Back to Journals » International Journal of Nanomedicine » Volume 13
Enhanced oral absorption of pemetrexed by ion-pairing complex formation with deoxycholic acid derivative and multiple nanoemulsion formulations: preparation, characterization, and in vivo oral bioavailability and anticancer effect
Authors Pangeni R ![]() , Choi JU
, Choi JU ![]() , Panthi VK, Byun Y, Park JW
, Panthi VK, Byun Y, Park JW ![]()
Received 12 March 2018
Accepted for publication 15 April 2018
Published 6 June 2018 Volume 2018:13 Pages 3329—3351
DOI https://doi.org/10.2147/IJN.S167958
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Rudra Pangeni,1,* Jeong Uk Choi,2,* Vijay Kumar Panthi,1 Youngro Byun,3 Jin Woo Park1
1College of Pharmacy and Natural Medicine Research Institute, Mokpo National University, Muan-gun, Jeonnam, Republic of Korea; 2College of Pharmacy, Seoul National University, Seoul, Republic of Korea; 3Department of Molecular Medicine and Biopharmaceutical Science, Graduate School of Convergence Science and Technology, College of Pharmacy, Seoul National University, Seoul, Republic of Korea
*These authors contributed equally to this work
Objective: The current study sought to design an oral delivery system of pemetrexed (PMX), a multitargeted antifolate antimetabolite, by enhancing its intestinal membrane permeability.
Materials and methods: PMX was ionically complexed with a permeation enhancer such as Nα-deoxycholyl-l-lysyl-methylester (DCK) and prepared as an amorphous solid dispersion by mixing with dispersants such as 2-hydroxypropyl-beta-cyclodextrin (HP-beta-CD) and poloxamer 188 (P188), forming an HP-beta-CD/PMX/DCK/P188; the complex was incorporated into multiple water-in-oil-in-water nanoemulsions in a supersaturated state (HP-beta-CD/PMX/DCK/P188-NE).
Results: After complex formation, the partition coefficient and in vitro membrane permeability of PMX were markedly increased, but it showed similar cytotoxic and inhibitory effects on cancer cell proliferation/migration. Furthermore, the intestinal membrane permeability and epithelial cell uptake of PMX were synergistically improved after HP-beta-CD/PMX/DCK/P188 was incorporated into a nanoemulsion with a size of 14.5±0.45 nm. The in vitro permeability of HP-beta-CD/PMX/DCK/P188-NE across a Caco-2 cell monolayer was 9.82-fold greater than that of free PMX, which might be attributable to the partitioning of PMX to the epithelial cells being facilitated via specific interaction of DCK with bile acid transporters, as well as the enhanced lipophilicity accompanied by surfactant-induced changes in the intestinal membrane structure and fluidity. Therefore, the oral bioavailability of HP-beta-CD/PMX/DCK/P188-NE in rats was evaluated as 26.8%±2.98% which was 223% higher than that of oral PMX. Moreover, oral HP-beta-CD/PMX/DCK/P188-NE significantly suppressed tumor growth in Lewis lung carcinoma cell-bearing mice, and the tumor volume was maximally inhibited by 61% compared with that in the control group.
Conclusion: These results imply that HP-beta-CD/PMX/DCK/P188-NE is an effective and promising delivery system for enhancing the oral absorption of PMX. Thus, there is the potential for new medical applications, including applications in metronomic cancer treatment.
Keywords: pemetrexed, deoxycholic acid derivative, multiple nanoemulsions, permeability, oral absorption, oral anticancer therapy
Introduction
Conventional chemotherapeutic drugs that are administered intravenously at the maximum tolerated dose, with the objective of optimum tumor cell killing, are selected based on the highest percentage of cure rate. However, this treatment strategy requires extended treatment-free scheduling to allow normal host cell recovery, which may also lead to resumption of the growth of vascular endothelial cells in the tumor beds, inducing regrowth of tumor cells.1,2 Thus, to minimize the limitations of conventional chemotherapy in tumor regrowth, the level of toxicity, and the need for supportive treatment, metronomic chemotherapy alone or in combination with other targeted therapies may cause the inhibition of angiogenesis or endothelial cell migration at a relatively low and minimal toxic dose.1,3,4 These common and low doses of chemotherapy as maintenance therapy with the optimum drug concentration in circulation for a prolonged duration can be achieved through painless and flexible oral administration, which prevents an initial rapid increase and subsequent decay of drug concentration in the blood and can also improve the patient’s quality of life.5–7 However, most anticancer drugs are not appropriate candidates for oral administration as they exhibit poor oral bioavailability due to low aqueous solubility, gastrointestinal (GI) instability, variable absorption, and high drug efflux through P-glycoprotein (P-gp) transporters.5,8,9
Pemetrexed (PMX, N-[4-[2-(2-amino-3,4-dihydro-4-oxo-7H-pyrrolo[2,3-d] pyrimidin-5-yl)-ethyl]-benzoyl]-L-glutamic acid) is a multitargeted antifolate antimetabolite drug that inhibits three folate-dependent enzymes: thymidylate synthase (TS), glycinamide ribonucleotide formyl transferase (GARFT), and dihydrofolate reductase (DHFR).10,11 This leads to the depletion of fully reduced folate, resulting in the disruption of nucleotide synthesis for both purines and pyrimidines. Thus, the potential antitumor activity and cytotoxic effect of PMX may be due to the inhibition of key folate-dependent enzymes.12 Clinically, PMX was approved by the US Food and Drug Administration (FDA) for the treatment of several stages of non-squamous non-small-cell lung cancer (NSCLC) as first-, second-, third-line, and maintenance therapy. Moreover, it is one of the recommended drugs for use in combination with platinum-based cisplatin or carboplatin for patients with stage IV non-squamous NSCLC.13–15 PMX is a polar compound exhibiting poor oral bioavailability because of low intestinal permeability.11
To address these problems, various strategies including the coadministration of a therapeutic agent and functional excipients (such as P-gp inhibitors), and/or enhancers of drug solubility or permeability, have been developed to circumvent the biological barriers in play, or to promote drug absorption across the GI tract by altering the physicochemical properties of the drugs.5,16,17 Taurocholic acid (a bile acid) was incorporated into a nanostructured lipid carrier (NLC) to mediate carrier uptake via the bile acid transporter. A curcumin-loaded NLC containing bile acid was of small size and exhibited high-level drug encapsulation, an improved absorption rate, and a higher permeability coefficient than curcumin alone. Oral bioavailability increased by 4.27-fold, which was attributable to increased drug solubility in the nanoparticle, elimination of drug degradation by gastric acid and/or digestive enzymes, and specific binding of NLCs containing taurocholic acid to bile acid transporters.18 In addition, polymeric or lipid-based drug carriers, such as polymeric nanoparticles, polymeric micelles, microemulsions, self-emulsifying drug delivery systems, carbon nanotubes, liposomes, lipid–drug conjugates, and nanocrystals, have been used to improve the oral bioavailability of drug.18–22 The intestinal uptake and oral bioavailability of gemcitabine were enhanced after formulation of the drug into poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles using a multiple emulsification solvent evaporation method; oral bioavailability increased by 21.5-fold.23 Kim et al24 showed that the intestinal paracellular absorption of doxorubicin improved after the formulation of medium-chain, glyceride-based colloidal drug systems. Furthermore, the in situ intestinal absorption and in vivo oral bioavailability of doxorubicin in rats were significantly enhanced, which was attributable to the lipids used in formulation. Recently, Yen et al25 reported enhanced oral bioavailability of andrographolide after formulation of the drug into nanoemulsion using α-tocopherol and ethanol as an oil phase, Cremophor EL as surfactant, and water as the aqueous phase, attributable to enhancement of drug solubility, intestinal permeability and a reduction in droplet size. To date, only one oral PMX formulation featuring lipid/drug conjugate nanoparticles has been reported.26 In this work, the nanoparticles were prepared using a solvent evaporation method after mixing PMX with stearic acid (a permeation enhancer).26 Thereafter, lipid/drug conjugates containing an oil and a surfactant were prepared using a cold, high-pressure homogenization technique to produce nanoparticles, which exhibited high-level drug entrapment and increased gut permeation, improving the oral absorption of PMX in rats, probably because of passive drug diffusion across the intestinal membrane by virtue of the enhanced lipophilicity. However, oral absorption of a lipid-based formulation can be readily affected by dietary intake, lipid digestion, lipid emulsifiers such as bile salts, and dilution effect in and the pH of GI fluids.27 In addition, many organic solvents were used to prepare the lipid/drug conjugate. However, when a drug is conjugated with a cell recognition moiety such as a bile transporter, in an effort to exploit a natural intestinal uptake process, the drug may be delivered via facilitated diffusion and/or an increase in the drug concentration gradient through the intestinal membrane; only minimal amounts of an enhancer may be required.28 Furthermore, synergistic improvement in oral absorption would be expected on incorporation of the drug conjugate into a nanoemulsion, because of improved drug lipophilicity upon encapsulation in an oil layer combined with a permeation-enhancing effect caused by surfactant-induced changes in intestinal membrane structure and fluidity.
In our previous studies, we prepared a cationic bile acid derivative, Nα-deoxycholyl-L-lysyl-methylester (DCK), by chemical linkage between deoxycholic acid (DOCA) and L-lysine, which is an intestinal permeation enhancer. These studies demonstrated an increase in the intestinal membrane permeability and oral bioavailability of polar and hydrophilic drugs, such as bisphosphonates, insulin, and platinum antitumor compound (oxaliplatin), after ion-pairing complex formation between the drug molecule and DCK.29–31 This effect was mainly due to the amphiphilicity of DCK, which can be recognized by the bile acid transporters in the GI lumen as well as avoidance of the dilution of DCK in the GI fluid after ion-pairing complex formation.32,33 DCK is also a natural component of the human body that is biocompatible, biodegradable, and has low toxicity. Furthermore, the formulation of multiple water-in-oil-in-water (w/o/w) nanoemulsions by the incorporation of an amphiphilic oxaliplatin/DCK complex significantly enhanced the oral absorption and tumor growth inhibition of oxaliplatin.34 Thus, our approach of preparing ion-pairing complex and multiple nanoemulsions can effectively improve oral drug absorption with a minimal quantity of enhancer compared with conventional penetration enhancers.
In the current study, the main objective was to design an oral delivery system for PMX based on multiple w/o/w nanoemulsions by incorporating the ion-pairing complex of PMX and DCK (PMX/DCK). This can be highly absorbed via interaction with bile acid transporters, as well as by enhanced cellular uptake of nanoemulsion systems, for the purpose of providing maintenance chemotherapy after conventional cyclic cancer treatments, as well as synergistic drug combinations with other oral anticancer drugs and immunotherapy. To achieve this goal, we prepared an ion-pairing complex of PMX using dispersants such as 2-hydroxypropyl-beta-cyclodextrin (HP-beta-CD) and poloxamer 188 (P188) and a permeation enhancer such as DCK, forming an HP-beta-CD/PMX/DCK/P188 complex, and further incorporated the complex into the w/o/w nanoemulsion in a supersaturated state. After investigating its efficacy in inhibiting cancer cell proliferation and migration, the in vitro permeability of the HP-beta-CD/PMX/DCK/P188-loaded nanoemulsion (HP-beta-CD/PMX/DCK/P188-NE) across an artificial intestinal membrane and Caco-2 cell monolayer was assessed. In addition, enhanced cellular uptake via interaction with bile acid transporters of the PMX complex-loaded nanoemulsion was demonstrated using Caco-2 and apical sodium bile acid transporter (ASBT)-transfected Madin–Darby canine kidney (MDCK) cells. Finally, the oral bioavailability of the PMX complex-loaded nanoemulsion in rats and its efficacy at inhibiting tumor growth following oral administration to tumor-bearing mice were evaluated.
Materials and methods
Materials
PMX disodium hemi-pentahydrate was purchased from Shilpa Medicare (Raichur, India). Caprylocaproyl macrogol-8-glycerides (Labrasol), diethylene glycol monoethyl ether (Transcutol HP), and propylene glycol monocaprylate (Capryol 90) were obtained from Gattefossé (Saint-Priest, France). Polyoxyethylene (160) polyoxypropylene (30) glycol (P188), and polyethoxylated castor oil (Cremophor EL) were provided by BASF (Ludwigshafen, Germany). DOCA, HP-beta-CD, phalloidin–tetramethylrhodamine B isothiocyanate, and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Antihuman ASBT antibody and Alexa Fluor 546-labeled secondary antibody were obtained from Abcam (Cambridge, MA, USA). 2,4-Diamino-N,10-methylpteroic acid 4-[N-(2,4-diamino-6-pterinidinyl-methyl)-N-methylamino] benzoic acid hemihydrochloride hydrate (DAMPA) was used as an internal standard (IS) and obtained from Sigma-Aldrich Co. N-hydroxysuccinimide ester-labeled fluorescein (NHS-fluorescein) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Chloroform and methanol were obtained from EMD Millipore (Billerica, MA, USA). Solvents for high-performance liquid chromatography (HPLC) and liquid chromatography/mass spectrometry (LC/MS) analysis were obtained from EMD Millipore and Thermo Fisher Scientific.
Animals
Sprague Dawley rats (males, 200–250 g) and C57BL/6 mice (females, 20–25 g) were purchased from Orientbio (Gwangju, Republic of Korea). The animals were housed under standard housing conditions of temperature (23°C±2°C), relative humidity (55%±10%), and light (12/12-h light/dark cycle). The animals had ad libitum access to a standard laboratory diet (Nestlé Purina, St Louis, MO, USA) and ion-sterilized tap water.
Ethical approval for this study was obtained from the Institutional Animal Care and Use Committee (IACUC) of Mokpo National University (Jeonnam, Republic of Korea). All animal experiments were performed according to the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals and the guidelines of the IACUC.
Preparation and characterization of HP-beta-CD/PMX/DCK/P188 complex
DCK was synthesized by the conjugation of DOCA with a positively charged lysine and used as an oral absorption enhancer as previously described.34 The ionic complex formation of PMX and DCK (PMX/DCK) was prepared to enhance the intestinal membrane permeability of PMX (Figure 1A). In addition, HP-beta-CD and P188 were added for the purpose of avoiding the aggregation of PMX/DCK complex during preparation as well as improving its re-dispersion in aqueous medium. Briefly, 32.66 mg of HP-beta-CD was dissolved in 2 mL of deionized water and added dropwise into 2 mL of PMX solution (5 mg/mL in deionized water) at a 1:1 molar ratio with stirring overnight. Separately, 10 mg of P188 was dissolved in 0.6 mL of deionized water and added dropwise into the HP-beta-CD/PMX mixture. Then, DCK solution in a 1:1 molar ratio with PMX (13.36 mg of DCK dissolved in 1.4 mL of deionized water) was added dropwise into the mixture of HP-beta-CD, PMX, and P188 with regular vortexing. Finally, the HP-beta-CD/PMX/DCK/P188 was freeze-dried at −70°C to remove the water completely.
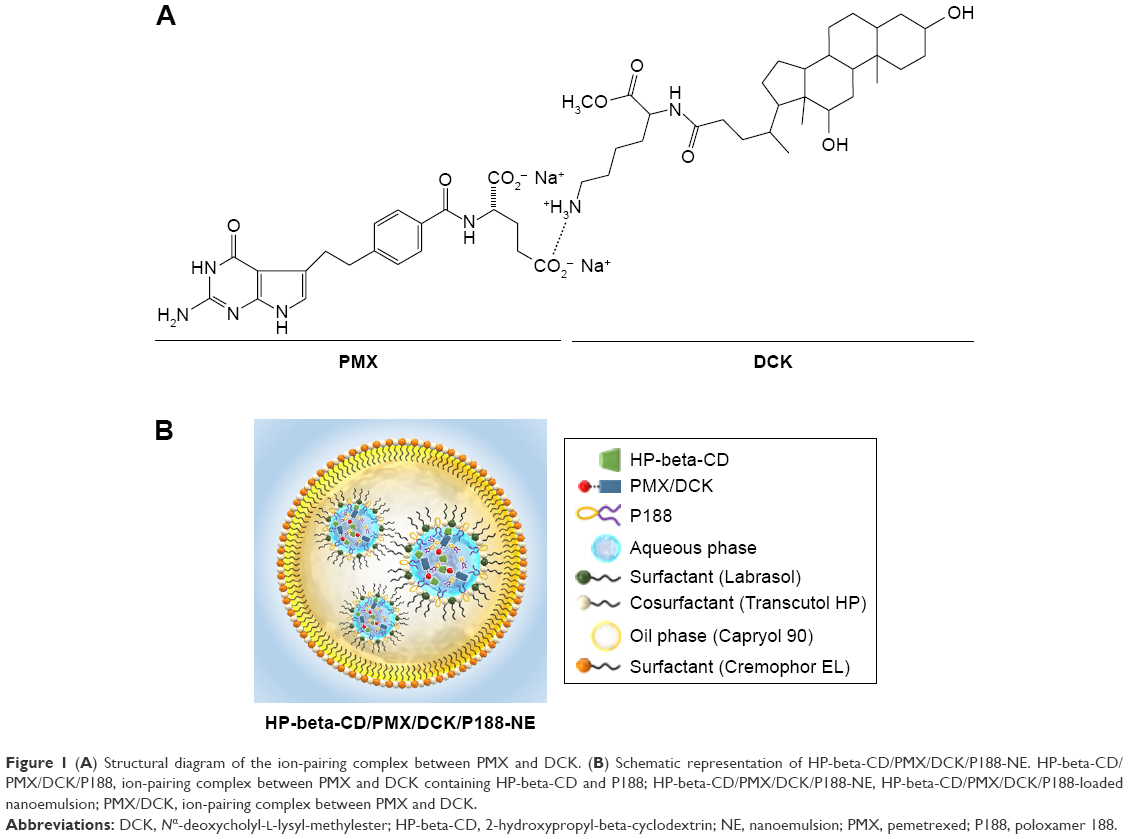 | Figure 1 (A) Structural diagram of the ion-pairing complex between PMX and DCK. (B) Schematic representation of HP-beta-CD/PMX/DCK/P188-NE. HP-beta-CD/PMX/DCK/P188, ion-pairing complex between PMX and DCK containing HP-beta-CD and P188; HP-beta-CD/PMX/DCK/P188-NE, HP-beta-CD/PMX/DCK/P188-loaded nanoemulsion; PMX/DCK, ion-pairing complex between PMX and DCK. |
To identify the complex formation between PMX and DCK, the characteristic crystalline properties of pure PMX and DCK, PMX/DCK, HP-beta-CD/PMX/DCK, and HP-beta-CD/PMX/DCK/P188 and a physical mixture of PMX, DCK, HP-beta-CD, and P188 were evaluated using powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC). The PXRD analysis of the powdered samples was performed using a D8 Advance diffractometer (Bruker AXS Inc., Madison, WI, USA) operating at 40 mA and 40 kV using copper (Cu-Kα1) radiation (λ=1.5418 Å). The powdered samples were deposited on an adhesive support with 0.5-mm thickness and analyzed using a diffractometer. The scanning region was set to step-scan mode in the range from 5° to 50° of the diffraction angle 2θ at a scanning rate of 0.02° per second. Moreover, DSC measurements were carried out to determine the thermal properties of each sample using a DSC Q1000 V9.9 Build 303 (TA Instrument Inc., New Castle, DE, USA). A few milligrams (1.5–2 mg) of each powder sample were non-hermetically trapped in a sealed aluminum pan and scanned at a heating rate of 5°C/min over the temperature range of 25°C–350°C under a nitrogen atmosphere.
Preparation and characterization of w/o/w nanoemulsion
A multiple nanoemulsion system was prepared by a two-step spontaneous emulsification method. First, the primary w/o nanoemulsion containing HP-beta-CD/PMX/DCK/P188 (based on 10 mg of PMX) was prepared based on the solubility of the complex in water by an oil phase titration method using Capryol 90, Labrasol, Transcutol HP, and deionized water as an oil, surfactant, cosurfactant, and aqueous phase, respectively. We prepared a transparent primary nanoemulsion featuring the smallest droplet size possible and the maximum aqueous content. We employed a 21.4% (w/w) aqueous solution of the HP-beta-CD/PMX/DCK/P188, a 50.0% (w/w) surfactant/cosurfactant mixture (Smix, 1; Labrasol:Transcutol HP, 1:2, w/w), and a 28.6% (w/w) oil phase. Second, the w/o/w nanoemulsion of HP-beta-CD/PMX/DCK/P188 was prepared by an aqueous phase titration method using the primary nanoemulsion, Cremophor EL, Transcutol HP, and deionized water as secondary oil phase, surfactant, cosurfactant, and aqueous phase, respectively (Figure 1B). We chose the optimum formulation for a w/o/w nanoemulsion entrapping HP-beta-CD/PMX/DCK/P188 (HP-beta-CD/PMX/DCK/P188-NE) by reference to the clear zone of the pseudo-ternary phase diagram based on relevant physicochemical properties, including droplet size and permeability of an artificial intestinal membrane in vitro. The composition was as follows: 16.7% (w/w) w/o nanoemulsion (oil phase), 50.0% (w/w) surfactant/cosurfactant mixture (Smix, 2; Cremophor EL:Transcutol HP, 1:1, w/w), and 33.3% (w/w) deionized water.
The optimized nanoemulsion was further characterized by average droplet size, polydispersity index (PDI), and zeta potential at 25°C using a dynamic laser light scattering analyzer (Malvern Zetasizer Nano ZS90; Malvern Instruments, Malvern, UK). The HP-beta-CD/PMX/DCK/P188-NE was diluted with deionized water (1:200) and sonicated for 1 min to minimize multiple scattering effects. The surface morphology and structure of the complex-loaded nanoemulsion were then evaluated using high-resolution transmission electron microscopy (TEM, JEM-200; JEOL, Tokyo, Japan). The optimized w/o/w nanoemulsion was diluted 100 times with deionized water, and a drop of nanoemulsion was placed on a copper grid. After removing the excess with filter paper, one drop of 2% aqueous solution of phosphotungstic acid was added onto the grid to allow negative staining. The excess was removed with filter paper, and the grid was observed by TEM.
In vitro inhibitory effect on cancer cell proliferation and migration
In vitro cytotoxic effect
The in vitro cytotoxic effects of free PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE were evaluated by a cell counting assay method (Cell Counting Kit-8 [CCK-8]; Dojindo Molecular Technologies, Rockville, MD, USA). Briefly, Lewis lung carcinoma (LLC) cells (ATCC® CRL-1642™) purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and human lung carcinoma (A549) cells (ATCC® CCL-185™) were seeded at 5×103 cells/well in 100-μL amounts of Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% (v/v) fetal bovine serum (FBS) or 100-μL amounts of Roswell Park Memorial Institute (RPMI) medium with 10% (v/v) FBS in 96-well plates, respectively, and cultured at 37°C for 24 h. The cells were then treated with serially diluted sample solutions at 0.01, 0.05, 0.1, 0.5, 1, 5, and 10 μg/mL PMX, PMX/DCK complexes, or HP-beta-CD/PMX/DCK/P188-NE in DMEM or RPMI. After drug loading, the cells were cultured for an additional 48 h. To evaluate the cell viability, a 10-μL WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt) solution was added to each well and incubated for 2 h. The absorbance was then measured using a microplate reader (PerkinElmer Multimode Plate Reader; PerkinElmer Inc., Waltham, MA, USA) at 450 nm. The obtained results for the treated cells were expressed as the percentage of viable cells compared with those of untreated cells.
In vitro wound-healing assay
Next, an in vitro wound-healing assay was performed to compare the efficacy of inhibition of cancer cell proliferation/migration after the complex formation with DCK as well as incorporation into the nanoemulsion. The LLC or A549 cells were seeded at a density of 3×104 cells/well in 200 μL of DMEM or RPMI medium containing 10% FBS on collagen-coated 96-well plates, respectively, and incubated at 37°C for 48 h to form a nearly confluent monolayer. Then, each well was carefully scratched to make a linear wound region (a cell-free zone) using a wound maker. The monolayer was washed twice with phosphate-buffered saline (PBS, pH 7.4) to remove the detached cells. Next, the cells were treated with free PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE in the respective media at concentrations equivalent to 0.01, 0.05, 0.1, 0.5, 1, 5, and 10 μg/mL PMX and cultured at 37°C for 48 h. The drug-treated wells were photographed, and cell migration was monitored every 12 h by phase-contrast imaging with an IncuCyte FLR microscope (Essen Bioscience, Ann Arbor, MI, USA). IncuCyte FLR image analysis software was used to detect the cell edges automatically and to generate an overlay mask, which was used to calculate the cell inhibition area.
Partition coefficient and membrane permeability
Partition coefficient
To determine the partition coefficients of pure PMX, HP-beta-CD/PMX, PMX/DCK, HP-beta-CD/PMX/DCK, and HP-beta-CD/PMX/DCK/P188, 1 mL of aqueous solution of PMX or each complex was mixed with 1 mL of octanol, and the mixture was vigorously vortexed for 10 min and agitated for 24 h in a thermostatic shaker bath at 25°C±0.1°C. After equilibration, the samples were centrifuged at 5,000× g for 15 min. Finally, the aqueous phase was collected and measured for the concentration of PMX using HPLC with a C18 column (4.6×250 mm, 5 μm, 100 Å; 20-μL sample injection) at 25°C. The mobile phase consisted of water (pH 3.5 adjusted with phosphoric acid)–acetonitrile (80:20, v/v) and was run at a flow rate of 1 mL/min. PMX or PMX/DCK complex was measured using an ultraviolet (UV) detector at 254 nm. The partition coefficient was calculated using the following equation:
| (1) |

where Co represents the concentration of PMX in octanol at equilibrium, and Cw represents the concentration of PMX in water at equilibrium.
In vitro artificial intestinal membrane permeability
The in vitro intestinal membrane permeabilities of free PMX, HP-beta-CD/PMX, PMX/DCK, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE were evaluated using a parallel artificial membrane permeability assay (PAMPA; BD Biosciences, San Jose, CA, USA) as described previously.34 Donor samples were prepared by the dilution of free PMX, PMX/DCK complexes, or HP-beta-CD/PMX/DCK/P188-NE with PBS (pH 6.8) at a concentration equivalent to 200 μg/mL PMX. Then, 200 μL of sample solution was added to each well of the donor plate, and 300 μL of PBS (pH 6.8) was added to each well of the acceptor plate. After 5 h of incubation at room temperature, the plate assembly was detached and samples were withdrawn from both the acceptor and the donor plates. The concentration of PMX that permeated through the membrane was then measured by HPLC at 254 nm as mentioned earlier. The effective permeability (Pe) of each drug was calculated using the following formula:
| (2) |

where Pe is the permeability (cm/s), A is the effective filter area (0.228 cm2), VD is the volume of the donor well (0.2 mL), VA is the volume of the receptor well (0.3 mL), t is the total time of incubation in seconds, CA(t) denotes the concentration of drug in the receptor well at time t, and Cequilibrium represents (CD[t] × VD + CA[t] × VA)/(VD + VA), where CD(t) denotes the concentration of drug in the donor well at time t.
In vitro permeability across a Caco-2 cell monolayer
To investigate the permeability of free PMX, HP-beta-CD/PMX, PMX/DCK, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE across a Caco-2 monolayer, Caco-2 cells (ATCC® HTB-37™) were grown on each 12-well Transwell® (pore size 0.4 μm, surface area 1.12 cm2; Corning Incorporated, Corning, NY, USA) at a density of 3×105 cells/well in DMEM containing 10% FBS and 1% penicillin/streptomycin for 21–29 days. When the cell monolayers exhibited transepithelial electrical resistance (TEER) of >350 Ω·cm2, the apical and basolateral compartments were stabilized with 0.5 and 1.5 mL of preheated (37°C) Hanks’ balanced salt solution (HBSS) for 20 min at 37°C, respectively. Then, HBSS in the apical compartment was replaced with 0.5 mL of free PMX, PMX/DCK complexes, or HP-beta-CD/PMX/DCK/P188-NE diluted with HBSS (equivalent to 100 μg/mL PMX), and the basolateral compartment was replaced with 1.5 mL of fresh HBSS. During incubation of the plate at 37°C, 100 μL of sample solution was withdrawn from each basolateral compartment and 100 μL of fresh HBSS was replaced at 0.5, 1, 2, 3, 4, and 5 h. The collected samples were then filtered through a membrane filter (0.45 μm, polyvinylidene fluoride), and the concentrations of PMX or PMX complex that permeated through the Caco-2 monolayer were determined using the HPLC system with a UV detector as mentioned earlier. The apparent permeability coefficient (Papp) of PMX or PMX complex was calculated according to the following equation:
| (3) |
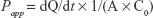
where dQ/dt indicates the linear appearance rate of mass in the basolateral sides (μmol/s), C0 is the initial concentration of PMX or PMX complex on the apical side (μmol/mL), and A is the surface area of the monolayer (cm2).
Cellular uptake into the Caco-2 and ASBT-transfected MDCK cells
To evaluate the improvement in cellular uptake of PMX after complex formation with DCK as well as incorporation into the nanoemulsion, cellular uptake of PMX/DCK or PMX/DCK-loaded nanoemulsion through Caco-2 cells was assessed by treating the cells with fluorescein isothiocyanate (FITC)-conjugated PMX or PMX/DCK (FITC-PMX and FITC-PMX/DCK, respectively) and their nanoemulsions (FITC-PMX-NE and FITC-PMX/DCK-NE, respectively) for 1 h at a concentration equivalent to 100 μg/mL PMX at 37°C. In addition, the actin filaments on the Caco-2 cells were stained with phalloidin–rhodamine (100 nM) for 1 h.
To investigate the effect of interaction between DCK and bile acid transporters on facilitating the intestinal membrane penetration of PMX/DCK complex, MDCK cells (ATCC® CCL-34™) were transfected with ASBT genes using Lipofectamine 2000® (Thermo Fisher Scientific) and were grown until the formation of a confluent monolayer in a 10-mm coverslip (Warner Instruments, Hamden, CT, USA) coated with Cell-Tak solution (Sigma-Aldrich Co.) for 20 min. After washing three times with 1 mL of ice-cold HBSS, 100 μL of FITC-PMX, FITC-PMX/DCK, FITC-PMX-NE, or FITC-PMX/DCK-NE diluted with DMEM (each containing 100 μg/mL PMX) was loaded in the dish and incubated for 1 h at 37°C. Then, the cells were washed three times with 1 mL of ice-cold HBSS, followed by fixing with 100 μL of 4% cold paraformaldehyde solution in PBS (pH 7.4) for 20 min at room temperature. Next, the samples were treated with 0.3% Triton X-100 in blocking solution made of 10% normal goat serum in PBS (pH 7.4) for 40 min and treated with antihuman ASBT antibody (1:500) diluted in blocking buffer overnight at 4°C. The primary antihuman ASBT antibody was then stained with 10 μg/mL Alexa Fluor 546-labeled secondary antibody for 1 h. The nucleus was also counterstained by DAPI (1 μg/mL) for 5 min. After several washes with PBS (pH 7.4), the cells were covered with a glass cover slip using mounting medium. Finally, fluorescence images were obtained using a confocal laser scanning microscope (CLSM; Carl Zeiss Meditec AG, Jena, Germany).
In vitro drug release
To assess the drug dissolution rate of free PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE, in vitro drug release studies were performed in 500 mL of medium containing 0.1 N HCl solution (pH 1.2) or phosphate buffer (pH 6.8) at 37°C±0.2°C, using an United States Pharmacopeia dissolution test apparatus I (basket) at 100 rpm. Then, 10 mg of PMX, each PMX complex (equivalent to 10 mg of PMX) or 800 mg of HP-beta-CD/PMX/DCK/P188-NE composed of the primary w/o nanoemulsion including HP-beta-CD/PMX/DCK/P188 equivalent to 10 mg of PMX and Smix, 2, was poured into a hard gelatin capsule of size #00. Each capsule was then subjected to a dissolution test, and 1 mL of sample solution was withdrawn at 15, 30, 45, 60, 90, and 120 min. After filtration, the amount of PMX or PMX complex in the sample was quantified by HPLC using a UV detector as described earlier.
In vivo pharmacokinetic study in rats
The pharmacokinetic properties of free PMX, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE after oral administration to rats were determined to evaluate the effects of ionic complex formation with DCK nanoemulsion formulation of PMX/DCK on the oral absorption of PMX. The rats received oral doses of 400 μL of an aqueous solution of free PMX (20 mg/kg), HP-beta-CD/PMX/DCK/P188 (equivalent to 20 mg/kg PMX), or HP-beta-CD/PMX/DCK/P188-NE (equivalent to 20 mg/kg PMX). In addition, 150 μL of aqueous solution of PMX (10 mg/kg) was injected via the tail vein to evaluate the oral bioavailability. After administration of the drug solution or formulation, blood samples were collected from a capillary in the retro-orbital plexus at predetermined time intervals and directly mixed with 50 μL of 3.8% sodium citrate solution. After the blood samples were immediately centrifuged (2,500× g, 15 min, 4°C), the plasma samples were isolated and stored at −70°C until analysis.
To measure the plasma concentration of PMX, the plasma sample was defrosted and centrifuged at 2,500× g for 5 min at 4°C. A total of 100 μL of each standard solution or plasma sample was then mixed with 10 μL of DAMPA (5 μg/mL, IS), and 300 μL of 2% NH4OH in water was added to each tube. The entire mixture was further subjected to solid-phase extraction using a Plexa Bond Elut PAX cartridge (30 mg, 1 mL; Agilent Technologies, Santa Clara, CA, USA) as recommended by the manufacturer. The cartridge was conditioned with 500 μL of methanol followed by 500 μL of deionized water. The standard or plasma sample solution diluted with 2% NH4OH was loaded into the cartridge and washed with 500 μL of deionized water followed by 500 μL of methanol to remove the impurities and unadsorbed samples. Then, the adsorbed samples were eluted with 2×250 μL of 5% formic acid prepared in methanol. After evaporating organic solvent in the collected samples using a centrifugal evaporator (Genevac Ltd., Ipswich, UK), the dried residue was resuspended with 100 μL of 5% formic acid prepared in methanol. The plasma concentration of PMX was determined using an Agilent 6120 quadruple LC/MS system with a Phenomenex Luna C18 column (100×2 mm, 3 μm), and the samples were chromatographed using isocratic mobile phase (acetonitrile:0.34% formic acid, 15:85, v/v) at a flow rate of 0.2 mL/min. A 5-μL sample was injected, and PMX was measured in positive ionization mode using an atmospheric pressure ionization-electron spray (API-ES) source. The following parameters were optimized for PMX analysis: capillary voltage, 3,500 V; drying gas flow rate, 3.1 L/min, and drying gas temperature, 300°C. The fragment ions at this capillary voltage were detected and quantified at ([M+H]+=428) and ([M+H]+=326.1) for PMX and DAMPA, respectively.
In vivo tumor growth inhibition effect
To evaluate the effect on tumor growth inhibition of orally administered HP-beta-CD/PMX/DCK/P188-NE, LLC cells were subcutaneously grafted into the flanks of C57BL/6 mice at a concentration of 1×106 cells/100 μL of PBS (pH 7.4). When each tumor became palpable (80–100 mm3), mice were randomly divided into four groups of 10 animals each: control (not treated), PMX-IV (once-weekly intravenous [IV] administration of 20 mg/kg PMX in water), PMX-S (once-a-day oral administration of 20 mg/kg PMX in water), and HP-beta-CD/PMX/DCK/P188-NE (once-a-day oral administration of HP-beta-CD/PMX/DCK/P188-loaded w/o/w nanoemulsion consisting of 20 mg/kg PMX). During the treatment, mice were fasted for 4 h before and 2 h after the oral administration. The growth rate of the tumor was measured every 3 days in two dimensions using a Vernier caliper, and tumor volumes were calculated as a2×b×0.52, where a is the width and b is the length. Body weight was also measured. At 21 days after administration, the isolated tumor masses were measured and tumor tissues were fixed in 10% formalin for histological evaluation. Angiogenesis in tumor tissues was assessed by CD31 vessel immunostaining, which presented brown staining of endothelial cells or their clusters. In addition, proliferating cells and apoptotic cell death in tissue sections were analyzed by staining with proliferating cell nuclear antigen (PCNA) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, respectively.
Pharmacokinetic and statistical analyses
Pharmacokinetic parameters were determined using a non-compartmental method in WinNonlin® software (ver. 5.3; Pharsight Corporation, St Louis, MO, USA). All data are expressed as mean ± SD. A P-value of <0.05 was considered to indicate statistical significance using a Student’s t-test between two mean values for unpaired data or one-way analysis of variance followed by Tukey’s multiple comparison test among three or more mean values for unpaired data.
Results and discussion
Preparation and characterization of HP-beta-CD/PMX/DCK/P188 complex
To confirm the ion-pairing complex formation between PMX and DCK, the PXRD spectra of PMX, DCK, and the physical mixtures were compared with those of PMX/DCK, HP-beta-CD/PMX/DCK, and HP-beta-CD/PMX/DCK/P188 (Figure 2A). Pure PMX presented characteristic crystalline peaks at 11.43°, 14.08°, 15.88°, 16.37°, 16.88°, 17.41°, 19.08°, 22.78°, 23.94°, 24.60°, 26.19°, 27.74°, 28.96°, and 30.07° over the 2θ range in the PXRD spectra. Crystalline peaks of PMX were also identified in the physical mixture, indicating that PMX remained in crystalline form. However, these peaks were not observed in the lyophilized powder forms of PMX/DCK, HP-beta-CD/PMX/DCK, or HP-beta-CD/PMX/DCK/P188. Therefore, PMX had become molecularly dispersed with DCK, HP-beta-CD, and P188, resulting in the formation of amorphous inclusion complexes. The disappearance of PMX crystallinity was confirmed by DSC (Figure 2B). The thermal curve of pure PMX exhibited a narrow, sharp endothermic peak at 115°C, characteristic of the crystalline form; this was absent in PMX/DCK, HP-beta-CD/PMX/DCK, and HP-beta-CD/PMX/DCK/P188. In addition, no clear endothermic PMX peak was evident in the thermogram of a physical mixture of HP-beta-CD, PMX, DCK, and P188, indicating that PMX was molecularly dispersed (thus no longer crystalline) after conversion into an amorphous state, probably due to the formation of a solid dispersion during heating.35
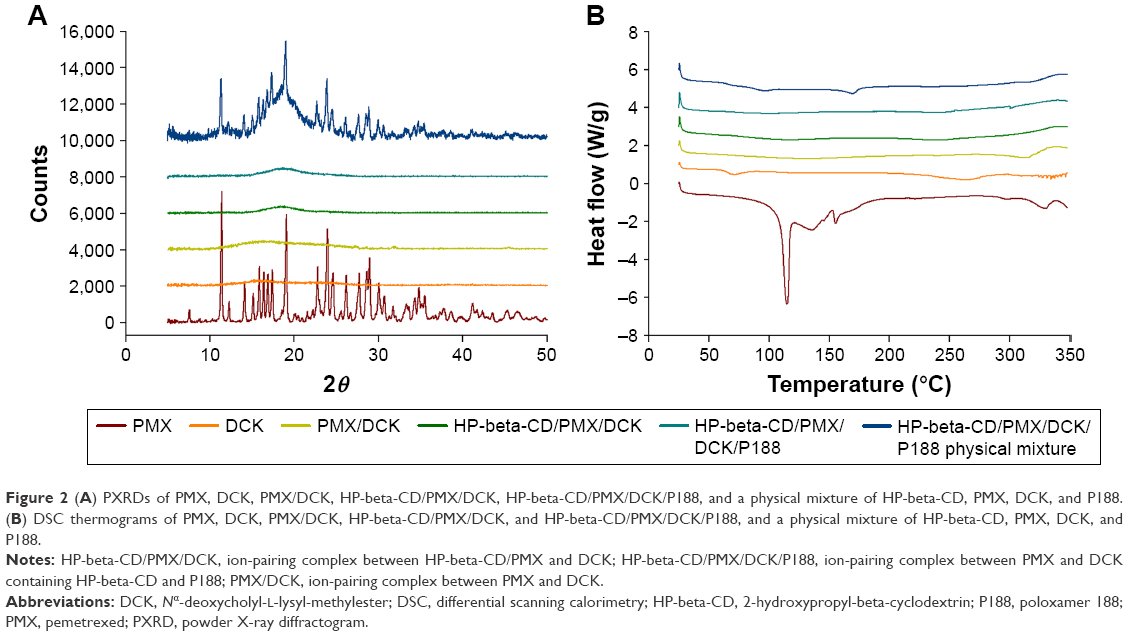 | Figure 2 (A) PXRDs of PMX, DCK, PMX/DCK, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and a physical mixture of HP-beta-CD, PMX, DCK, and P188. (B) DSC thermograms of PMX, DCK, PMX/DCK, HP-beta-CD/PMX/DCK, and HP-beta-CD/PMX/DCK/P188, and a physical mixture of HP-beta-CD, PMX, DCK, and P188. |
Previous studies revealed that the amorphous solid dispersion of a poorly water-soluble drug can represent a way of overcoming the trade-off regarding an increase in the apparent solubility of the drug leading to a loss of its apparent permeability; this is because an amorphous solid dispersion comprising drug and solubilizing agent can retain the supersaturated state over a prolonged time in which solubility–permeability functions independently, leading to improved oral absorption.36–39 Similarly, the membrane permeability of a freely aqueous soluble PMX can be enhanced by ion-pairing complex formation with the lipophilic DCK molecule without loss of its aqueous solubility because the PMX exists as an amorphous form in DCK complexes.
Preparation and characterization of w/o/w nanoemulsion
Components for w/o/w nanoemulsion were determined based on the miscibility and stability of each excipient. Subsequently, pseudo-ternary phase diagrams were constructed to identify the self-nanoemulsifying region and optimize the concentrations of water, surfactant, cosurfactant, and oil.40 The formulation with maximum nanoemulsion area that appeared to be clear, transparent, and free flowing was selected and analyzed for particle size, zeta potential, and membrane permeability (data not shown). Furthermore, the amphiphilic HP-beta-CD/PMX/DCK/P188 could be incorporated into the w/o primary nanoemulsion as a supersaturated form, which may be possible due to the amorphous state of the PMX/DCK and dispersing agents (HP-beta-CD and P188). Thus, HP-beta-CD/PMX/DCK/P188 may be located in the aqueous phase as well as the oil–water interface along with Smix, 1. The optimum multiple nanoemulsion system was then chosen from the formulations that can undergo self-nanoemulsification without phase separation as well as drug complex precipitation when subjected to infinite dilution with GI fluids. As a result, the optimum w/o/w nanoemulsion exhibited a droplet size and PDI of 14.5±0.45 and 0.13±0.05 nm, respectively (Figure 3A). A TEM image also confirmed the formation of nano-sized droplets that were uniformly dispersed, with a narrow size distribution of diameter <50 nm, according to the findings measured by a particle size analyzer (Figure 3B). These results implied that the surfactants and cosurfactants were preferentially adsorbed at the oil–water interface, resulting in a thermodynamically stable spontaneous dispersion form.22,41 The optimized Smix ratio increased the dispersion entropy and interfacial area, which was accompanied by a reduction in the interfacial tension and free energy of the oral nanoemulsion system at its minimum concentration value.42,43 Moreover, the Smix to oil phase ratio was also optimized to form a more closely packed surfactant layer at the oil–water interface, allowing reductions in the interfacial energy and droplet size as well as acting as a mechanical barrier to flocculation.44,45 The zeta potential of the optimized nanoemulsion was −3.15±0.89 mV, indicating that the fatty acids and esters in the oil layer of the w/o primary emulsion were completely covered with surfactant/cosurfactant in the w/o/w double emulsion. Furthermore, the drug content of the optimized nanoemulsion was maintained at >95%; we recorded no precipitation, phase separation, or significant change in either droplet size or the PDI 3 months after the storage of samples at 25°C±2°C (65%±5% relative humidity) or 40°C±2°C (75%±5% relative humidity; data not shown).
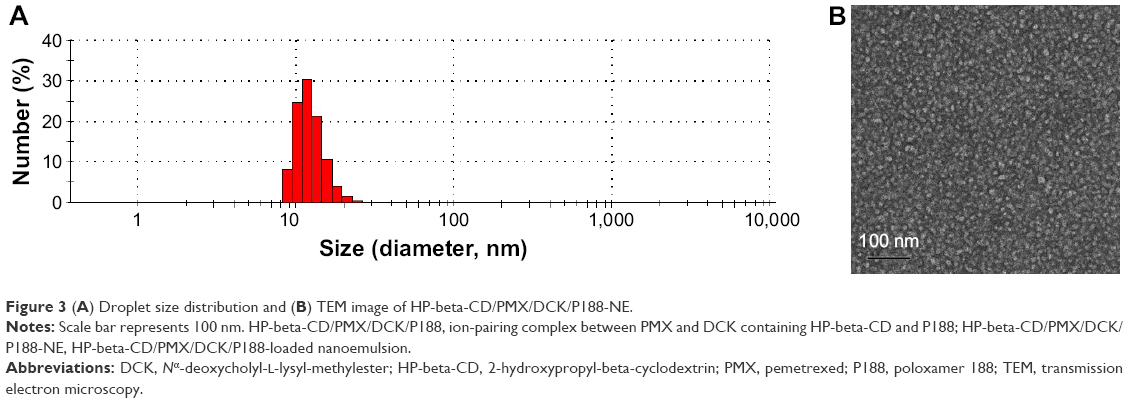 | Figure 3 (A) Droplet size distribution and (B) TEM image of HP-beta-CD/PMX/DCK/P188-NE. |
Cytotoxic effect
The in vitro cytotoxic effects of PMX or PMX/DCK complexes and PMX/DCK complex-loaded nanoemulsion were evaluated by cell viability assays in LLC and A549 cells, after treatment with various concentrations of PMX or PMX complexes. At >0.05 μg/mL (0.08 μM) PMX, the LLC cell viability was decreased to <80% and was 45.4%±3.74% at 0.5 μg/mL (0.84 μM). As the PMX concentration increased, the cell viability decreased and reached 29.8%±3.82% at 5 μg/mL PMX (Figure 4A). The cell viability of HP-beta-CD/PMX or PMX/DCK complexes also decreased as the PMX concentration increased, but there was no significant change in the LLC cell viability compared with the effects of PMX at the same molar concentration; the cell viability values of HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, and HP-beta-CD/PMX/DCK/P188 were evaluated as 47.4%±6.65%, 45.4%±6.63%, and 50.7%±4.49%, respectively, at 0.5 μg/mL (0.84 μM) PMX. The HP-beta-CD/PMX/DCK/P188-NE also displayed dose-dependent cytotoxicity; the cell viability value was 39.8%±8.98% at 0.5 μg/mL (0.84 μM) PMX in nanoemulsion. However, the cell viability of HP-beta-CD/PMX/DCK/P188-NE was decreased significantly compared with that of free PMX when the PMX concentration in the nanoemulsion was higher than 5 μg/mL (8.37 μM). At the same molar concentrations, the cell viability after the administration of HP-beta-CD/PMX/DCK/P188-NE was 45.6% less than that after the addition of PMX at 5 μg/mL (8.37 μM), possibly because of increased cellular uptake of the nano-sized droplets of the nanoemulsion. The cell viability associated with the administration of nanoemulsion alone was 98.5%±2.06%.
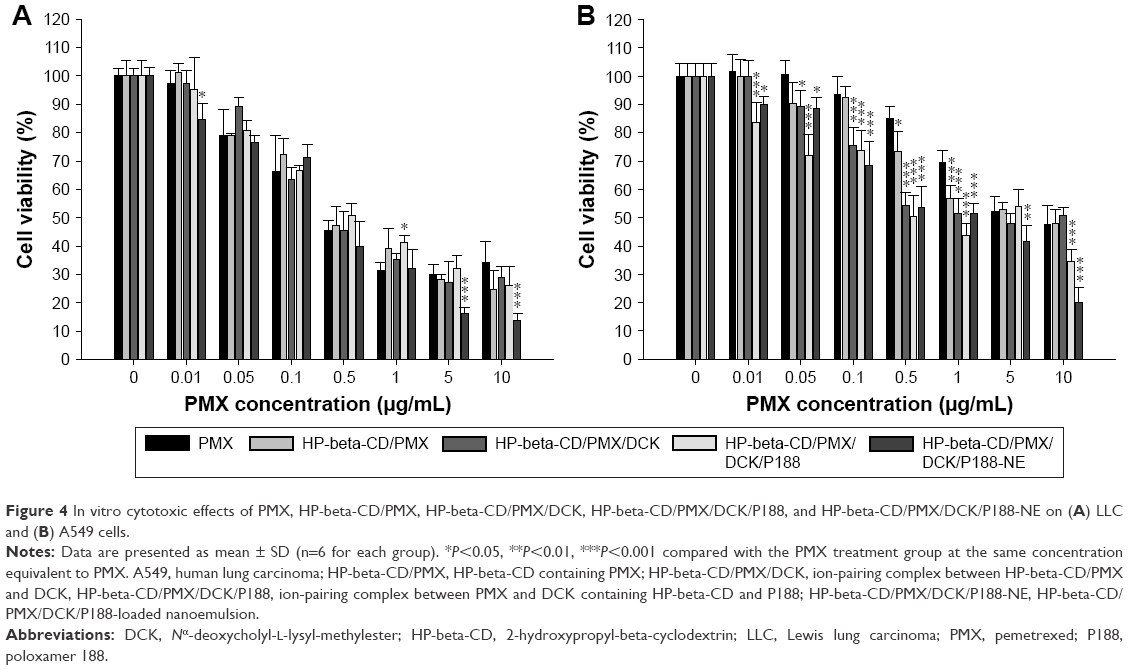 | Figure 4 In vitro cytotoxic effects of PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE on (A) LLC and (B) A549 cells. |
PMX also showed a dose-dependent cytotoxic effect on A549 cells when the drug concentration was higher than 0.5 μg/mL (0.84 μM); the A549 cell viability was decreased to 52.2%±5.25% at 5 μg/mL (8.37 μM; Figure 4B). The cell viability levels of the HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE were also decreased as the PMX concentration increased and were <80% when the concentration of PMX was higher than 0.1 μg/mL (0.17 μM). Moreover, the cytotoxicity of PMX on A549 cells was significantly increased after complex formation with DCK or by incorporation into the nanoemulsion; cell viability values of HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE at a concentration equivalent to 0.5 μg/mL (0.84 μM) PMX were decreased to 54.3%±4.56%, 50.4%±7.42%, and 53.4%±7.60%, respectively. In addition, the cell viabilities after the addition of HP-beta-CD/PMX/DCK/P188 and HP-beta-CD/PMX/DCK/P188-NE at concentrations equivalent to 10 μg/mL (16.8 μM) PMX were 28.0% and 57.7% lower than that after the addition of PMX alone, respectively. However, the cell viabilities after the addition of HP-beta-CD, DCK, P188, and nanoemulsion alone were all >80% of that when the same molar concentration of PMX was administered. Therefore, the enhanced cytotoxic effects of the PMX/DCK complexes or HP-beta-CD/PMX/DCK/P188-NE may be attributable to improved membrane permeability, implying that the cytotoxic efficacy of PMX was enhanced by complex formation with DCK followed by incorporation into the nanoemulsion.
The PMX/DCK complex and HP-beta-CD/PMX/DCK/P188-NE were more cytotoxic than PMX toward A549 than LLC cells, perhaps because LLC cells absorbed the drug more effectively than did A549 cells. In other studies, LLC cells commonly exhibit better uptake than A549 cells under identical conditions, possibly attributable to internalization of drugs or micelles that effectively transport drugs into cells.46,47 Therefore, the enhancement in membrane permeability caused by complex or nanoemulsion formulation created a more marked cytotoxic effect toward A549 than LLC cells.
Inhibition of cancer cell proliferation and migration
Once we confirmed the dose-dependent cytotoxic effects of PMX/DCK complexes and HP-beta-CD/PMX/DCK/P188-NE, we performed a wound-healing assay to investigate their inhibitory efficacy in cancer cell proliferation/migration in comparison with PMX. More than 90% of the scratched wound area in the control LLC cells was recovered at 48 h after treatment (Figure 5A). On the other hand, the proliferation/migration of LLC cells treated with more than 0.05 μg/mL (0.08 μM) PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE was significantly suppressed in a dose-dependent manner. The process of wound closure, driven by the proliferation/migration of LLC cells, is illustrated by representative images in Figure 5B and C. While the wounded area in the control cells was almost completely recovered at 48 h, the wound recovery rates were significantly decreased 24 h after treatment with PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, or HP-beta-CD/PMX/DCK/P188-NE equivalent to 0.5 μg/mL (0.84 μM) PMX, resulting in sizeable wound area gaps in the monolayer still being present; at 48 h after treatment with PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, or HP-beta-CD/PMX/DCK/P188-NE, the relative wound recovery values were 57.5%±5.76%, 52.4%±2.22%, 55.7%±6.50%, 53.3%±7.94%, and 45.2%±2.57%, respectively. Ion-pairing complex formation between PMX and DCK and the addition of HP-beta-CD and P188 did not have a further inhibitory effect on LLC cell proliferation/migration. However, in the LLC cells treated with HP-beta-CD/PMX/DCK/P188-NE containing more than 0.5 μg/mL (0.84 μM) PMX, there was a greater inhibitory effect on cell proliferation/migration than that for PMX. Therefore, after 48 h of treatment, the relative recovery of the cells treated with HP-beta-CD/PMX/DCK/P188-NE equivalent to 1 μg/mL (1.68 μM) PMX was 1.55-fold lower than that of PMX and further decreased to 2.76%±1.64% after the cells were treated with HP-beta-CD/PMX/DCK/P188-NE equivalent to 10 μg/mL (16.8 μM) PMX (41.7%±5.83%), and this may be because of the higher cancer cell permeability of PMX/DCK complex after incorporation into the nanoemulsive system.
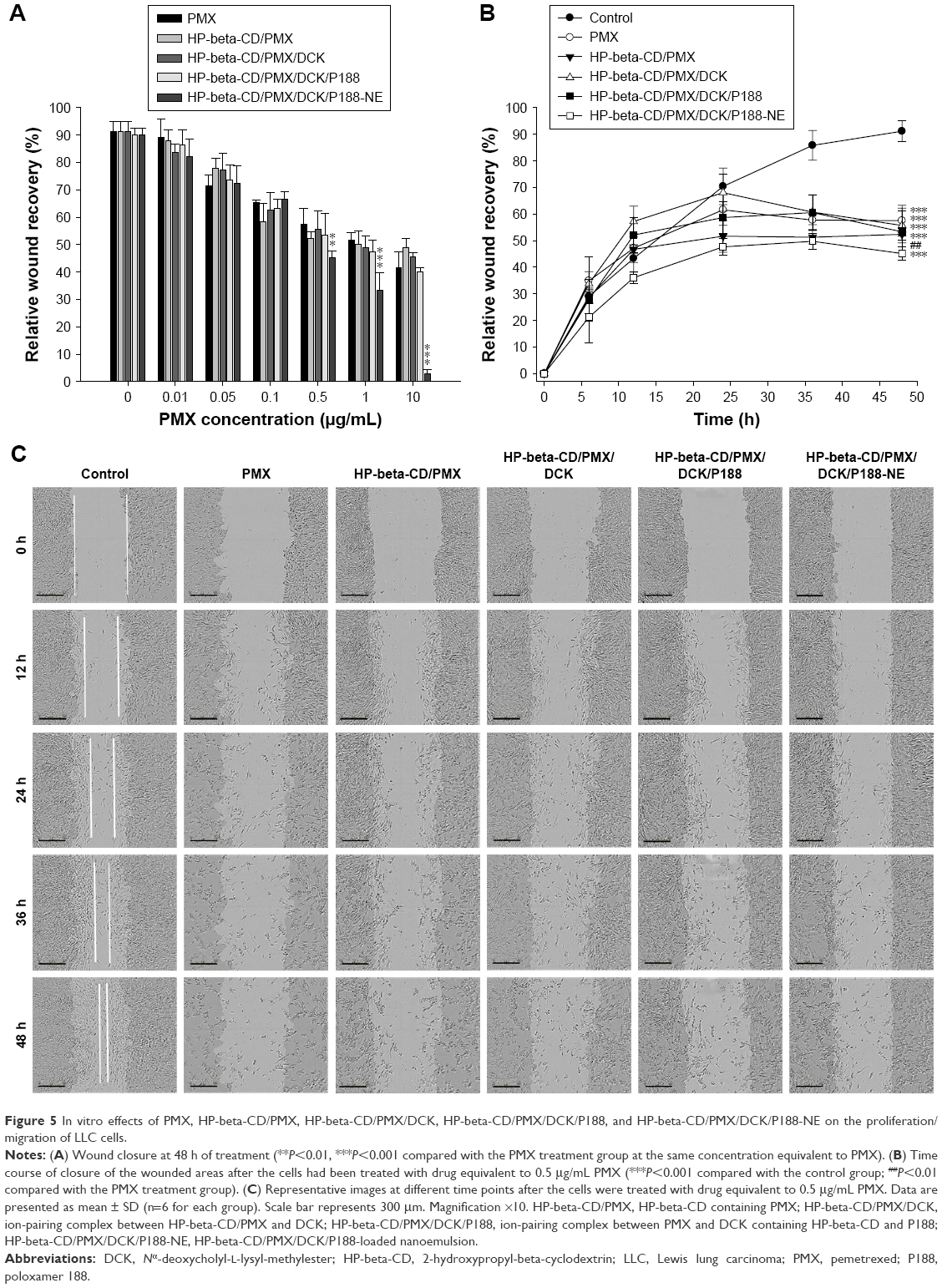 | Figure 5 In vitro effects of PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE on the proliferation/migration of LLC cells. |
Next, we examined the inhibitory effects of PMX, PMX/DCK complexes, and HP-beta-CD/PMX/DCK/P188-NE on A549 cell proliferation/migration. The cell proliferation/migration was significantly inhibited when the concentration of PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, or HP-beta-CD/PMX/DCK/P188-NE was more than 0.1 μg/mL (0.17 μM) based on PMX (Figure 6A). At 48 h, the cells treated with PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, or HP-beta-CD/PMX/DCK/P188-NE equivalent to 0.5 μg/mL (0.84 μM) PMX presented 1.28-, 1.52-, 1.43-, 1.50-, and 1.75-fold higher inhibition of cell proliferation/migration than that of the control cells, respectively. Figure 6B and C shows the wound recovery rates of A549 cells at different time points. A549 cell proliferation/migration was also significantly inhibited after 24 h of drug treatment, and the wound recovery rates at 48 h after the cells were treated with PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, or HP-beta-CD/PMX/DCK/P188-NE equivalent to 1 μg/mL (1.67 μM) PMX were evaluated as 54.0%±5.09%, 54.3%±4.29%, 55.3%±4.25%, 49.6%±2.13%, and 45.9%±6.20%, respectively. Thus, PMX/DCK complexes and HP-beta-CD/PMX/DCK/P188-NE can be applied as an effective anticancer therapy as well as delivery carriers for PMX due to their dose-dependent cytotoxic and inhibitory activity on cancer cell proliferation/migration.
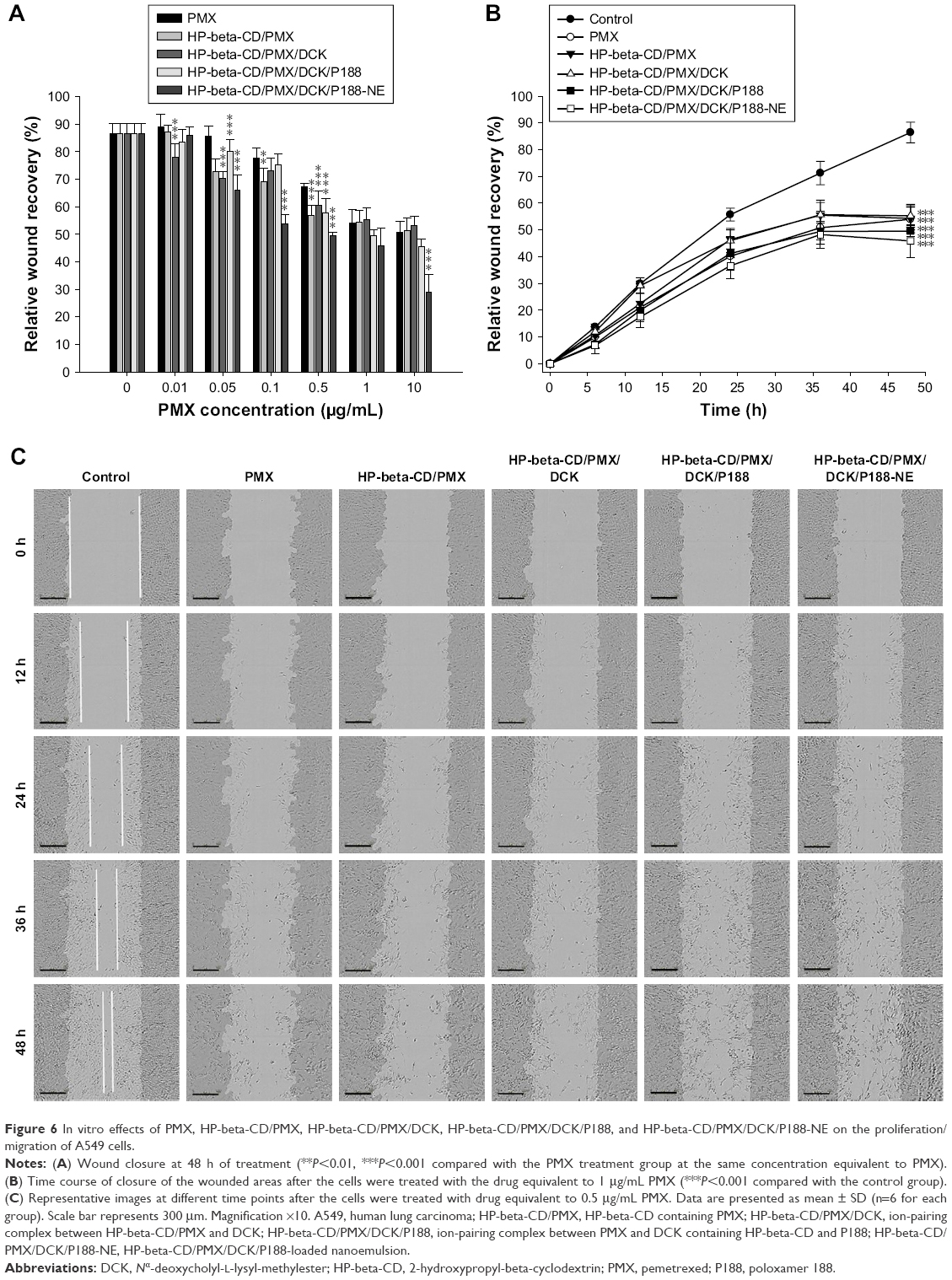 | Figure 6 In vitro effects of PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE on the proliferation/migration of A549 cells. |
Partition coefficient and membrane permeability
The effects of a permeation enhancer (DCK) and surfactants (HP-beta-CD and P188) on the octanol/water partitioning of PMX were investigated (Table 1). The addition of PMX to HP-beta-CD did not increase lipophilicity, but the partition coefficients of PMX and HP-beta-CD/PMX rose by 1.67- and 2.40-fold after ion-pairing complex formation with DCK, respectively, perhaps attributable to neutralization of the PMX carboxylic acid and a surfactant effect of DCK. The DOCA of DCK is a hydrophobic, secondary bile acid featuring an amphiphilic steroid nucleus with a hydrophilic α-side and a lipophilic β-side, thus serving as a solubilizing agent that forms weak micelles.32,48,49 Moreover, the partition coefficient of HP-beta-CD/PMX was further increased by adding P188 as a dispersing agent of the DCK complex, resulting in the partition coefficient of HP-beta-CD/PMX/DCK/P188 being 5.78-fold greater than that of pure PMX.
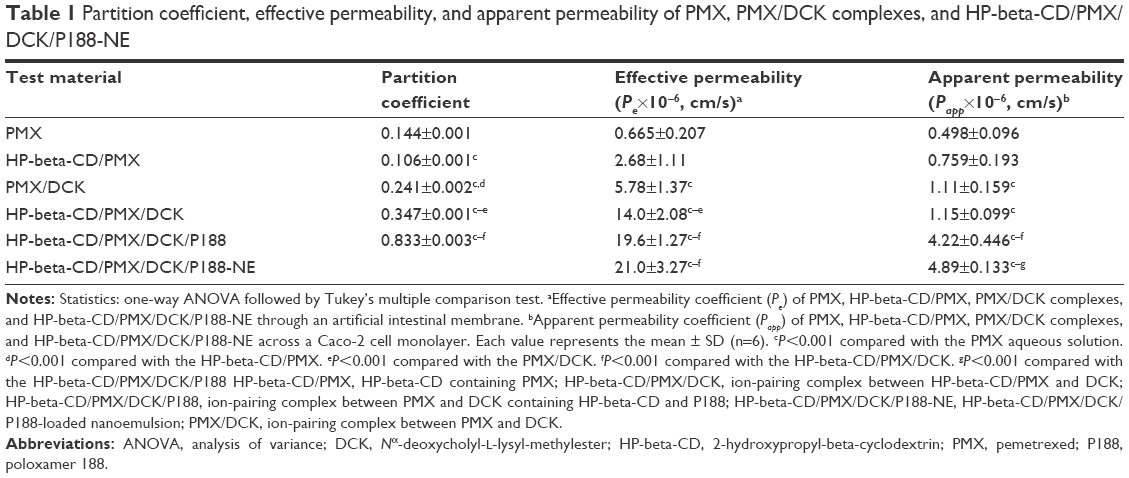 | Table 1 Partition coefficient, effective permeability, and apparent permeability of PMX, PMX/DCK complexes, and HP-beta-CD/PMX/DCK/P188-NE |
Next, the absorption potential of PMX through an artificial intestinal membrane was evaluated after increasing its lipophilicity by complex formation as HP-beta-CD/PMX/DCK/P188 and entrapping the complex into a nanoemulsive system (Table 1). The permeability of PMX after the addition of HP-beta-CD was increased by 4.03-fold compared with that of free PMX, which may have been caused by the surfactant property of HP-beta-CD. On the other hand, the effective permeability of PMX/DCK was 8.69-fold greater than that of PMX, and it was further increased by 142% and 239% by the inclusion of HP-beta-CD or HP-beta-CD and P188 in the PMX/DCK complex, respectively, resulting in the permeability of HP-beta-CD/PMX/DCK/188 being 29.5-fold higher than that of free PMX. In this study, the enhanced amphiphilicity of PMX can be ascribed to the synergistic activity of DCK, HP-beta-CD, and P188. Furthermore, the membrane permeability of HP-beta-CD/PMX/DCK/P188 was further increased by 15.9% after incorporation into the nanoemulsion and the effective permeability of HP-beta-CD/PMX/DCK/P188-NE was 31.6-fold greater than that of free PMX. This result demonstrated the positive role of nanoemulsions in promoting the passive absorption of HP-beta-CD/PMX/DCK/P188 through the phospholipid layer, which may be possible through the presence of the surfactant mixture in the nanoemulsion. The surfactant mixture can increase the affinity between the oil phase of nanoemulsion and the intestinal membrane, which is followed by enhancing partitioning into the lipophilic membrane as well as disrupting the structural integrity of the lipid bilayer.50
The in vitro permeabilities of PMX, PMX complexes, and HP-beta-CD/PMX/DCK/P188-NE across a Caco-2 cell monolayer are presented in Table 1. The apparent permeability (Papp) of PMX was increased by 1.52- and 2.23-fold after adding HP-beta-CD or ion-pairing complex formation with DCK, respectively. In addition, the permeability value of HP-beta-CD/PMX/DCK was 2.31-fold higher than that of PMX, and this value was further increased by 267% after the addition of P188 during complex formation. This increase may have been due to the permeation-enhancing activity of DCK and P188 in addition to their surfactant effects. DOCA is known to modulate or increase intestinal paracellular permeability via epithelial growth factor receptor (EGFR) auto-phosphorylation, occludin dephosphorylation, and rearrangement at the tight junction level.34,51 It was also reported that P188 can change the lateral packing density of membranes by insertion into the lipid layers.52 Furthermore, nanoemulsions entrapping the HP-beta-CD/PMX/DCK/P188 demonstrated a significant increase in Papp across a Caco-2 cell monolayer by 1.15- and 9.82-fold compared with those of HP-beta-CD/PMX/DCK and free PMX, respectively, further implying enhanced oral bioavailability. The increase in Papp could have been due to enhanced contact with the biological membrane because of an increase in the surface area of the droplets.53 Nonionic surfactants and cosurfactants of the self-nanoemulsifying formulation, such as Labrasol, Transcutol HP, and Cremophor EL, may also have an impact on the membrane fluidity and thus alter the epithelial barrier property. Mechanistic study demonstrated that Labrasol can disrupt tight junctions at the molecular level, including the loss of junction integrity via changes in F-actin ring and redistribution of zonula occludens-1 (ZO-1), leading them to act as potential absorption enhancers of hydrophilic drugs.54,55 Based on the results of the in vitro artificial intestinal membrane and Caco-2 cell monolayer permeability analyses, further experiments, including those on the in vivo oral absorption in rats and tumor growth suppression efficacy in mice, were carried out using HP-beta-CD/PMX/DCK/P188-NE.
Uptake into Caco-2 and ASBT-expressing MDCK cells
The intracellular uptake of PMX or PMX/DCK complex into the Caco-2 cells as an in vitro model that simulated intestinal epithelial physiology was studied using CLSM. FITC-PMX/DCK showed a tendency for an increase in cellular uptake compared with FITC-PMX, which was confirmed by the more intense fluorescence signals (Figure 7A). This may be attributable to the fact that DCK, which is a bile acid derivative, is efficiently absorbed through ASBT in the ileum and enters the portal blood, enhancing the oral bioavailability of poorly permeable drugs.56–59 FITC-PMX-NE also penetrated through the cells, but FITC-PMX/DCK-NE exhibited significantly higher cellular uptake than found in other treatment groups. This enhanced cellular uptake might have been due to the synergistic effect of surfactant and cosurfactant mixtures in improving drug bio-accessibility as well as the increased intracellular uptake of nano-sized oil droplets caused by permeation enhancers and other formulation components, such as surfactants and cosurfactants.60
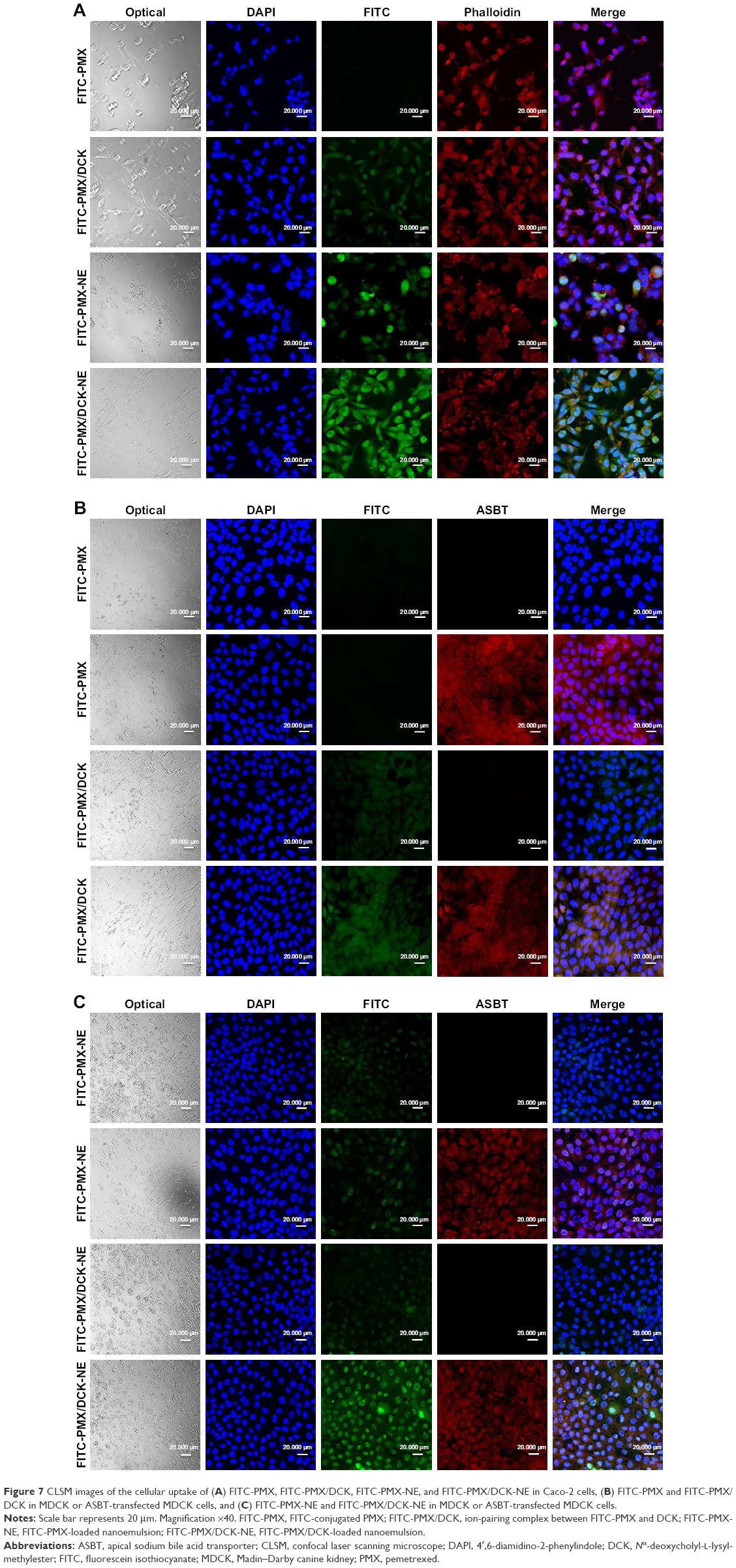 | Figure 7 CLSM images of the cellular uptake of (A) FITC-PMX, FITC-PMX/DCK, FITC-PMX-NE, and FITC-PMX/DCK-NE in Caco-2 cells, (B) FITC-PMX and FITC-PMX/DCK in MDCK or ASBT-transfected MDCK cells, and (C) FITC-PMX-NE and FITC-PMX/DCK-NE in MDCK or ASBT-transfected MDCK cells. |
To evaluate the absorption of PMX/DCK complex through the bile acid transporter-mediated route, the cellular uptake of FITC-PMX or FITC-PMX/DCK was investigated in normal or ASBT-gene-transfected MDCK cell lines. FITC-PMX was rarely present in both normal and ASBT-expressing MDCK cells (Figure 7B). In contrast, FITC-PMX/DCK complex was observed in the normal MDCK cells and was more abundant in the ASBT-expressing MDCK cells, which was attributable to the specific interaction between the complexed DCK molecules and bile acid transporters present on the cell membrane. FITC-PMX-NE could be transported regardless of the presence of ASBT in the MDCK cells (Figure 7C). However, FITC-PMX/DCK complex more extensively penetrated into the ASBT-expressing MDCK cells after incorporation in the nanoemulsion. This was consistent with the results from in vitro membrane permeability studies, implying the synergistic effect in enhancing penetration through the membrane. This was achieved by establishing high concentration gradients of both the PMX/DCK complex and enhancer via interaction with bile acid transporters as well as surfactant-induced modification of cell membrane fluidity, followed by enhancing the cellular permeability of the drug complex after capture in the nanoemulsive system.61,62
In vitro drug dissolution
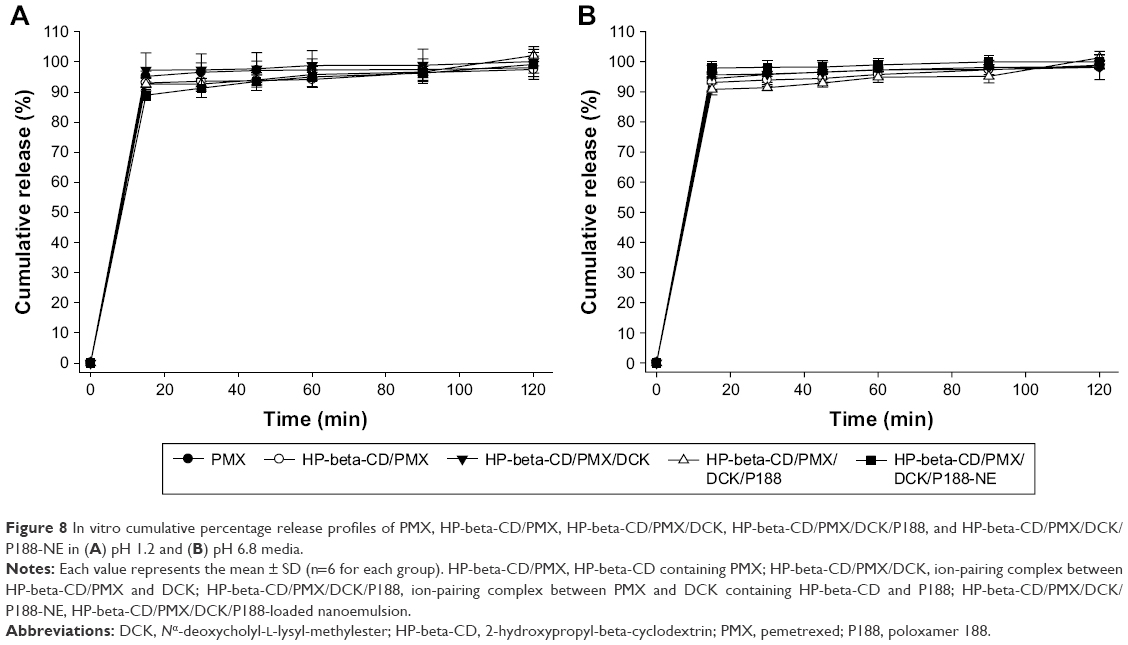
Figure 8 shows the drug dissolution properties of PMX/DCK complexes and HP-beta-CD/PMX/DCK/P188-NE in the simulated gastric (pH 1.2) and intestinal (pH 6.8) media. More than 90% of free PMX and PMX/DCK complex was dissolved within 15 min in the medium at pH 1.2 (Figure 8A). In addition, the release of PMX/DCK complex from the nanoemulsion was similar to that of free PMX (>90% within 15 min) in the medium at pH 6.8 (Figure 8B). Thus, there was no significant change in the dissolution rate of drug molecules after ion-pairing complex formation with DCK as well as incorporation of the complex into the w/o/w nanoemulsion, indicating that HP-beta-CD/PMX/DCK/P188-NE successfully formed a self-nanoemulsification without phase separation, aggregation of the DCK molecules, or delayed action caused by the oil phase in the nanoemulsion after dispersion in both conditions.
 | Figure 8 In vitro cumulative percentage release profiles of PMX, HP-beta-CD/PMX, HP-beta-CD/PMX/DCK, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE in (A) pH 1.2 and (B) pH 6.8 media. |
In vivo oral absorption in rats
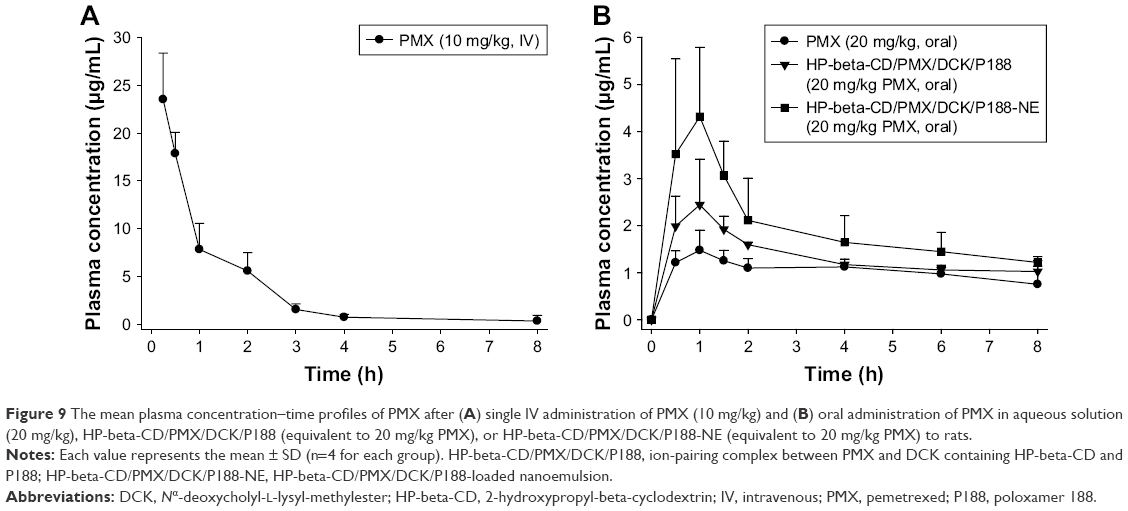
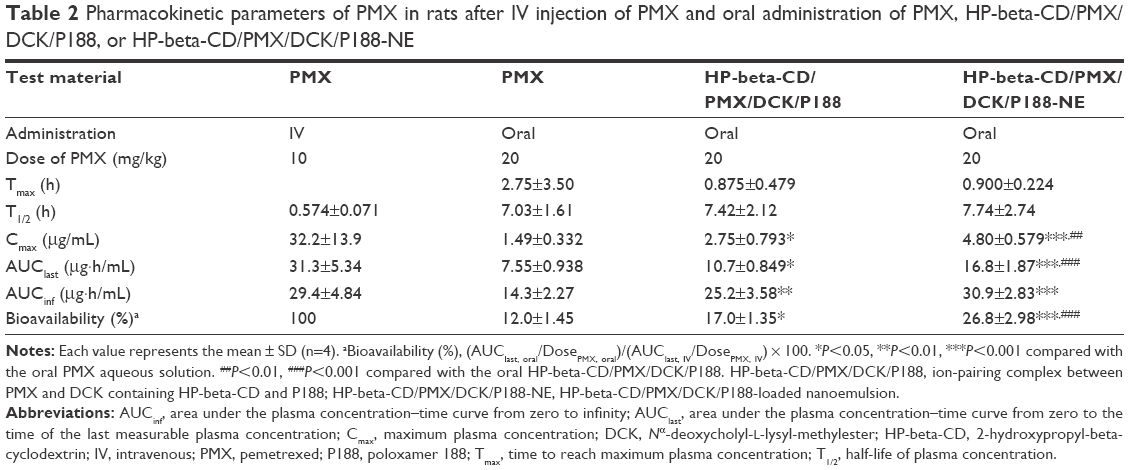
Regarding the in vivo oral absorption in rats, the PMX in plasma concentration–time profiles following IV and oral administration are shown in Figure 9. After oral administration of 20 mg/kg PMX alone, the maximum plasma concentration (Cmax) attained was 1.49±0.332 μg/mL, the area under the plasma concentration–time curve (AUClast) was 7.55±0.938 μg·h/mL, and the oral bioavailability (compared with the IV dose) was 12.0%±1.45%. However, compared with the oral administration of PMX alone, oral HP-beta-CD/PMX/DCK/P188 (equivalent to 20 mg/kg PMX) afforded a 1.85-fold increase in Cmax (2.75±0.793 μg/mL) and a 1.42-fold increase in AUClast (10.7±0.849 μg·h/mL), resulting in a 1.42-fold increase in oral bioavailability (17.0%±1.35%; Table 2). In addition, the Cmax and AUClast values of oral HP-beta-CD/PMX/DCK/P188-NE (equivalent to 20 mg/kg PMX) were 1.75- and 1.57-fold greater than that of oral HP-beta-CD/PMX/DCK/P188, respectively, and 3.22- and 2.23-fold greater than that of oral PMX solution (Figure 9B). The oral bioavailability of HP-beta-CD/PMX/DCK/P188-NE was thus 2.23-fold higher than that of PMX solution (26.8%±2.98%; Table 2). Therefore, intestinal membrane permeability and PMX absorption in rats were significantly improved by ion-pair complex formation with DCK and incorporation of the complex in a nanoemulsive system. The time to maximum concentration (Tmax) values were 2.75±3.50 and 0.900±0.224 h after oral administration of free PMX or HP-beta-CD/PMX/DCK/P188-NE, respectively, implying that the PMX/DCK complex was quickly released from the nanoemulsion to permeate through the intestinal membrane. Such rapid absorption may be attributable to the enhanced membrane permeability of the PMX/DCK complex, without any decrease in the solubility of PMX, after incorporation of HP-beta-CD/PMX/DCK/P188 into nano-sized droplets. However, the terminal elimination half-lives (T1/2 values) after oral administration of PMX, HP-beta-CD/PMX/DCK/P188, and HP-beta-CD/PMX/DCK/P188-NE (alone) were significantly longer than those after IV administration. The differences in half-lives after IV and oral administration of PMX suggest that absorption may still occur during the elimination phase after oral intake. Flip-flop occurs when the rate of absorption is slower than the rate of elimination. Modified dosage formulations commonly cause flip-flop; however, formulation characteristics, such as the drugs per se or the excipients, can also trigger flip-flop.63 Drugs, the oral absorption of which is limited by the permeability rate, are most likely to exhibit flip-flop pharmacokinetics.64 Thus, oral HP-beta-CD/PMX/DCK/P188-NE would be expected to exhibit rapid onset action and a sustained therapeutic effect compared with IV or oral PMX alone. However, a longer duration of sampling may be necessary to confirm that bi-exponential elimination is in play.
 | Figure 9 The mean plasma concentration–time profiles of PMX after (A) single IV administration of PMX (10 mg/kg) and (B) oral administration of PMX in aqueous solution (20 mg/kg), HP-beta-CD/PMX/DCK/P188 (equivalent to 20 mg/kg PMX), or HP-beta-CD/PMX/DCK/P188-NE (equivalent to 20 mg/kg PMX) to rats. |
 | Table 2 Pharmacokinetic parameters of PMX in rats after IV injection of PMX and oral administration of PMX, HP-beta-CD/PMX/DCK/P188, or HP-beta-CD/PMX/DCK/P188-NE |
The enhanced oral absorption of PMX in HP-beta-CD/PMX/DCK/P188-NE might be attributable to the following factors: 1) DCK complex increased the lipophilicity, partition coefficient, and apparent liposolubility of the drug in the intestinal membrane. 2) DOCA in the DCK can disrupt the intestinal barrier via occludin dephosphorylation and cytoskeletal rearrangement at the epithelial lining tight junction level, followed by promotion of the paracellular transport of drug; moreover, selective interaction with the bile acid transporters on the intestinal membrane may facilitate transcellular transport of the drug.18,51,65 3) Nano-sized droplets solubilizing the HP-beta-CD/PMX/DCK/P188 in a supramolecular state also improved the oral absorption of the drug by increasing the dispersibility of the drug complex and protecting the complex form from dissociation, thereby facilitating its diffusion across the unstirred water layer and access to the brush border membrane of enterocytes.66 4) Surfactant-induced membrane fluidity and thus permeability changes potentially afforded the enhanced drug absorption.62,67
In vivo tumor growth inhibition effect
The efficacy of oral HP-beta-CD/PMX/DCK/P188-NE, in terms of inhibiting tumor growth in mice, was compared with that of an equivalent dose of PMX administered IV. In a preliminary study, we observed tumor suppression in LLC cell-bearing mice after once-weekly IV administration of PMX at >20 mg/kg (data not shown). In addition, HP-beta-CD/PMX/DCK/P188-NE exhibited 23.8%–29.8% of the oral bioavailability of PMX. Thus, we compared the anticancer effects after once-daily oral HP-beta-CD/PMX/DCK/P188-NE (equivalent to 20 mg/kg PMX) with those after once-weekly IV administration of 20 mg/kg PMX. Tumor growth was significantly suppressed after once-weekly IV administration of 20 mg/kg PMX (Figure 10A); the tumor volume after 3-week IV administration of PMX was 49% and 46% lower than those of the control and oral PMX groups, respectively. Once-a-day oral administration of HP-beta-CD/PMX/DCK/P188-NE (20 mg/kg as PMX) also significantly suppressed tumor growth and, consequently, the tumor volume was decreased by 61% and 59% compared with those of the control and oral PMX groups, respectively. Meanwhile, the oral administration of HP-beta-CD/PMX/DCK/P188-NE did not affect the body weights of tumor-bearing mice (Figure 10B). At 21 days after administration, the isolated tumor masses of the mice treated with oral HP-beta-CD/PMX/DCK/P188-NE were reduced by 55% and 56% compared with those from the control and oral PMX treatment groups, respectively, implying that oral HP-beta-CD/PMX/DCK/P188-NE was as effective as intravenously administered PMX (Figure 10C and D). In contrast, the oral PMX group did not show significant reductions in the tumor volume and mass, observed in the control mice (Figure 10A and C).
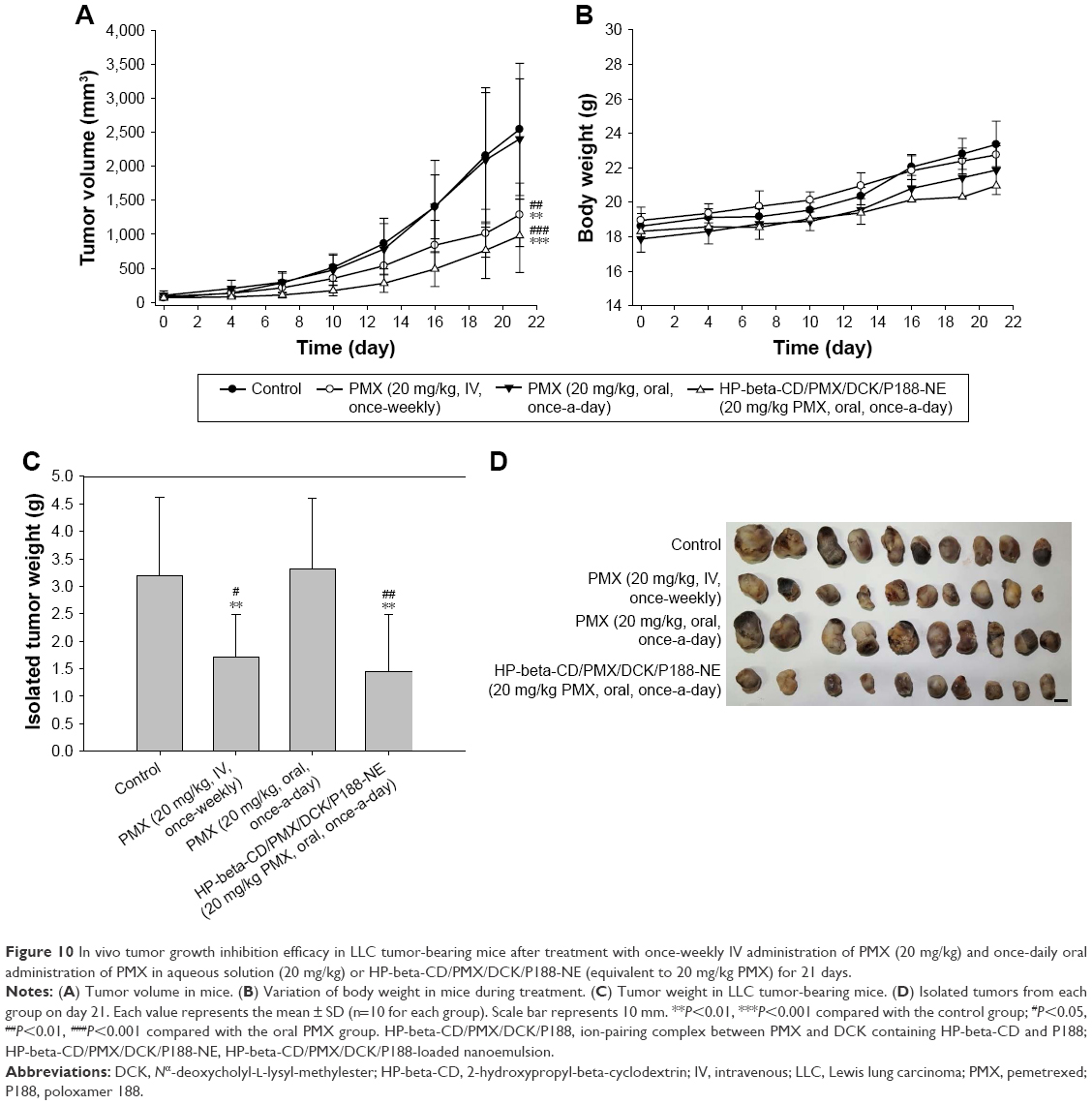 | Figure 10 In vivo tumor growth inhibition efficacy in LLC tumor-bearing mice after treatment with once-weekly IV administration of PMX (20 mg/kg) and once-daily oral administration of PMX in aqueous solution (20 mg/kg) or HP-beta-CD/PMX/DCK/P188-NE (equivalent to 20 mg/kg PMX) for 21 days. |
The same tendency was observed in the histological sections from the tumor tissues stained with anti-CD31 antibody, PCNA, and TUNEL (Figure 11). The oral HP-beta-CD/PMX/DCK/P188-NE treatment was associated with a lower density of anti-CD31-positive microvessels and decreased proliferating cell density, as shown by PCNA immunostaining in comparison with those of the control and oral PMX groups. Moreover, the oral treatment of HP-beta-CD/PMX/DCK/P188-NE had more apoptotic potential than that found in the tumors of the control and oral PMX treatment groups.
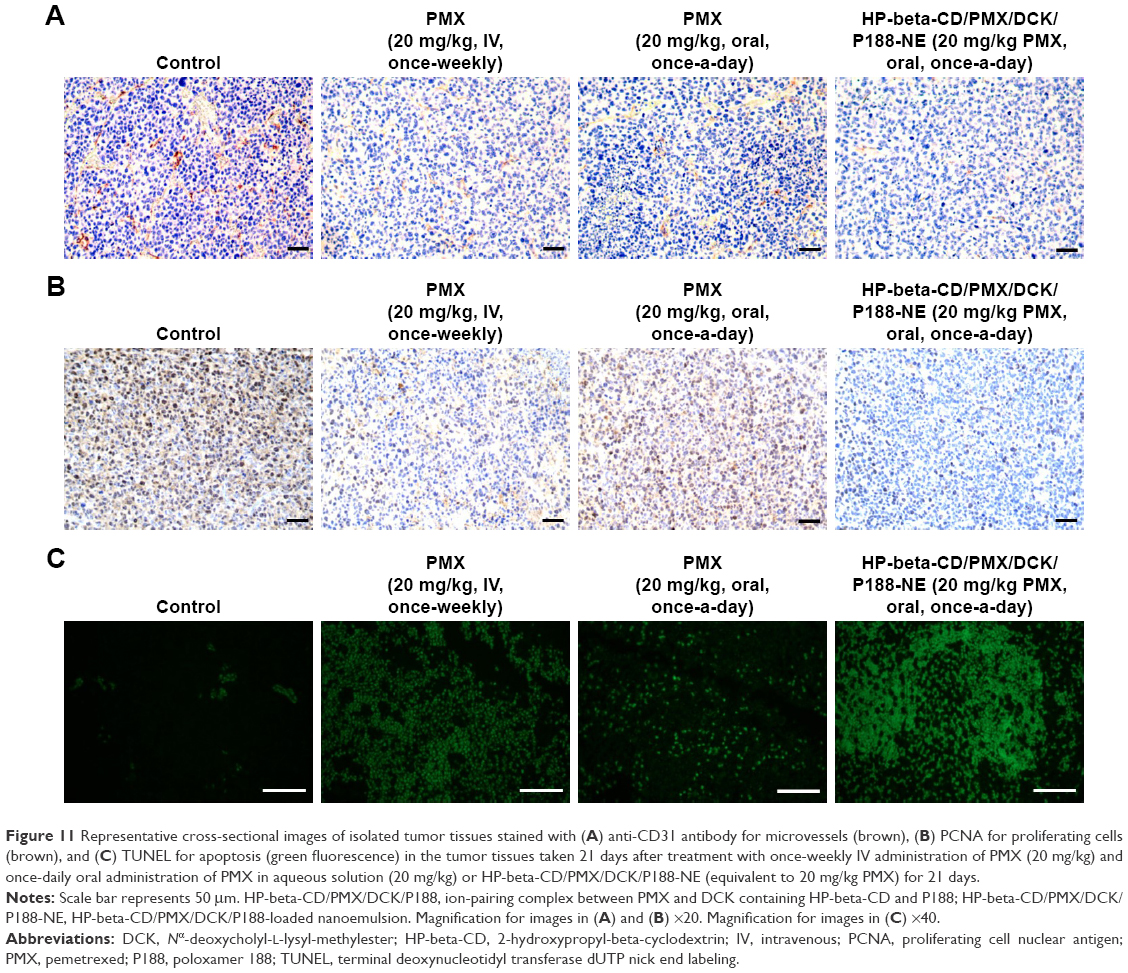 | Figure 11 Representative cross-sectional images of isolated tumor tissues stained with (A) anti-CD31 antibody for microvessels (brown), (B) PCNA for proliferating cells (brown), and (C) TUNEL for apoptosis (green fluorescence) in the tumor tissues taken 21 days after treatment with once-weekly IV administration of PMX (20 mg/kg) and once-daily oral administration of PMX in aqueous solution (20 mg/kg) or HP-beta-CD/PMX/DCK/P188-NE (equivalent to 20 mg/kg PMX) for 21 days. |
Based on these results, the oral administration of nanoemulsion comprising HP-beta-CD/PMX/DCK/P188 exhibited enhanced oral absorption properties accompanying an equivalent tumor growth inhibitory effect of PMX administered by injection, confirming the improvement in the oral bioavailability of PMX. Therefore, HP-beta-CD/PMX/DCK/P188-NE may be preferred, to deliver PMX effectively via the oral route, opening the way for new medical applications, including metronomic chemotherapy. However, further studies are required to determine the optimum oral doses of HP-beta-CD/PMX/DCK/P188-NE in various cancer cell-bearing animal models.
Conclusion
PMX/DCK complex-loaded multiple nanoemulsions, HP-beta-CD/PMX/DCK/P188-NE, were designed for oral anticancer PMX therapy. PMX was ionically combined with positively charged DCK as an absorption enhancer, followed by the inclusion of HP-beta-CD and P188 to improve dispersibility. HP-beta-CD/PMX/DCK/P188 was further incorporated into the multiple w/o/w nanoemulsions. After the complex formation, the partition coefficient and intestinal membrane permeability of HP-beta-CD/PMX/DCK/P188 were increased by 478% and 2,847% compared with those of free PMX, respectively, with similar dose-dependent cytotoxic and inhibitory effects on LLC and A549 cell proliferation/migration. The in vitro permeability across a Caco-2 cell monolayer and in vivo oral absorption of PMX were markedly increased after entrapping the complex into the nanoemulsive system; this might be attributable to the facilitation of partitioning of PMX/DCK to the epithelial cells via specific interaction with bile acid transporters, as well as enhanced lipophilicity accompanied by surfactant-induced changes in the intestinal membrane structure and fluidity. Thus, the oral bioavailability of PMX in rats was increased by 2.23-fold compared with that of free PMX, and orally administered HP-beta-CD/PMX/DCK/P188-NE maximally suppressed the tumor growth by 61% compared with that of the control group in the LLC cell-bearing mice. Therefore, HP-beta-CD/PMX/DCK/P188-NE might be a preferred alternative system for delivering PMX effectively via the oral route and is expected to provide new medical applications for PMX, including applications in metronomic chemotherapy for the prophylaxis of recurrence and in combinational chemotherapy with other anticancer agents.
Acknowledgments
This study was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government Ministry of Science and ICT (MSIT; NRF-2017M3A9F5029656).
Disclosure
The authors report no conflicts of interest in this work.
References
Browder T, Butterfield CE, Kraling BM, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60(7):1878–1886. | ||
Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4(6):423–436. | ||
Malik PS, Raina V, Andre N. Metronomics as maintenance treatment in oncology: time for chemo-switch. Front Oncol. 2014;4:1–7. | ||
Soriano JL, Batista N, Santiesteban E, et al. Metronomic cyclophosphamide and methotrexate chemotherapy combined with 1E10 anti-idiotype vaccine in metastatic breast cancer. Int J Breast Cancer. 2011;2011:1–6. | ||
Thanki K, Gangwal RP, Sangamwar AT, Jain S. Oral delivery of anticancer drugs: challenges and opportunities. J Control Release. 2013;170(1):15–40. | ||
DeVita VT Jr, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68(21):8643–8653. | ||
Deng L, Dong H, Dong A, Zhang J. A strategy for oral chemotherapy via dual pH-sensitive polyelectrolyte complex nanoparticles to achieve gastric survivability, intestinal permeability, hemodynamic stability and intracellular activity. Eur J Pharm Biopharm. 2015;97(Pt A):107–117. | ||
Shin SC, Choi JS, Li X. Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int J Pharm. 2006;313(1–2):144–149. | ||
Kuppens IE, Bosch TM, van Maanen MJ, et al. Oral bioavailability of docetaxel in combination with OC144-093 (ONT-093). Cancer Chemother Pharmacol. 2005;55(1):72–78. | ||
Adjei AA. Pharmacology and mechanism of action of pemetrexed. Clin Lung Cancer. 2004;5:S51–S55. | ||
Hanauske AR, Chen V, Paoletti P, Niyikiza C. Pemetrexed disodium: a novel antifolate clinically active against multiple solid tumors. Oncologist. 2001;6(4):363–373. | ||
Russo F, Bearz A, Pampaloni G; Investigators of Italian Pemetrexed Monotherapy of NSCLC Group. Pemetrexed single agent chemotherapy in previously treated patients with locally advanced or metastatic non-small cell lung cancer. BMC Cancer. 2008;8:216. | ||
Fuld AD, Dragnev KH, Rigas JR. Pemetrexed in advanced non-small-cell lung cancer. Expert Opin Pharmacother. 2010;11(8):1387–1402. | ||
Tomasini P, Barlesi F, Mascaux C, Greillier L. Pemetrexed for advanced stage nonsquamous non-small cell lung cancer: latest evidence about its extended use and outcomes. Ther Adv Med Oncol. 2016;8(3):198–208. | ||
Lal R, Hillerdal GN, Shah RN, et al. Feasibility of home delivery of pemetrexed in patients with advanced non-squamous non-small cell lung cancer. Lung Cancer. 2015;89(2):154–160. | ||
Aungst BJ. Absorption enhancers: applications and advances. AAPS J. 2012;14(1):10–18. | ||
Bansal T, Akhtar N, Jaggi M, Khar RK, Talegaonkar S. Novel formulation approaches for optimising delivery of anticancer drugs based on P-glycoprotein modulation. Drug Discov Today. 2009;14(21–22):1067–1074. | ||
Tian C, Asghar S, Wu Y, et al. Improving intestinal absorption and oral bioavailability of curcumin via taurocholic acid-modified nanostructured lipid carriers. Int J Nanomed. 2017;12:7897–7911. | ||
Kalepu S, Manthina M, Padavala V. Oral lipid-based drug delivery systems – an overview. Acta Pharmaceutica Sinica B. 2013;3(6):361–372. | ||
Guan P, Lu Y, Qi J, et al. Enhanced oral bioavailability of cyclosporine A by liposomes containing a bile salt. Int J Nanomed. 2011;6:965–974. | ||
Dian L, Yu E, Chen X, et al. Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale Res Lett. 2014;9(1):1–11. | ||
Sigward E, Mignet N, Rat P, et al. Formulation and cytotoxicity evaluation of new self-emulsifying multiple W/O/W nanoemulsions. Int J Nanomedicine. 2013;8:611–625. | ||
Joshi G, Kumar A, Sawant K. Enhanced bioavailability and intestinal uptake of gemcitabine HCl loaded PLGA nanoparticles after oral delivery. Eur J Pharm Sci. 2014;60:80–89. | ||
Kim JE, Yoon IS, Cho HJ, Kim DH, Choi YH, Kim DD. Emulsion-based colloidal nanosystems for oral delivery of doxorubicin: improved intestinal paracellular absorption and alleviated cardiotoxicity. Int J Pharm. 2014;464(1):117–126. | ||
Yen CC, Chen YC, Wu MT, Wang CC, Wu YT. Nanoemulsion as a strategy for improving the oral bioavailability and anti-inflammatory activity of andrographolide. Int J Nanomedicine. 2018;13:669–680. | ||
Soni K, Mujtaba A, Kohli K. Lipid drug conjugate nanoparticle as a potential nanocarrier for the oral delivery of pemetrexed diacid: formulation design, characterization, ex vivo, and in vivo assessment. Int J Biol Macromol. 2017;103:139–151. | ||
Rezhdo O, Speciner L, Carrier R. Lipid-associated oral delivery: mechanisms and analysis of oral absorption enhancement. J Control Release. 2016;240:544–560. | ||
Park JW, Kim SJ, Kwag DS, et al. Multifunctional delivery systems for advanced oral uptake of peptide/protein drugs. Curr Pharm Des. 2015;21(22):3097–3110. | ||
Mahmud F, Jeon OC, Al-Hilal TA, et al. Absorption mechanism of a physical complex of monomeric insulin and deoxycholyl-l-lysyl-methylester in the small intestine. Mol Pharm. 2015;12(6):1911–1920. | ||
Jeon OC, Byun Y, Park JW. Preparation of oxaliplatin-deoxycholic acid derivative nanocomplexes and in vivo evaluation of their oral absorption and tumor growth suppression. J Nanosci Nanotechnol. 2016;16(2):2061–2064. | ||
Jeon OC, Seo DH, Kim HS, Byun Y, Park JW. Oral delivery of zoledronic acid by non-covalent conjugation with lysine-deoxycholic acid: in vitro characterization and in vivo anti-osteoporotic efficacy in ovariectomized rats. Eur J Pharm Sci. 2016;82:1–10. | ||
Sun S, Liang N, Kawashima Y, Xia D, Cui F. Hydrophobic ion pairing of an insulin-sodium deoxycholate complex for oral delivery of insulin. Int J Nanomedicine. 2011;6:3049–3056. | ||
Miller JM, Dahan A, Gupta D, Varghese S, Amidon GL. Enabling the intestinal absorption of highly polar antiviral agents: ion-pair facilitated membrane permeation of zanamivir heptyl ester and guanidino oseltamivir. Mol Pharm. 2010;7(4):1223–1234. | ||
Pangeni R, Choi SW, Jeon OC, Byun Y, Park JW. Multiple nanoemulsion system for an oral combinational delivery of oxaliplatin and 5-fluorouracil: preparation and in vivo evaluation. Int J Nanomed. 2016;11:6379–6399. | ||
De La Torre P, Torrado S, Torrado S. Preparation, dissolution and characterization of praziquantel solid dispersions. Chem Pharm Bull. 1999;47(11):1629–1633. | ||
Dahan A, Beig A, Ioffe-Dahan V, Agbaria R, Miller JM. The twofold advantage of the amorphous form as an oral drug delivery practice for lipophilic compounds: increased apparent solubility and drug flux through the intestinal membrane. AAPS J. 2013;15(2):347–353. | ||
Frank KJ, Rosenblatt KM, Westedt U, et al. Amorphous solid dispersion enhances permeation of poorly soluble ABT-102: true supersaturation vs. apparent solubility enhancement. Int J Pharm. 2012;437(1–2):288–293. | ||
Beig A, Miller JM, Dahan A. Accounting for the solubility-permeability interplay in oral formulation development for poor water solubility drugs: the effect of PEG-400 on carbamazepine absorption. Eur J Pharm Biopharm. 2012;81(2):386–391. | ||
Moes J, Koolen S, Huitema A, Schellens J, Beijnen J, Nuijen B. Development of an oral solid dispersion formulation for use in low-dose metronomic chemotherapy of paclitaxel. Eur J Pharm Biopharm. 2013;83(1):87–94. | ||
Pangeni R, Sharma S, Mustafa G, Ali J, Baboota S. Vitamin E loaded resveratrol nanoemulsion for brain targeting for the treatment of Parkinson’s disease by reducing oxidative stress. Nanotechnology. 2014;25(48):1–13. | ||
Tadros T, Izquierdo P, Esquena J, Solans C. Formation and stability of nano-emulsions. Adv Colloid Interface Sci. 2004;10(8–109):303–318. | ||
Gao Y, Wang Y, Ma Y, et al. Formulation optimization and in situ absorption in rat intestinal tract of quercetin-loaded microemulsion. Colloids Surf B Biointerfaces. 2009;71(2):306–314. | ||
Bali V, Ali M, Ali J. Study of surfactant combinations and development of a novel nanoemulsion for minimising variations in bioavailability of ezetimibe. Colloids Surf B Biointerfaces. 2010;76(2):410–420. | ||
Silva HD, Cerqueira MA, Vicente AA. Influence of surfactant and processing conditions in the stability of oil-in-water nanoemulsions. J Food Eng. 2015;167:89–98. | ||
Patel AR, Vavia PR. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. AAPS J. 2007;9(3):E344–E352. | ||
Hu FQ, Wu XL, Du YZ, You J, Yuan H. Cellular uptake and cytotoxicity of shell crosslinked stearic acid-grafted chitosan oligosaccharide micelles encapsulating doxorubicin. Eur J Pharm Biopharm. 2008;69(1):117–125. | ||
Melguizo C, Cabeza L, Prados J, et al. Enhanced antitumoral activity of doxorubicin against lung cancer cells using biodegradable poly(butylcyanoacrylate) nanoparticles. Drug Des Devel Ther. 2015;9:6433–6444. | ||
Mrestani Y, Bretschneider B, Hartl A, Neubert RH. In-vitro and in-vivo studies of cefpirom using bile salts as absorption enhancers. J Pharm Pharmacol. 2003;55(12):1601–1606. | ||
Hofmann AF, Mysels KJ. Bile salts as biological surfactants. Colloids and Surfaces. 1987;30(1):145–173. | ||
Gupta S, Kesarla R, Omri A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharm. 2013;2013:1–16. | ||
Raimondi F, Santoro P, Barone MV, et al. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294(4):G906–G913. | ||
Seelig A, Gerebtzoff G. Enhancement of drug absorption by noncharged detergents through membrane and P-glycoprotein binding. Expert Opin Drug Metab Toxicol. 2006;2(5):733–752. | ||
Fofaria NM, Qhattal HS, Liu X, Srivastava SK. Nanoemulsion formulations for anti-cancer agent piplartine – characterization, toxicological, pharmacokinetics and efficacy studies. Int J Pharm. 2016;498(1–2):12–22. | ||
Sposito PA, Mazzeti AL, de Oliveira Faria C, et al. Ravuconazole self-emulsifying delivery system: in vitro activity against trypanosoma cruzi amastigotes and in vivo toxicity. Int J Nanomedicine. 2017;12:3785–3799. | ||
Sha X, Yan G, Wu Y, Li J, Fang X. Effect of self-microemulsifying drug delivery systems containing labrasol on tight junctions in Caco-2 cells. Eur J Pharm Sci. 2005;24(5):477–486. | ||
Balakrishnan A, Polli JE. Apical sodium dependent bile acid transporter (ASBT, SLC10A2): a potential prodrug target. Mol Pharm. 2006;3(3):223–230. | ||
Lazaridis KN, Pham L, Tietz P, et al. Rat cholangiocytes absorb bile acids at their apical domain via the ileal sodium-dependent bile acid transporter. J Clin Invest. 1997;100(11):2714–2721. | ||
Park JW, Kim SK, Al-Hilal TA, Jeon OC, Moon HT, Byun Y. Strategies for oral delivery of macromolecule drugs. Biotechnol Bioprocess Eng. 2010;15(1):66–75. | ||
Jeon OC, Hwang SR, Al-Hilal TA, et al. Oral delivery of ionic complex of ceftriaxone with bile acid derivative in non-human primates. Pharm Res. 2013;30(4):959–967. | ||
Choudhury H, Gorain B, Karmakar S, et al. Improvement of cellular uptake, in vitro antitumor activity and sustained release profile with increased bioavailability from a nanoemulsion platform. Int J Pharm. 2014;460(1–2):131–143. | ||
Dai WG, Dong LC, Song Y. Enhanced bioavailability of poorly absorbed hydrophilic compounds through drug complex/in situ gelling formulation. Int J Pharm. 2013;457(1):63–70. | ||
Wan K, Sun L, Hu X, et al. Novel nanoemulsion based lipid nanosystems for favorable in vitro and in vivo characteristics of curcumin. Int J Pharm. 2016;504(1–2):80–88. | ||
Yanez JA, Remsberg CM, Sayre CL, Forrest ML, Davies NM. Flip-flop pharmacokinetics – delivering a reversal of disposition: challenges and opportunities during drug development. Ther Deliv. 2011;2(5):643–672. | ||
Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. | ||
Forsgard RA, Korpela R, Stenman LK, Osterlund P, Holma R. Deoxycholic acid induced changes in electrophysiological parameters and macromolecular permeability in murine small intestine with and without functional enteric nervous system plexuses. Neurogastroenterol Motil. 2014;26(8):1179–1187. | ||
Gao F, Zhang Z, Bu H, et al. Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: performance and mechanism. J Control Release. 2011;149(2):168–174. | ||
Heredia RM, Boeris PS, Biasutti MA, Lopez GA, Paulucci NS, Lucchesi GI. Coordinated response of phospholipids and acyl components of membrane lipids in Pseudomonas putida A (ATCC 12633) under stress caused by cationic surfactants. Microbiology. 2014;160(Pt 12):2618–2626. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.