good anticancer effect while was less toxic to normal cells. Taken together, MSN-DPH showed strong potential for targeted cancer therapy.
 | Figure 5 Relative viability of (A) Cos-7 and (B) MGC-803 cells incubated with MSN and MSN-DPH (consist of Dox loaded, TPP attached, HA capped mesoporous silica nanoparticles) for 48 hrs. Data were presented as mean±SD (n=3). Abbreviation: MSN, mesoporous silica nanoparticle. |
 | Figure 6 Live/dead images of MGC-803 cells incubated with MSN and MSN-DPH (consist of Dox loaded, TPP attached, HA capped mesoporous silica nanoparticles). Live cells and dead cells were stained by AM (green) and PI (red), respectively. Scale bars denote 25 μm.Abbreviation: MSN, mesoporous silica nanoparticle. |
Conclusion
In this study, a MSN-based enzyme-responsive DDS capable of multistage targeting cancer cells and mitochondria was developed. The capping of HA on the surface of MSN provided effective sealing capacity in normal cell while enhanced selective uptake by cancer cells via CD44 receptor-mediated endocytosis pathway. Moreover, MSN-DPH exhibited enzyme-responsive Dox release under the degradation of the overexpressed HAase in cancer cell. Additionally, TPP decoration endowed DDS the ability to target mitochondria and release Dox at subcellular organelle. MSN-DPH exhibited favorable antitumor activity while much lower cytotoxicity to normal cells. Therefore, MSN-DPH had strong potential for targeted cancer therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation of Jiangsu Province (BK20170181, BK20170202, BK20170203) and National Natural Science Foundation of China (21574059, 31700708). We also thank the Fundamental Research Funds for the Central Universities (JUSRP11748), National First-class Discipline Program of Light Industry Technology and Engineering (LITE2018-20) and Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (No. PPZY2015B146).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Battogtokh G, Gotov O, Kang JH, et al. Triphenylphosphine-docetaxel conjugate-incorporated albumin nanoparticles for cancer treatment. Nanomedicine. 2018;13(3):325–338. doi:10.2217/nnm-2017-0274
2. Youn YS, Bae YH. Perspectives on the past, present, and future of cancer nanomedicine. Adv Drug Delivery Rev. 2018;130:3–11. doi:10.1016/j.addr.2018.05.008
3. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi:10.1038/nrc.2016.108
4. Chen G, Xie Y, Peltier R, et al. Peptide-decorated gold nanoparticles as functional nano-capping agent of mesoporous silica container for targeting drug delivery. ACS Appl Mater Interfaces. 2016;8(18):11204–11209. doi:10.1021/acsami.6b02594
5. Dong D-W, Xiang B, Gao W, Yang -Z-Z, Li J-Q, Qi X-R. pH-responsive complexes using prefunctionalized polymers for synchronous delivery of doxorubicin and siRNA to cancer cells. Biomaterials. 2013;34(20):4849–4859. doi:10.1016/j.biomaterials.2013.03.018
6. Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release. 2015;200:138–157. doi:10.1016/j.jconrel.2014.12.030
7. Bhaskara RM, Linker SM, Voegele M, Koefinger J, Hummer G. Carbon nanotubes mediate fusion of lipid vesicles. ACS Nano. 2017;11(2):1273–1280. doi:10.1021/acsnano.6b05434
8. Feng S, Zhang H, Yan T, et al. Folate-conjugated boron nitride nanospheres for targeted delivery of anticancer drugs. Int J Nanomedicine. 2016;11:4573–4582. doi:10.2147/IJN.S110689
9. Feng S, Zhang H, Zhi C, Gao X-D, Nakanishi H. pH-responsive charge-reversal polymer-functionalized boron nitride nanospheres for intracellular doxorubicin delivery. Int J Nanomedicine. 2018;13:641–652. doi:10.2147/IJN.S153476
10. Merino S, Martin C, Kostarelos K, Prato M, Vazquez E. Nanocomposite hydrogels: 3D polymer-nanoparticle synergies for on-demand drug delivery. ACS Nano. 2015;9(5):4686–4697. doi:10.1021/acsnano.5b01433
11. Pattni BS, Chupin VV, Torchilin VP. New developments in liposomal drug delivery. Chem Rev. 2015;115(19):10938–10966. doi:10.1021/acs.chemrev.5b00046
12. Ulbrich K, Hola K, Subr V, Bakandritsos A, Tucek J, Zboril R. Targeted drug delivery with polymers and magnetic nanoparticles: covalent and noncovalent approaches, release control, and clinical studies. Chem Rev. 2016;116(9):5338–5431. doi:10.1021/acs.chemrev.5b00589
13. Yao X, Niu X, Ma K, et al. Graphene quantum dots-capped magnetic mesoporous silica nanoparticles as a multifunctional platform for controlled drug delivery, magnetic hyperthermia, and photothermal therapy. Small. 2017;13(2). doi:10.1002/smll.201602225.
14. Yu X, Zhu Y. Preparation of magnetic mesoporous silica nanoparticles as a multifunctional platform for potential drug delivery and hyperthermia. Sci Technol Adv Mat. 2016;17(1):229–238. doi:10.1080/14686996.2016.1178055
15. Chen C, Sun W, Wang X, Wang Y, Wang P. pH-responsive nanoreservoirs based on hyaluronic acid end-capped mesoporous silica nanoparticles for targeted drug delivery. Int J Biol Macromol. 2018;111:1106–1115. doi:10.1016/j.ijbiomac.2018.01.093
16. Huang L, Liu J, Gao F, et al. A dual-responsive, hyaluronic acid targeted drug delivery system based on hollow mesoporous silica nanoparticles for cancer therapy. J Mater Chem B. 2018;6(28):4618–4629. doi:10.1039/C8TB00989A
17. Tao C, Zhu Y. Magnetic mesoporous silica nanoparticles for potential delivery of chemotherapeutic drugs and hyperthermia. Dalton Trans. 2014;43(41):15482–15490. doi:10.1039/c4dt01984a
18. Tian Z, Yu X, Ruan Z, Zhu M, Zhu Y, Hanagata N. Magnetic mesoporous silica nanoparticles coated with thermo-responsive copolymer for potential chemo- and magnetic hyperthermia therapy. Microporous Mesoporous Mater. 2018;256:1–9. doi:10.1016/j.micromeso.2017.07.053
19. de la Torre C, Agostini A, Mondragon L, et al. Temperature-controlled release by changes in the secondary structure of peptides anchored onto mesoporous silica supports. Chem Commun. 2014;50(24):3184–3186. doi:10.1039/C3CC49421G
20. Qiao L, Wang X, Gao Y, et al. Laccase-mediated formation of mesoporous silica nanoparticle based redox stimuli-responsive hybrid nanogels as a multifunctional nanotheranostic agent. Nanoscale. 2016;8(39):17241–17249. doi:10.1039/c6nr05943k
21. Sun Y-L, Zhou Y, Li Q-L, Yang Y-W. Enzyme-responsive supramolecular nanovalves crafted by mesoporous silica nanoparticles and choline-sulfonatocalix 4 arene 2 pseudorotaxanes for controlled cargo release. Chem Commun. 2013;49(79):9033–9035. doi:10.1039/c3cc45216f
22. Zeng X, Liu G, Tao W, et al. A drug-self-gated mesoporous antitumor nanoplatform based on pH-sensitive dynamic covalent bond. Adv Funct Mater. 2017;27(11):1605985.
23. Zhang Q, Liu F, Kim Truc N, et al. Multifunctional mesoporous silica nanoparticles for cancer-targeted and controlled drug delivery. Adv Funct Mater. 2012;22(24):5144–5156. doi:10.1002/adfm.201201316
24. Cheng W, Nie J, Xu L, et al. pH-sensitive delivery vehicle based on folic acid-conjugated polydopamine-modified mesoporous silica nanoparticles for targeted cancer therapy. ACS Appl Mater Interfaces. 2017;9(22):18462–18473. doi:10.1021/acsami.7b02457
25. Oezalp VC, Schaefer T. Aptamer-based switchable nanovalves for stimuli-responsive drug delivery. Chem-Eur J. 2011;17(36):9893–9896. doi:10.1002/chem.201101403
26. Sun X, Luo Y, Huang L, Yu B-Y TJ. A peptide-decorated and curcumin-loaded mesoporous silica nanomedicine for effectively overcoming multidrug resistance in cancer cells. RSC Adv. 2017;7(27):16401–16409. doi:10.1039/C7RA01128H
27. Zhang Y, Guo J, Zhang X-L, et al. Antibody fragment-armed mesoporous silica nanoparticles for the targeted delivery of bevacizumab in ovarian cancer cells. Int J Pharm. 2015;496(2):1026–1033. doi:10.1016/j.ijpharm.2015.10.080
28. Chen Z, Li Z, Lin Y, Yin M, Ren J, Qu X. Bioresponsive hyaluronic acid-capped mesoporous silica nanoparticles for targeted drug delivery. Chem-Eur J. 2013;19(5):1778–1783. doi:10.1002/chem.201202038
29. Liu DC, Pearlman E, Diaconu E, et al. Expression of hyaluronidase by tumor cells induces angiogenesis in vivo. Proc Natl Acad Sci U S A. 1996;93(15):7832–7837.
30. Stern R, Jedrzejas MJ. Hyaluronidases: their genomics, structures, and mechanisms of action. Chem Rev. 2006;106(3):818–839. doi:10.1021/cr050247k
31. Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4(7):528–539. doi:10.1038/nrc1391
32. Chamberlain GR, Tulumello DV, Kelley SO. Targeted delivery of doxorubicin to mitochondria. ACS Chem Biol. 2013;8(7):1389–1395. doi:10.1021/cb400095v
33. Xi J, Li M, Jing B, et al. Long-circulating amphiphilic doxorubicin for tumor mitochondria-specific targeting. ACS Appl Mater Interfaces. 2018;10:43482–43492. doi:10.1021/acsami.8b17399
34. Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci U S A. 2012;109(40):16288–16293. doi:10.1073/pnas.1210096109
35. Chakrabortty S, Agrawalla BK, Stumper A, et al. Mitochondria targeted protein-ruthenium photosensitizer for efficient photodynamic applications. J Am Chem Soc. 2017;139(6):2512–2519. doi:10.1021/jacs.6b13399
36. Smith RAJ, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003;100(9):5407–5412. doi:10.1073/pnas.0931245100
37. Zhang X, Yan Q, Mulatihan DN, et al. Pharmaceutical micelles featured with singlet oxygen-responsive cargo release and mitochondrial targeting for enhanced photodynamic therapy. Nanotechnology. 2018;29(25):255101. doi:10.1088/1361-6528/aabbdb
38. Lv X, Zhang L, Xing F, Lin H. Controlled synthesis of monodispersed mesoporous silica nanoparticles: particle size tuning and formation mechanism investigation. Microporous Mesoporous Mater. 2016;225:238–244. doi:10.1016/j.micromeso.2015.12.024
39. Nairi V, Magnolia S, Piludu M, et al. Mesoporous silica nanoparticles functionalized with hyaluronic acid. Effect of the biopolymer chain length on cell internalization. Colloids Surf B. 2018;168:50–59. doi:10.1016/j.colsurfb.2018.02.019
40. Xu Y, Claiden P, Zhu Y, Morita H, Hanagata N. Effect of amino groups of mesoporous silica nanoparticles on CpG oligodexynucleotide delivery. Sci Technol Adv Mat. 2015;16(4):045006. doi:10.1088/1468-6996/16/4/045006
41. Li S-D, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. doi:10.1021/mp800049w
42. Zhou J, Hao N, De Zoyza T, Yan M, Ramstrom O. Lectin-gated, mesoporous, photofunctionalized glyconanoparticles for glutathione-responsive drug delivery. Chem Commun. 2015;51(48):9833–9836. doi:10.1039/C5CC02907D
43. Chen W-H, Luo G-F, Qiu W-X, et al. Mesoporous silica-based versatile theranostic nanoplatform constructed by layer-by-layer assembly for excellent photodynamic/chemo therapy. Biomaterials. 2017;117:54–65. doi:10.1016/j.biomaterials.2016.11.057
44. Jiang H, Peterson RS, Wang WH, Bartnik E, Knudson CB, Knudson W. A requirement for the CD44 cytoplasmic domain for hyaluronan binding, pericellular matrix assembly, and receptor-mediated endocytosis in COS-7 cells. J Biol Chem. 2002;277(12):10531–10538. doi:10.1074/jbc.M108654200
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.