Back to Journals » International Journal of Nanomedicine » Volume 12
Enhanced anticancer efficacy of paclitaxel through multistage tumor-targeting liposomes modified with RGD and KLA peptides
Authors Sun J, Jiang L, Lin Y, Gerhard EM, Jiang X, Li L, Yang J, Gu Z
Received 21 September 2016
Accepted for publication 4 January 2017
Published 27 February 2017 Volume 2017:12 Pages 1517—1537
DOI https://doi.org/10.2147/IJN.S122859
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Linlin Sun
Jiawei Sun,1 Lei Jiang,2 Yi Lin,3 Ethan Michael Gerhard,4 Xuehua Jiang,1 Li Li,3 Jian Yang,4 Zhongwei Gu3
1West China School of Pharmacy, Sichuan University, Chengdu, Sichuan, 2Department of Pharmaceutics, China Pharmaceutical University, Nanjing, Jiangsu, 3National Engineering Research Center for Biomaterials, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 4Department of Biomedical Engineering Materials Research Institute, The Huck Institutes of The Life Sciences, The Pennsylvania State University, University Park, PA, USA
Abstract: Mitochondria serve as both “energy factories” and “suicide weapon stores” of cells. Targeted delivery of cytotoxic drugs to the mitochondria of tumor cells and tumor vascular cells is a promising strategy to improve the efficacy of chemotherapy. Here, multistage tumor-targeting liposomes containing two targeted peptide-modified lipids, cRGD-PEG2000-DSPE and KLA-PEG2000-DSPE, were developed for encapsulation of the anticancer drug paclitaxel (PTX, RGD-KLA/PTX-Lips). Compared with Taxol (free PTX), RGD/PTX-Lips and KLA/PTX-Lips, the half-maximal inhibitory concentration (IC50) value of RGD-KLA/PTX-Lips in vitro was 1.9-, 36.7- and 22.7-fold lower with 4T1 cells, respectively, because of higher levels of cellular uptake. Similar results were also observed with human umbilical vascular endothelial cells (HUVECs). An apoptosis assay showed that the total apoptotic ratio of RGD-KLA/PTX-Lips was the highest because of the mitochondria-targeted drug delivery and the activation of mitochondrial apoptosis pathways, as evidenced by visible mitochondrial localization, decreased mitochondrial membrane potential, release of cytochrome c and increased activities of caspase-9 and caspase-3. The strongest tumor growth inhibition (TGI; 80.6%) and antiangiogenesis effects without systemic toxicity were also observed in RGD-KLA/PTX-Lip-treated 4T1 tumor xenograft BALB/c mice. In conclusion, these multistage tumor-targeting liposomes represent a promising anticancer drug delivery system (DDS) capable of maximizing anticancer therapeutic efficacy and minimizing systemic toxicity.
Keywords: multistage tumor-targeting liposome, mitochondria, paclitaxel, anticancer, antiangiogenesis
Introduction
Cancer is one of the leading causes of death globally. The World Health Organization estimates that 8.2 million people died of cancer in 2012, and this figure is expected to increase to 13 million annually within the next 2 decades.1 The ultimate goal of cancer therapeutics is to prolong the survival time and improve the quality of life of patients by minimizing systemic toxicity and maximizing the anticancer efficacy of chemotherapy.2,3 Combining anticancer therapies with antiangiogenic therapies is becoming increasingly important since this approach can kill tumor cells directly and cut off their oxygen and nutrient supplies.4,5 Investigators have reported that dual-targeting nanoparticles that simultaneously target both tumor vascular endothelial cells and tumor cells inhibit the process of angiogenesis and kill the tumor cells, resulting in an additive effect on anticancer efficacy compared with either therapy alone.6–8 However, the efficacy of combined therapy is still limited due to the evasion of cell death and drug resistance.9–11
Organelle-targeted drug delivery that can directly induce cell death is an advantageous anticancer strategy.12 The mitochondrion is a good candidate for organelle targeting because it is implicated in multiple aspects of tumorigenesis and tumor progression, such as the regulation of cellular differentiation, growth, energy production and programmed cell death.13,14 As a crucial regulator of apoptosis, the mitochondrion releases proapoptotic proteins from the mitochondrial intermembrane space to the cytosol to activate apoptosis.15 The permeabilization of the mitochondrial outer membrane is regarded as a potent way to induce intrinsic apoptosis.16
Paclitaxel (PTX), a broad-spectrum anticancer drug, is usually considered to disrupt the microtubule dynamics that are required for cell division and vital interphase processes.17,18 However, some reports have found that PTX also has a direct effect on mitochondria isolated from cancer cells, resulting in the apoptosis of cancer cells.19,20 PTX can bind to the Bcl-2 antiapoptotic protein in mitochondria by mimicking Nur77, an orphan nuclear receptor, thereby changing the function of Bcl-2 from antiapoptotic to proapoptotic and resulting in the apoptosis of cancer cells.21,22 Other studies have shown that PTX depolarizes mitochondria and promotes the induction of the mitochondrial permeability transition pore, which then activates death signal proteins such as caspases.23,24 Therefore, it can be assumed that delivery of PTX to the mitochondria of tumor cells and tumor vascular endothelial cells is a promising strategy to improve the specificity of PTX and decrease the evasion of cell death.
A drug delivery system (DDS) that can efficiently transport PTX from the injection site to mitochondria to maximize anticancer efficacy should possess the following three targeting capabilities: 1) effectively encapsulate drugs, avoid capture by the reticuloendothelial system (RES) during blood circulation and accumulate in the tumor extracellular matrix; 2) specifically bind onto tumor cells (anticancer) and tumor vascular endothelial cells (antiangiogenesis) and 3) bind to and subsequently destroy the mitochondrial membranes to trigger programmed cell apoptosis. Despite outstanding advances, most previous reports have only addressed one or two of these criteria.7,8,14,25–27 Therefore, a multistage targeted DDS capable of satisfying the above requirements is highly desirable.
Here, two peptide-conjugated PEGylated phospholipid derivatives (cRGD-polyethylene glycol-1,2-distearoyl-sn-glycerol-3-phosphoethanolamine [cRGD-PEG2000-DSPE] and D-(KLAKLAK)2-polyethylene glycol-1,2-distearoyl-sn-glycerol-3-phosphoethanolamine [KLA-PEG2000-DSPE]) were synthesized as tumor and vascular endothelial cell- and mitochondria-targeting molecules, respectively. PTX liposomes were modified using these two conjugates for the first time. It was hypothesized that the targeted PTX liposomes could overcome physiological and biological barriers between the injection site and the mitochondria (Scheme 1). First, the nanoscaled liposomes (in the size range of 20–200 nm) could be loaded with anticancer drugs and accumulated in tumor tissue via the “enhanced permeability and retention (EPR) effect,” discovered by Maeda and Matsumura.28 PEG chains on the surface of liposomes help them evade clearance by the RES in the blood, consequently prolonging their circulation time and raising the local concentration of the drug in the tumor tissue.3 Second, cyclic derivatives of RGD (Arg-Gly-Asp) oligopeptides in the liposomes can selectively bind to the αvβ3 integrin because of the receptor–ligand interaction.29,30 Some tumor cells such as breast cancer, lung cancer, and activated vascular endothelial cells have been found to highly express the dimeric transmembrane integrin αvβ3, while other endothelial cells and most noncancerous organs rarely express αvβ3.31 RGD has been shown to be a good targeting peptide for vascular endothelial cells in many cases.32 Third, D-(KLAKLAK)2 (KLA), a specific mitochondria-targeting sequence with a positive charge, has the ability to target mitochondria and disrupt the mitochondrial membrane when a threshold concentration (10 μmol) is reached.33–35 Previous research conducted by this research group demonstrated that KLA-modified nanocarriers could effectively deliver drugs to the mitochondria of both sensitive and drug-resistant cancer cells.26 Moreover, compared with triphenylphosphonium, a lipophilic cation, which targets mitochondria with a nonspecific toxicity, KLA-modified nanocarriers, may exhibit higher safety.36 The combination of cRGD and KLA peptides is expected to reduce off target effects and increase anticancer potency. In the present study, the physicochemical properties, cytotoxicity, cellular uptake, mitochondrial targeting, mitochondria-mediated apoptosis, and anticancer efficacy in breast tumor-bearing mice of PTX-loaded multistage-targeted liposomes and single targeted liposomes were evaluated and compared in detail.
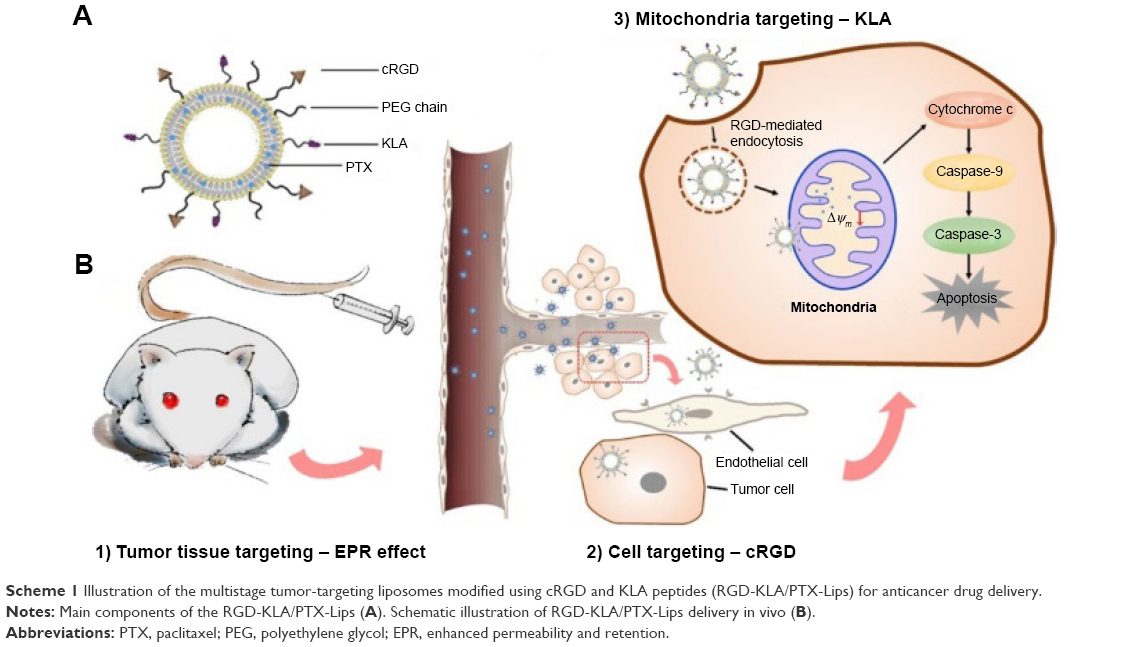 | Scheme 1 Illustration of the multistage tumor-targeting liposomes modified using cRGD and KLA peptides (RGD-KLA/PTX-Lips) for anticancer drug delivery. |
Materials and methods
Materials and measurements
cRGD peptide with a terminal cysteine (cRGD-(D-Tyr)-K, MW =722.84, cRGD-SH) and KLA peptide with a terminal cysteine (D-(KLAKLAK)2-Cys, MW =1,627, KLA-SH) were purchased from SciLight Biotechnology, LLC (Beijing, China). Soybean phosphatidylcholine (SPC) was purchased from Lipoid GmbH (Ludwigshafen, Germany). Cholesterol, amiloride, chlorpromazine and filipin were obtained from Sigma-Aldrich (Louis, MO, USA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy-(polyethylene glycol)-2000] (DSPE-mPEG2000) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide-(polyethylene glycol)-2000] (DSPE-mPEG2000-mal) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). 1,10-Dioctadecyl-3,3,30,30-tetramethylindocarbocyanine perchlorate (DIL) was purchased from Fanbo Biochemicals, Co., Ltd. (Beijing, China). LysoTracker® green and Mitotracker® Green FM were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). A Cell Counting Kit-8 assay (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). PTX was purchased from Meilun Biology Technology Company (Dalian, China). Taxol was purchased from Sichuan Taiji Pharmaceutical Company (Chengdu, China). Characterization and structural identification were performed using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS, Autoflex MALDI-TOF/TOF). Particle size and zeta potential measurements were performed in MilliQ water using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK).
Synthesis of DSPE-PEG-RGD and DSPE-PEG-KLA
DSPE-mPEG2000-Mal (100 mg, 37 μmol) was dissolved in chloroform and then cRGD-SH (40.20 mg, 55.5 μmol) dissolved in methanol was added into the DSPE-PEG-Mal solution. The mixed solution was then stirred under nitrogen for ~24 h in darkness at room temperature. After confirming the disappearance of DSPE-PEG-Mal using thin-layer chromatography, the organic solvent was removed using rotary evaporation. The white precipitate was washed using methanol three times, collected through filtration, and then confirmed using MALDI-TOF MS. DSPE-PEG-KLA was synthesized using the same method as that used for DSPE-PEG-cRGD. The feed molar ratio of DSPE-mPEG2000-Mal to KLA-SH was 37:55.5. The resultant product was confirmed using MALDI-TOF MS.
Preparation and characterization of liposomes
PTX-loaded liposomes were prepared using the thin-film hydration method.26 Briefly, SPC, cholesterol, DSPE-mPEG2000 and PTX (90:10:5:10, molar ratio) were dissolved in chloroform:methanol (2:1, v:v) in a round-bottomed flask. After the organic solvent was removed using rotary evaporation, the dried lipid films were hydrated in phosphate-buffered saline (PBS, pH 7.4) or ultrapure water at 37°C to achieve a final phospholipid concentration of 3.0 mg/mL. The liposomes were sonicated in a sonication bath at 37°C for 15 min and then further sonicated using a probe sonicator (Sonics & Materials Inc., Danbury, CT, USA) at 40 W for 80 s to form PTX-loaded PEGylated liposomes (PEG/PTX-Lips). The unloaded PTX was removed from the liposomes through centrifugation at 1,000 rpm for 10 min.
Peptide-modified liposomes were also prepared as per the above procedure. The components of the cRGD-modified liposomes (RGD/PTX-Lips) were SPC, cholesterol, DSPE-mPEG2000, DSPE-PEG2000-cRGD and PTX (90:10:2.5:2.5:10, molar ratio). The components of the KLA-modified liposomes (KLA/PTX-Lips) were SPC, cholesterol, DSPE-mPEG2000, DSPE-PEG2000-KLA and PTX (90:10:2.5:2.5:10, molar ratio). The components of the multistage tumor-targeting liposomes (RGD-KLA/PTX-Lips) were SPC, cholesterol, DSPE-PEG2000-cRGD, DSPE-PEG2000-KLA and PTX (90:10:2.5:2.5:10, molar ratio). DIL-loaded liposomes were prepared as per the same protocol as that used for the PTX-loaded liposomes, but PTX was replaced with DIL. The preparation of blank liposomes followed the same procedure as that used to prepare the PTX-loaded liposomes, without the addition of PTX.
The encapsulation efficiency (EE) was calculated as follows: EE = W/W0×100%. W was the weight of PTX loaded in the liposomes after centrifugation. W0 was the weight of PTX initially added during liposome preparation. Briefly, PTX-containing liposomal solution was centrifuged at 1,000 rpm for 10 min to remove any PTX already released from the liposomes. The supernatant was evaporated to dryness under vacuum, redissolved in methanol and analyzed using high-performance liquid chromatography (HPLC; Agilent Technologies, Santa Clara, CA, USA). The average particle size and zeta potentials of the various liposomes were measured at 25°C using a Zetasizer Nano ZS (Malvern Instruments). The liposomes (100 μL; phospholipid concentration 3 mg/mL) were DILuted in 1 mL of PBS for these measurements.
Cell lines and cell culture
NIH 3T3 fibroblast cells and 4T1 cells (murine breast cancer cells) were purchased from the Chinese Academy of Science Cell Bank for Type Culture Collection (Shanghai, People’s Republic of China). NIH 3T3 fibroblasts cells were cultured in Dulbecco’s Modified Eagle’s Medium, while 4T1 cells were cultured in RPMI-1640, both supplemented with 10% fetal bovine serum and 100 μg/mL streptomycin, at 37°C under 5% CO2. Human umbilical vascular endothelial cells (HUVECs) were purchased from Sciencell™ Research Laboratories (Carlsbad, CA, USA) and cultured at 37°C under 5% CO2 in endothelial cell medium, supplemented with 5% fetal bovine serum, 1% endothelial cell growth supplement and 1% penicillin/streptomycin solution. Cells were harvested using 0.25% trypsin-EDTA and rinsed with PBS. The resulting cell suspension was used for in vitro cell experiments.
Cellular uptake and mitochondrial targeting
Cellular uptake efficiency
The cellular uptake by HUVECs and 4T1cells of DIL-loaded liposomes was measured using flow cytometry (FCM; Cytomics™ FC 500; Beckman Coulter, Miami, FL, USA). HUVECs or 4T1 cells were seeded in six-well plates at a density of 4×105 cells/well in 2 mL of growth medium. After incubation for 12 h at 37°C and 5% CO2, the cells were treated with various DIL formulations, which were DILuted in culture medium to reach a final DIL concentration of 2 μM. Control experiments were performed by adding blank medium. After 1 or 4 h of incubation, the collected cell pellets were washed twice using cold PBS and then resuspended in 0.3 mL of PBS. The fluorescence intensity of 1×104 cells was measured using FCM with an excitation wavelength of 549 nm and an emission wavelength of 565 nm. Each assay was repeated in triplicate.
Endocytosis pathways for RGD-KLA-Lips
To explore the endocytosis pathways for various liposomes, cellular uptake of DIL-loaded liposomes by 4T1 cells in the presence of endocytosis inhibitors was measured using FCM. The cells (4T1 cells) were seeded in six-well plates at a density of 4×105/well in 2 mL of growth medium. After incubation for 12 h at 37°C and 5% CO2, cells were preincubated with various inhibitors of endocytosis, including a caveolin-mediated endocytosis inhibitor: filipin (5 μg/mL); a micropinocytosis inhibitor: amiloride (1.48 mg/mL) and a clathrin-mediated endocytosis inhibitor: chlorpromazine (20 μg/mL), for 0.5 h at 37°C. Then, 4T1 cells were treated with DIL-loaded liposomes. After a further 2 h incubation, the collected cell pellets were washed twice using cold PBS, and the fluorescence intensity was determined using FCM.
Endosomal escape
The endosomal escape capacity of RGD-KLA-Lips from 4T1 cells was assessed using a confocal laser scanning microscope (CLSM; Leica TCP SP5, Mannheim, Germany). The cells (1×104 4T1 cells/well) were seeded in a glass-bottomed dish (Φ 15 mm; NEST, China). After 24 h of incubation, the cells were treated using DIL-loaded RGD-KLA/DIL-Lips (2 μM) for 2 h, and the medium was then replaced with fresh medium. Cells were cultivated for an additional 1, 2, 3 or 4 h. Subsequently, the cells were washed twice using cold PBS and stained using LysoTracker Green (100 nM) at 37°C for 30 min. Stained cells were rinsed three times using PBS to remove free tracking agent and then observed using CLSM.
Drug content in the isolated mitochondrial fraction
Drug accumulation in the isolated mitochondrial fractions in 4T1 cells was detected using a flow cytometer. The cells seeded in six-well plates were cultured for 24 h and then treated with DIL-loaded liposomes of different formulations at a 2 μM concentration of DIL for another 8 h. Cells were washed using cold PBS twice, and mitochondria were isolated using a cell mitochondria isolation kit (Beyotime Institute of Biotechnology, China). Briefly, the cells were solubilized using lysis buffer, cooled in an ice bath for 30 min and stirred in a homogenizer. The lysates were centrifuged for 10 min at 600× g. The supernatant was collected and centrifuged at 11,000× g for 10 min to collect the mitochondria from the precipitates. The uptake of liposomes in the mitochondria was detected using FCM, using the fluorescence intensity of the loaded DIL. Each assay was performed in triplicate.
Mitochondrial targeting
Mitochondrial localization of various DIL-loaded liposomal formulations in HUVECs and 4T1 cells was observed using a CLSM. Cells were seeded at a density of 1×104 cells/well in a glass-bottomed dish (Φ 15 mm; NEST). After 24 h of incubation, the cells were treated with RGD/DIL-Lips, KLA/DIL-Lips and RGD-KLA/DIL-Lips (DIL concentration of 2 μM) for 8 h. Subsequently, the cells were washed twice with cold PBS and stained using Mitotracker Green FM (75 nM) at 37°C for 30 min. Stained cells were rinsed three times with PBS to remove free tracking agent and then observed using CLSM.
Cytotoxicity assay
A cytotoxicity assay of the blank RGD-KLA-Lips was conducted using NIH 3T3 fibroblasts. NIH 3T3 cells were seeded into a 96-well plate at a density of 1×104 cells/well. After 24 h of incubation at 37°C and 5% CO2, cells were treated with fresh medium containing a series concentration of RGD-KLA-Lips. Cells treated with blank culture media served as a control. After an additional 24 h, 10 μL of CCK-8 was added into each well and cells were further incubated for 2 h at 37°C. The absorbance of the samples was measured at 450 nm using a microplate reader (Thermo Scientific, Waltham, MA, USA). The cytotoxicity of PTX formulations, including Taxol (free PTX), RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips, was evaluated using HUVECs and 4T1 cells. Cells were seeded into 96-well plates at a density of 1×104 cells/well and incubated for 24 h at 37°C and 5% CO2. Cells were then treated using different concentrations of the PTX formulations and evaluated using a CCK-8 assay. The half-maximal inhibitory concentration (IC50) of each treatment was also calculated using Graph Pad Prism 5 software (GraphPad Software, Inc., San Diego, USA).
Mitochondria-mediated cell apoptosis pathway
Mitochondrial targeting
Mitochondrial localization of various DIL-loaded liposomal formulations in HUVECs and 4T1 cells was observed using CLSM. Cells were seeded at a density of 1×104 cells/well in a glass-bottomed dish (Φ 15 mm; NEST). After 24 h of incubation, the cells were treated with RGD/DIL-Lips, KLA/DIL-Lips and RGD-KLA/DIL-Lips (DIL concentration of 2 μM) for 8 h. Subsequently, the cells were washed twice with cold PBS and stained using Mitotracker Green FM (75 nM) at 37°C for 30 min. Stained cells were rinsed three times with PBS to remove free tracking agent and then observed using CLSM.
Mitochondrial depolarization
The change in mitochondrial membrane potential (ΔΨm) was detected using FCM after incubation with JC-1 (Beyotime Institute of Biotechnology) using the protocol recommended by the manufacturer. Briefly, 4T1 cells were seeded in six-well plates at a density of 2×105 cells/well followed by a 12 h incubation. Then, various PTX formulations (Taxol, RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips) were added to each well to give a final PTX concentration of 4 μg/mL and incubated for 10 h. Cells were harvested and washed using PBS. The cells were then suspended in binding buffer and incubated with JC-1 (5 μg/mL) at 37°C in the dark. After 20 min, the cells were washed twice with cold PBS and analyzed immediately using FCM. Carbonyl cyanide 3-chlorophenylhydrazone-treated cells were used as a positive control, and untreated cells (without liposomes) were used as a negative control. Decreased ΔΨm =100%× green fluorescence/red fluorescence. Each assay was repeated in triplicate.
Release of cytochrome c from mitochondria
Release of cytochrome c from mitochondria was detected using a streptavidin–peroxidase immunohistochemical kit (Zhongshan Goldenbridge Biotechnology, Co., Ltd., Beijing, China). Briefly, 4T1 cells at a density of 2×105/well were seeded in six-well plates with coverslips at the bottom of each well and incubated for 12 h. Then Taxol and PTX-loaded liposomes (RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips) were added to each well to give a final PTX concentration of 4 μg/mL and cultured for 10 h. Culture medium without PTX was used for a negative control. The cells were fixed in 4% paraformaldehyde for 10 min, soaked in 3% H2O2 for 5 min and blocked using 5% bull serum albumin. Cells were then incubated with an anticytochrome c primary antibody solution (Nanjing KeyGen Biotechnology, Co., Ltd., Nanjing, China), a secondary antibody solution and a streptavidin horseradish peroxidase (HRP) conjugate. The cells were stained using hematoxylin and an acetic bath and observed using an optical microscope.
Caspase activity assay
The activity of caspase-9 and −3 was determined using colorimetric assays included in caspase-9 and caspase-3 activation kits (Beyotime Institute of Biotechnology). Briefly, 4T1 cells were seeded in six-well plates at a density of 2×105 cells/well for a 12 h incubation. Then, Taxol and PTX-loaded liposomes (RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips) were added to each well to give a final PTX concentration of 4 μg/mL and incubated for 10 h. Cells incubated in culture medium without PTX were used as a negative control. Cells were harvested, washed with PBS and suspended in 100 μL of the supplied lysis buffer for 30 min in an ice bath. Obtained cell lysates were centrifuged at 16,000× g for 15 min at 4°C. The supernatant was collected and incubated with caspase-9 or caspase-3 substrates at 37°C for 4 h in the dark. The activity of caspase-9 and caspase-3 was then detected by measuring the absorbance at 405 nm using a microplate reader. Each assay was repeated in triplicate.
Apoptosis assay
Apoptosis induced by various PTX formulations was measured using an Annexin V-FITC Apoptosis Detection kit (Qcbio Science & Technologies, Shanghai, China) and analyzed using FCM. Briefly, 4T1 cells were seeded in six-well plates at a density of 2×105 cells/well for a 12 h incubation. Then Taxol and PTX-loaded liposomes (RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips) were added to each well at a final PTX concentration of 4 μg/mL and cultured for another 12 h. Blank RGD-KLA-Lips, with a KLA peptide concentration of 2.76 μM, which is equal to RGD-KLA/PTX-Lips at a concentration of 4 μg/mL of PTX, were also assessed in the assay. Cells were harvested, washed with cold PBS, suspended in binding buffer and stained using Annexin V-FITC and propidium iodide (PI, provided in kit) for 20 min and were analyzed using FCM with excitation wavelengths of 488 and 535 nm and emission wavelengths of 525 and 615 nm for FITC and PI-DNA, respectively.
In vivo antitumor efficacy
Six-week old female BALB/c mice were purchased from Dossy Biological Technology, Co., Ltd. (Chengdu, China) and maintained on the premises under standard animal house conditions. Animal welfare and experimental procedures were carried out strictly in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care and Office of Laboratory Animal Welfare guidelines under the direction of the Institutional Animal Care and Use Committee. All animal experiments were performed under the approval of the Experimental Animal Ethical Committee of Sichuan University. For the establishment of xenograft tumors, 4T1 cells (1×106) suspended in PBS (50 μL) were subcutaneously injected into the right-hand side of the back of female BALB/c mice. Tumor volume (V) was calculated as V = L × W2/2, where the length (L) and width (W) were measured. The drug was administered when tumors grew to a uniform size of ~50–100 mm3.
To investigate antitumor efficacy in vivo, 4T1 tumor-bearing mice were randomly divided into five groups (n=6). Saline, Taxol, RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips (200 μL) were injected via the tail vein in each group at days 9, 12, 15 and 18 posttumor inoculation, and the injected PTX dosage was normalized to 7.5 mg/kg. The tumor volume and body weight of mice were measured every 3 days. At day 22 after inoculation, the mice were euthanized, and tumors were collected and weighed. Liver, heart, spleen, lung and kidney were also collected. The collected tissues were fixed in 4% formaldehyde overnight, embedded in paraffin and cut using a microtome (5 μm sections) for hematoxylin and eosin (H&E) staining and were then analyzed using an optical microscope.
Immunohistochemical detection
A Ki67 staining method was used to quantify the proliferative cells in the tumors. Briefly, the tissue sections (5 μm thick) were deparaffinized using xylene, rehydrated using alcohol and were incubated in 1% H2O2 to inactivate endogenous peroxidase. The tissue sections were then incubated with a Ki67 antibody (Abcam, Cambridge, MA, USA) at 4°C overnight. After incubation with biotinylated goat antirabbit antibodies (ZSGB-BIO, Beijing, China) as secondary antibodies (DILution ratio, 1:100) for 30 min at 37°C, the sections were incubated with HRP (ZSGB-BIO; DILution ratio, 1:100) for 30 min and then counterstained using hematoxylin. The Ki67 index was calculated as the ratio of proliferative cells to total cells in each field, choosing five random fields with an optical microscope.
To quantify the apoptotic cells, the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay was conducted. Tissue sections (5 μm thick) were incubated with proteinase K (Merck Millipore, Bedford, MA, USA) at 37°C for 25 min and washed several times in PBS. After incubation with equilibration buffer and TdT enzyme, the tissue sections were treated with POD-conjugated antifluorescein (conjugated with HRP; Roche Applied Science, Penzberg, Germany) and 3,30-diaminobenzidine. All sections were analyzed using an optical microscope. The apoptotic index was calculated as the ratio of apoptotic cells to total cells in each field, choosing five random fields.
A CD31 staining assay was used to quantify the microvessel density (MVD) in the tumor tissues using an experimental method similar to that of the Ki67 staining assay. MVD was assessed in accordance with the international consensus report. The slides were examined under 100× magnification to identify the highest vascular density area (so called “hot spots”). Ten areas of highest MVD were selected for counting by two independent pathologists under 400× magnification and averaged to obtain the MVD level for each group.
PTX quantification using HPLC
PTX was measured via HPLC using an Agilent HPLC system consisting of a 1260-pump and a 1260-ultraviolet detector (Agilent Technologies). An Agilent ODS3 column (250×4.6 mm, 5 μm; Agilent Technologies) and a detector wavelength of 227 nm were used. A solvent mixture of methanol–water (50:50, v/v) was used as an eluent, and the flow rate was 1.5 mL/min.
Statistical analysis
All data are presented as mean ± SD. The Student’s t-test or one-way analysis of variance (ANOVA) was used to determine statistical significance. Statistical significance was set at *P<0.05, and **P<0.01 indicates highly significant differences.
Results and discussion
Synthesis of DSPE-PEG-RGD and DSPE-PEG-KLA
The synthetic routes of DSPE-PEG-RGD and DSPE-PEG-KLA are presented in Scheme S1. The resulting products were synthesized by conjugating DSPE-PEG-Mal to the cysteine residue on the cRGD or KLA peptides. The synthesis process was monitored using thin-layer chromatography, and the excessive peptide was removed via washing with methanol. The MALDI-TOF results (Figure S1A and B) show that the molecular weights of DSPE-PEG-RGD and DSPE-PEG-KLA are ~4,527 and 3,624 Da, respectively, which is consistent with the theoretical molecular weights (4,527 and 3,624 Da).
Preparation and characterization of liposomes
All the PTX-loaded liposomes had a similar diameter ranging from 109.97 to 134.90 nm and a similar polydispersity index ranging from 0.240 to 0.268, suggesting that they can enter leaky vasculature via the EPR effect because their diameter is in the range of 20–200 nm, as shown in Table 1.3 The PEG/PTX-Lips and RGD/PTX-Lips exhibited a moderately negative surface charge (−6.14±0.62 and −11.11±1.65 mV, respectively). The KLA/PTX-Lips exhibited a positive surface charge of 9.17±0.40 mV, mainly because of the six alkaline lysine residues on the KLA peptide, whereas the RGD-KLA/PTX-Lips were slightly negatively charged with a zeta potential value of −1.19 mV, as expected. It has been reported that nanoparticles with a slightly negative surface charge exhibit preferential uptake at the tumor site but not at normal organs,37 which could be beneficial for extending the duration that the nanoparticles are in circulation because of efficient evasion of renal elimination.38,39 All the liposomes had high encapsulation efficiencies, >80%. The size and zeta distributions of the RGD-KLA/PTX-Lips are shown in Figure 1A and B, and the size distributions of the other liposomes are presented in Figure S2.
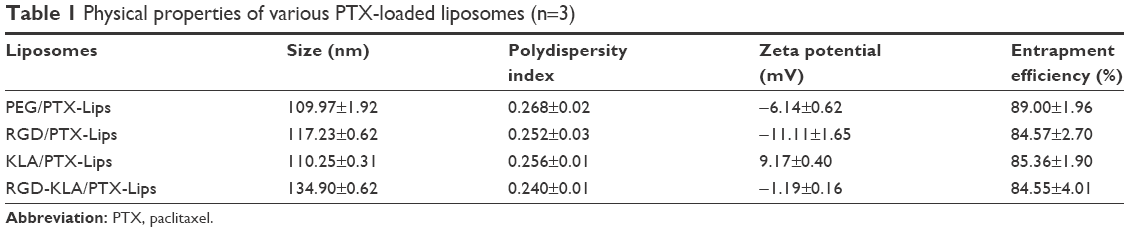 | Table 1 Physical properties of various PTX-loaded liposomes (n=3) |
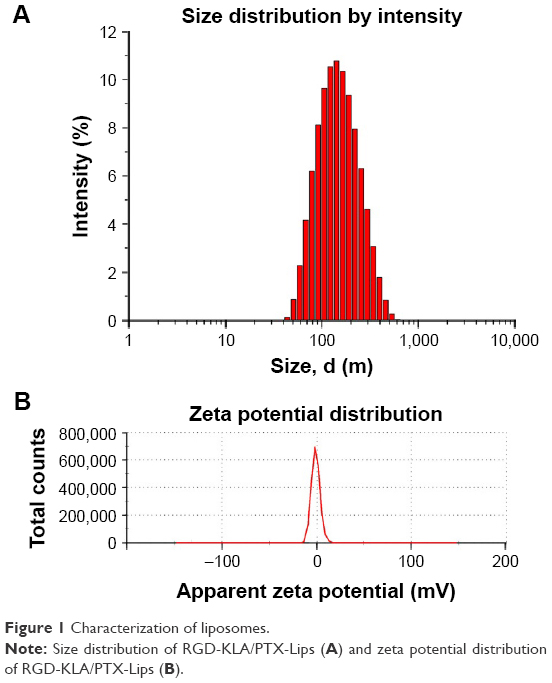 | Figure 1 Characterization of liposomes. |
Cellular uptake and endocytosis pathways
The cellular uptake of the liposomes by HUVECs and 4T1 cells was measured using FCM. Fluorescence intensities were used to indicate uptake efficiencies. After incubation with HUVECs for 1 h (Figure 2A), few PEG/DIL-Lips were internalized by cells and the fluorescence intensity was only 2.83±0.04. In contrast, the cellular uptake of RGD/DIL-Lips was 8.7-fold higher than that of PEG/DIL-Lips mainly because of the high binding affinity between cRGD and integrin αvβ3, which is highly expressed in HUVECs.40,41 KLA/DIL-Lips could also deliver more DIL into cells with a 9.2-fold higher fluorescence level in comparison with PEG/DIL-Lips. The enhanced uptake of KLA/DIL-Lips is attributed in part to the interaction with cell membranes via electrostatic attraction and hydrogen bonding.42,43 It is worth noting that the RGD-KLA/DIL-Lips achieved the highest cellular uptake among all the liposomes (103.5-fold higher than the PEG-Lips), suggesting that cRGD and KLA peptides have a synergistic effect on the internalization of liposomes. This synergistic effect can be attributed to the identification of the αvβ3 integrin by the RGD motifs, allowing liposomes to come into close contact with the cell surface, which facilitated the participation of KLA in electrostatic interactions with the cell surface.44 As a result, the cellular uptake of RGD-KLA/DIL-Lips was significantly increased. It is also related to the conformation of the two ligands, which caused heteromultivalency of the liposomes.45 Such heteromultivalent constructs have been reported to enhance binding affinity and selectivity compared with single modified liposomes.46–48 Moreover, the cellular uptake increased in a time-dependent manner. With an extension of incubation time, the cellular uptake of liposomes increased quickly, but the relative uptake of the various liposome formulations did not change. This time-dependent behavior could be explained by the presence of an active endocytosis process.8 Similar phenomena were also observed in 4T1 cells (Figure 2B). RGD-KLA/DIL-Lips showed an increased cellular uptake as compared with PEG/DIL-Lips (47.6-fold higher for 1 h and 62.2-fold higher for 4 h). The above results demonstrate that cRGD and KLA peptides can work together to facilitate the uptake of liposomes.
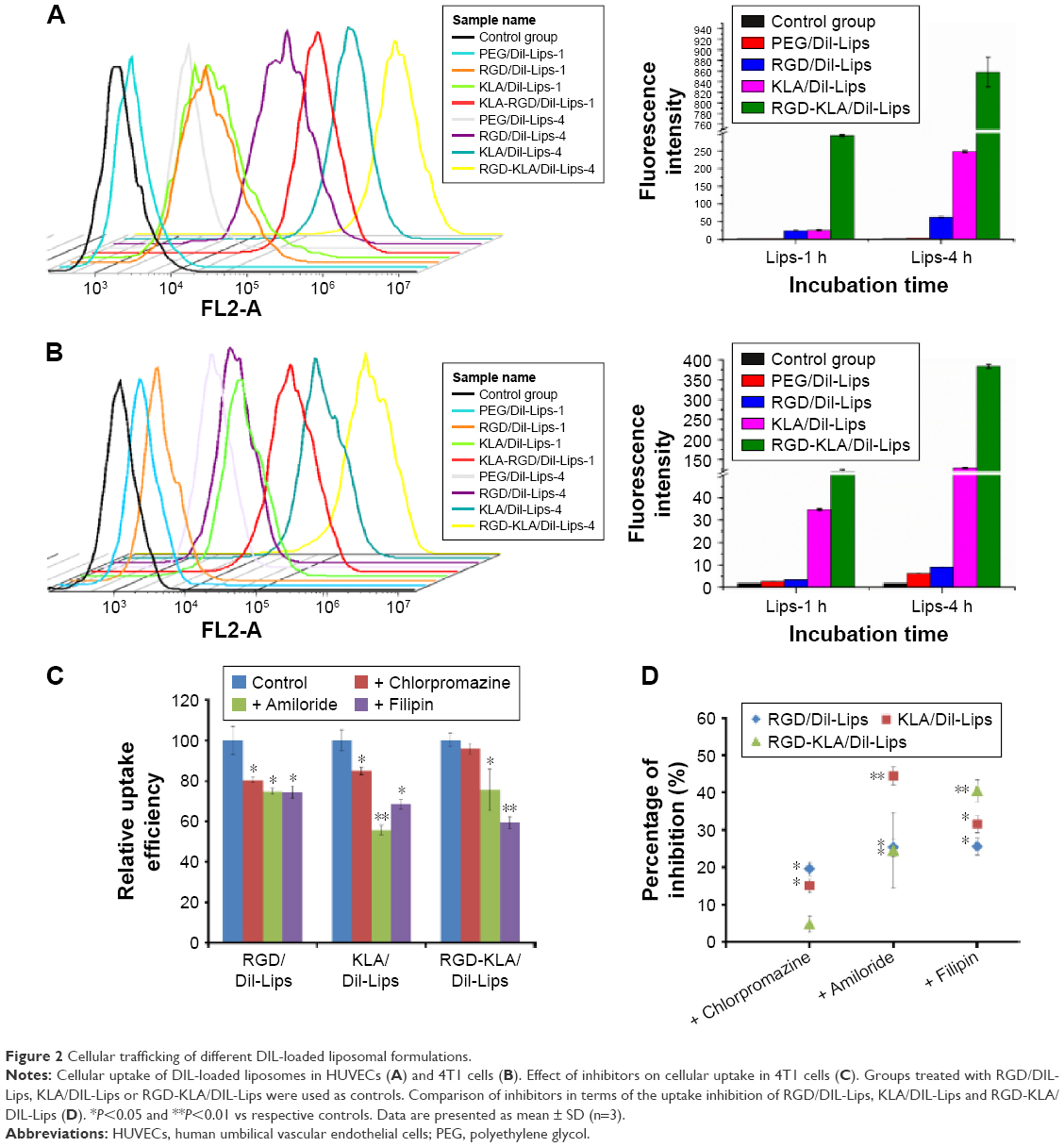 | Figure 2 Cellular trafficking of different DIL-loaded liposomal formulations. |
The internalization mechanism of RGD-KLA/DIL-Lips was also evaluated using various inhibitors of endocytosis pathways. Chlorpromazine, filipin and amiloride were used to block clathrin-, caveolin- and macropinocytosis-mediated endocytosis, respectively.49,50 Figure 2C and D demonstrate that the cellular uptake efficiencies of RGD/DIL-Lips were reduced by ~20% (P<0.05) in the presence of each of the endocytosis inhibitors, indicating that all three endocytic pathways are involved in the internalization of RGD/DIL-Lips in 4T1 cells, and they play almost equally important roles in the process. This result is consistent with those reported in previous studies.30,51 Similarly, KLA/DIL-Lips entered cells through macropinocytosis, clathrin and caveolin-dependent endocytosis. However, among them, caveolin-dependent endocytosis is the main endocytosis pathway since amiloride could significantly block the uptake of KLA/DIL-Lips with an inhibition of 44.5% (P<0.01). In the case of RGD-KLA/DIL-Lips, amiloride and filipin decreased the cellular uptake by 24.5% (P<0.05) and 40.6% (P<0.01), respectively. The results reveal that liposomes modified with cRGD and KLA demonstrated increased cellular uptake through caveolin-dependent endocytosis and macropinocytosis. These two independent pathways did not interfere with each other but rather promoted the cellular uptake of the nanoparticles.
Endosomal escape and mitochondrial targeting
To demonstrate the potential of the prepared mitochondria-targeted liposomes (RGD-KLA/DIL-Lips) for cancer therapy, endosomal escape and drug accumulation in isolated mitochondrial fractions were explored using confocal fluorescence microscopy and FCM. As shown in Figure 3A, with an extension of incubation time, more particles were released into the cytosol (red dots in merged images), indicating that liposomes escaped from lysosomes. The KLA conjugate contains six lysine residues and an alanine residue, which could produce a proton sponge effect similar to that produced by polyethylenimine, leading to swelling and disruption of endosomes and the liberation of intact liposomes.52,53 It has also been reported that internalization through macropinocytosis is more favorable for drug delivery than that through clathrin-mediated endocytosis, since the porous membrane structure of macropinosomes releases the internalized particles to the cytosol more easily (Figure 2C and D), resulting in a low level of lysosomal degradation.49,54 The above results suggest that multistage tumor-targeting liposomes are more easily internalized by cells through caveolin-dependent endocytosis and macropinocytosis and that they have the ability to escape from lysosomes. Figure 3B and C shows that the average fluorescence intensity of the RGD-KLA/DIL-Lip-treated group was 809.00±14.11, about 3-fold higher than that of the KLA/DIL-Lips group (256.67±8.33) and 17-fold higher than that of the RGD/DIL-Lips group (46.40±1.77). These data suggest that RGD-KLA/DIL-Lips could successfully target to mitochondria, with drug accumulation in the mitochondrial fractions and that KLA played an essential role in the targeting process.26,55
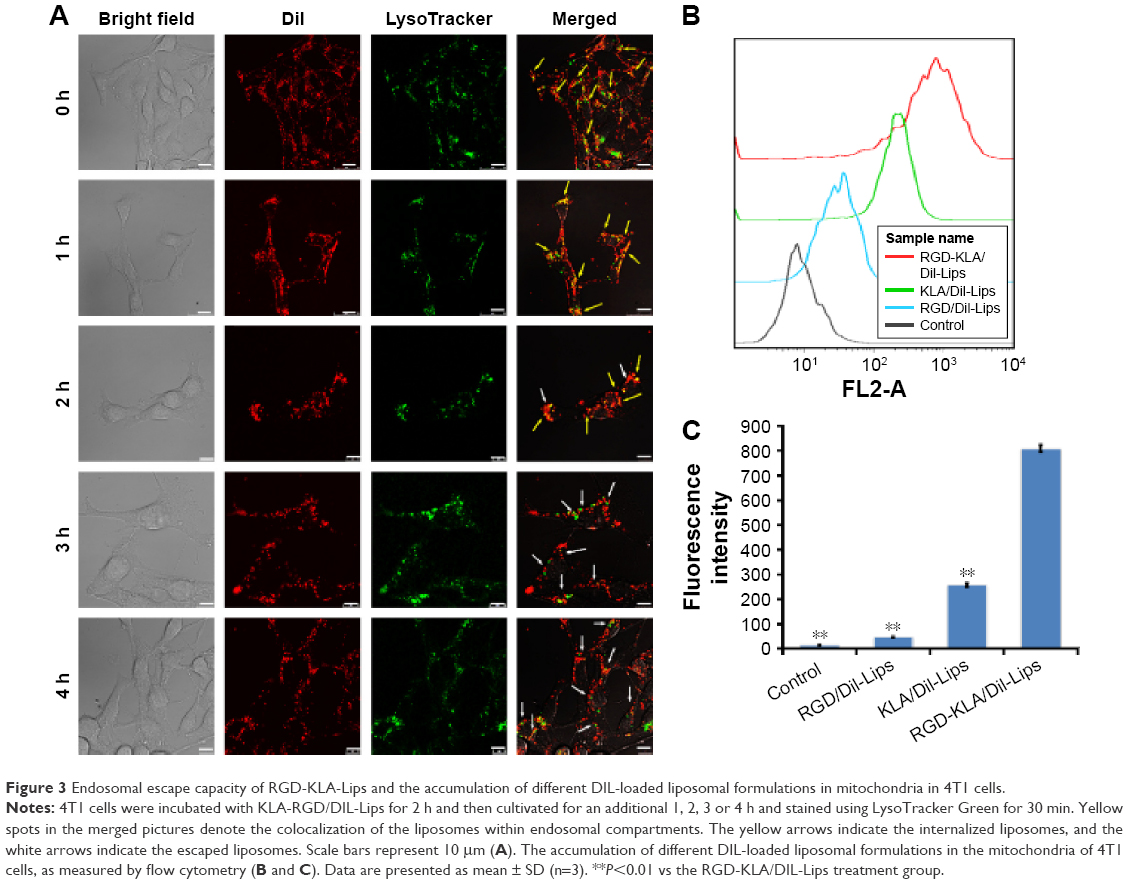 | Figure 3 Endosomal escape capacity of RGD-KLA-Lips and the accumulation of different DIL-loaded liposomal formulations in mitochondria in 4T1 cells. |
To determine whether the enhanced anticancer and antiangiogenesis effects of RGD-KLA/PTX-Lips resulted from the mitochondria-mediated apoptosis pathway, mitochondrial localization in HUVECs and 4T1 cells was first examined using a CLSM. Liposomes were tracked using a red fluorescent dye (DIL) and mitochondria were stained using Mitotracker Green FM. Yellow dots indicated the colocalization of red fluorescence from DIL-loaded liposomes and green fluorescence from Mitotracker Green FM. Figure 4A and B shows that both KLA/DIL-Lips and RGD-KLA/DIL-Lips selectively accumulate in mitochondria. RGD/DIL-Lips without the KLA segment did not show mitochondrial localization.
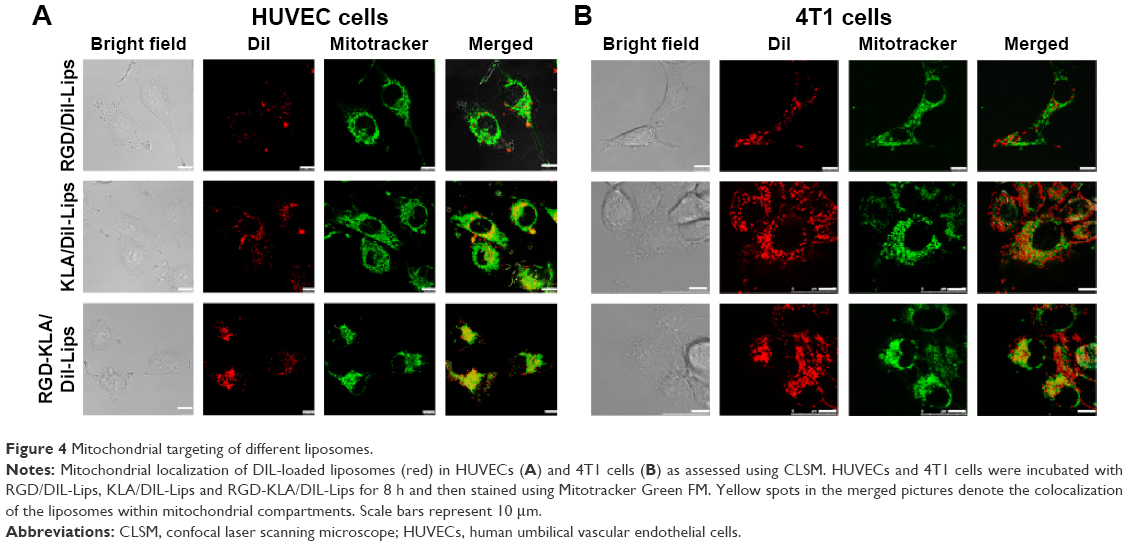 | Figure 4 Mitochondrial targeting of different liposomes. |
In vitro cytotoxicity study
The cytotoxicity of blank RGD-KLA-Lips against NIH 3T3 cells was evaluated using various lipid concentrations. Blank RGD-KLA-Lips had no influence on the proliferation of NIH 3T3 cells even at a high concentration of lipids (Figure S3), which indicates that RGD-KLA-Lips may be a safe drug carrier. Subsequently, the cytotoxicity of PTX formulations including Taxol, RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips against HUVECs and 4T1 cells was examined. Figure S4 and Table 2 show that the IC50 values of the PTX formulations against HUVECs are smaller than those against the 4T1 cells, indicating that HUVECs are quite sensitive to PTX and can be killed by PTX even at a very low concentration (Table 2). This result is consistent with other reports.56,57 PTX has been found to have antiangiogenic potential and interferes with the division of cells, but not specifically tumor cells, as the division of endothelial cells in tumors is also inhibited.57,58 Among the PTX formulations, RGD-KLA/PTX-Lips exhibited the lowest IC50 value (8.2-fold lower than Taxol), followed by RGD/PTX-Lips (2.3-fold lower than Taxol), which was caused by the enhanced cellular uptake. For 4T1 cells, the IC50 of RGD-KLA/PTX-Lips was 36.7-fold lower than that of RGD/PTX-Lips, 22.7-fold lower than that of KLA/PTX-Lips and 1.9-fold lower than that of Taxol. A strong cytotoxicity was observed in the Taxol (free PTX) group. Free PTX could be rapidly transported into cells via passive diffusion due to the high concentration gradient under in vitro conditions and immediately inhibit cell growth without a drug release process.59 The multistage tumor-targeting RGD-KLA/PTX-Lips exhibited the strongest cytotoxic effect on both cell lines. This could be a result of the high intracellular uptake and the initiation of the intrinsic apoptotic pathway by targeting mitochondria.
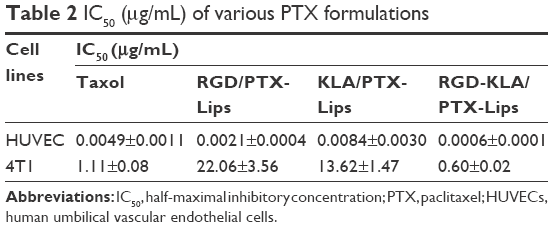 | Table 2 IC50 (μg/mL) of various PTX formulations |
Mitochondria-mediated apoptosis pathway
The destabilization of the mitochondrial membrane is considered an early event in mitochondria-mediated apoptosis, followed by the release of mitochondrial proteins, such as apoptosis inducing factor and cytochrome c, and the activation of caspases.60,61 To confirm whether intrinsic apoptosis is induced by mitochondrial targeting, various apoptotic markers in 4T1 cells were measured after incubation with Taxol, RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips.
JC-1 staining is often used to evaluate membrane depolarization. The stain specifically accumulates in the mitochondria as JC-1 aggregates emitting red fluorescence or exists in the cytoplasm as monomers emitting green fluorescence. During the process of apoptosis, the decrease in mitochondrial membrane potential results in the leakage of JC-1 from the mitochondria to the cytoplasm with an increase in green fluorescence. The reduced ratio of red to green fluorescence intensity can be calculated as ΔΨm, which indicates mitochondrial depolarization.62,63 As shown in Figure 5A and B, the ΔΨm of the blank RGD-KLA-Lip-treated group was 99.08%±1.67% of control (culture medium only), indicating that the blank RGD-KLA-Lips had no effect on the mitochondria. After treatment with Taxol, RGD/PTX-Lips, KLA/PTX-Lips or RGD-KLA/PTX-Lips for 10 h, the ΔΨm was 67.99%±6.78%, 73.65%±1.92%, 39.91%±0.40% and 19.73%±1.27% of control, respectively. RGD-KLA/PTX-Lips reduced ΔΨm the most, suggesting that RGD-KLA/PTX-Lips produced the most significant disruption of negatively charged mitochondrial membranes and triggered the highest depolarization.
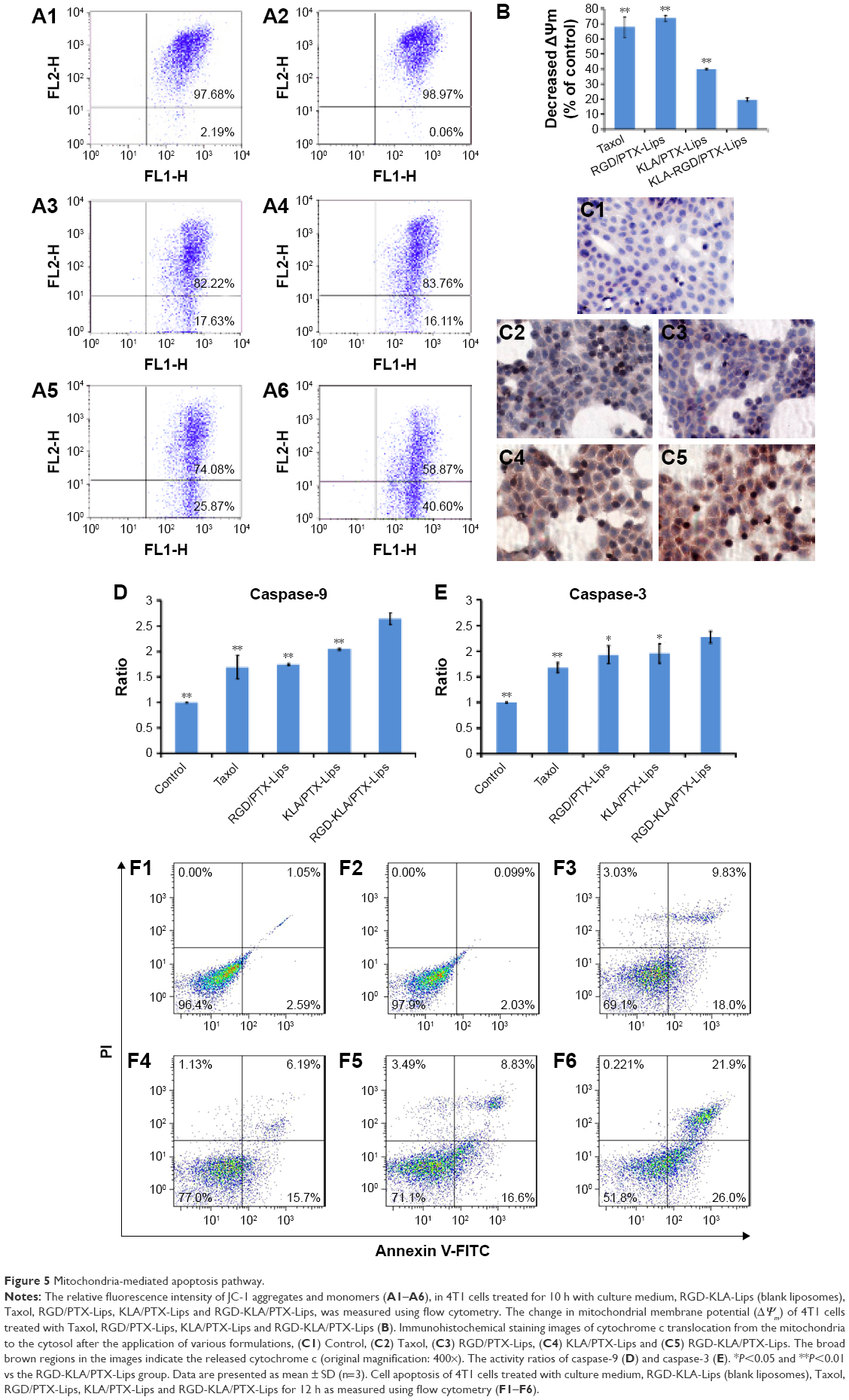 | Figure 5 Mitochondria-mediated apoptosis pathway. |
Cytochrome c, an important apoptotic marker, can form an apoptosome complex, which activates the effector caspases.64 The release of cytochrome c from the mitochondria to the cytosol was assessed. Figure 5C shows images of immunohistochemically stained samples and demonstrates the released cytochrome c, as evidenced by brown staining. No cytochrome c was observed in the control group (Figure 5C1). Only a slight cytochrome c release was observed in the Taxol group (Figure 5C2) and the RGD/PTX-Lips group (Figure 5C3). In contrast, KLA/PTX-Lips (Figure 5C4) produced a slightly increased release of cytochrome c, and RGD-KLA/PTX-Lips (Figure 5C5) produced the highest release. These results indicate the activation of mitochondria-mediated apoptosis pathways.
Cells commit irreversibly to apoptosis by activating caspases.65,66 Activation of the main caspases involved in cell apoptosis was assessed (Figure 5D and E). The activities of caspase-9 and caspase-3 were remarkably increased after treatment with RGD-KLA/PTX-Lips. The levels of caspase-9 and caspase-3 were ~2.6-fold and 2.3-fold higher than control, respectively. There was statistically significant difference between the RGD-KLA/PTX-Lip group and the other groups, whereby RGD-KLA/PTX-Lips initiated greater levels of irreversible cell apoptosis.
Apoptosis in 4T1 cells was quantified using Annexin V/PI staining and FCM (Figure 5F). After treatment with blank RGD-KLA-Lips, the total apoptotic ratio was 3.40%±1.20%, which is comparable with the negative control (3.26%±0.51%). Therefore, blank RGD-KLA-Lips did not induce cell apoptosis. The total apoptotic ratios of the Taxol, RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips groups were 29.68%±1.79%, 23.36%±1.35%, 24.29%±1.25% and 44.13%±3.43%, respectively. RGD-KLA/PTX-Lips demonstrated the strongest apoptosis-inducing effect. Based on the above results, RGD-KLA/PTX-Lips induced cell apoptosis through the mitochondria-dependent pathway, which enhanced the antitumor effect of PTX. Previous reports indicate that combining angiogenesis inhibitors with conventional anticancer drugs results in additive or even synergistic antitumor effects.4 However, there are still important issues to be addressed, such as inherent/acquired resistance to angiogenesis inhibitors like bevacizumab, enhanced invasiveness and how to achieve the most effective combinations.10,67 The multistage tumor-targeting liposomes (RGD-KLA/PTX-Lips) developed in the current study can deliver PTX to the mitochondria of both tumor vascular endothelial cells and tumor cells. Here, PTX was exploited to implement a promising therapeutic strategy to control tumor growth by inhibiting angiogenesis and killing tumor cells at the same time.
In vivo antitumor efficacy
4T1 tumor-bearing mice were treated with different PTX formulations when tumors averaged 50–100 mm3 in size. As shown in Figure 6A, the tumor volume of the saline group rapidly increased, and the tumor volume of mice treated with free PTX was not significantly decreased compared with the normal saline group, in which the tumor volume grew to 750.35±201.45 mm3. Mice treated with RGD/PTX-Lips or KLA/PTX-Lips displayed a similar tumor suppression, which was better than PEG/PTX-Lips. As expected, RGD-KLA/PTX-Lips significantly inhibited tumor growth, with statistical significance obtained on day 21.
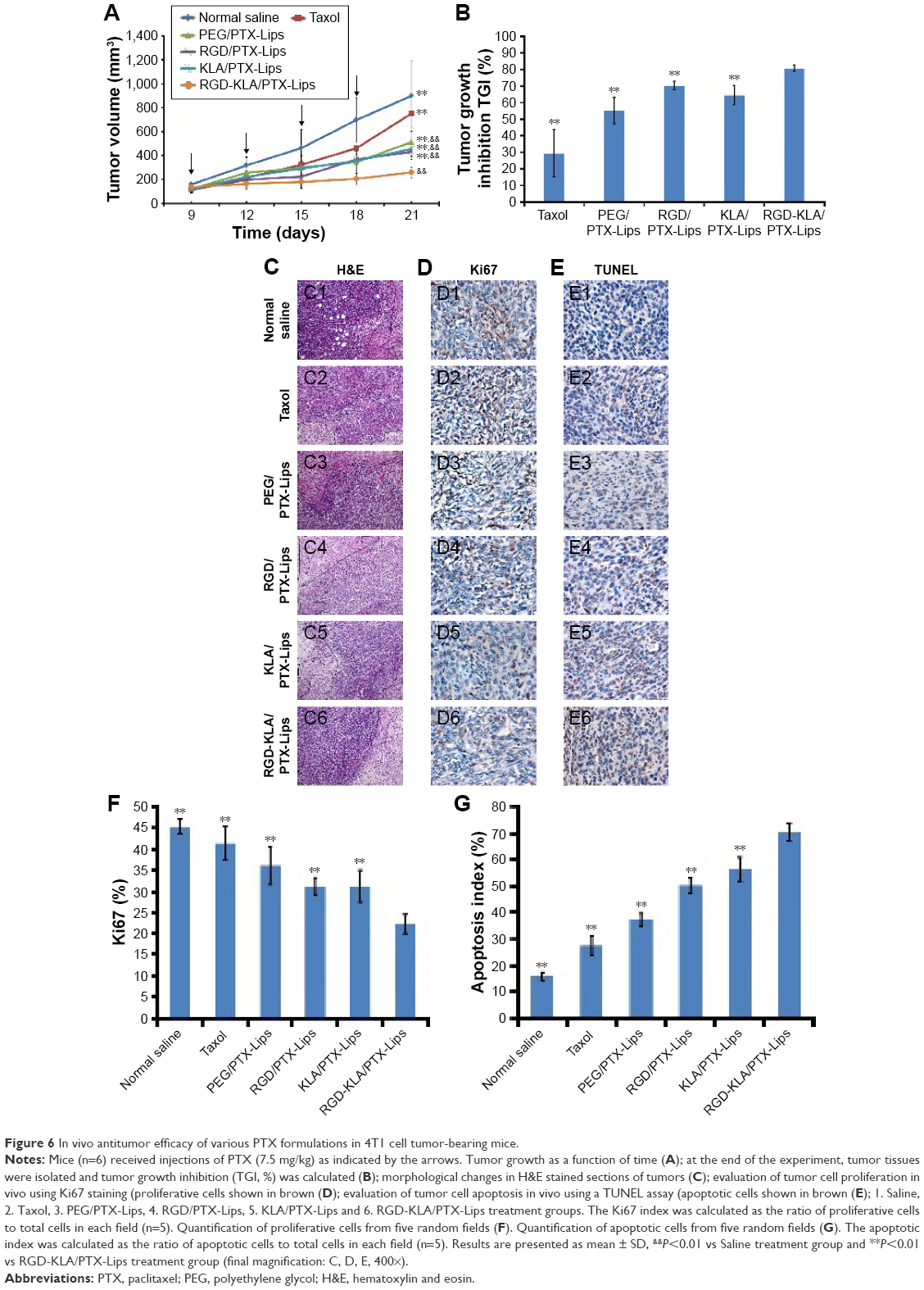 | Figure 6 In vivo antitumor efficacy of various PTX formulations in 4T1 cell tumor-bearing mice. |
After euthanizing the animals, tumor tissues were collected and weighed, and the tumor growth inhibition (TGI) rate was calculated (Figure 6B). The TGI rate of the Taxol, PEG/PTX-Lips, KLA/PTX-Lips and RGD/PTX-Lips groups was 29.41%, 55.21%, 70.16% and 64.54%, respectively, which demonstrates the importance of targeted therapies. In the RGD-KLA/PTX-Lips group, the tumor growth was suppressed by 80.62%, indicating that the multistage tumor-targeting liposome was a more efficient DDS than single-target liposomes. H&E staining confirmed that the ratio of apoptosis and necrosis was highest in the RGD-KLA/PTX-Lips group (Figure 6C). Ki-67 staining was performed to assess the inhibition of tumor cell proliferation. The RGD-KLA/PTX-Lip-treated group showed the lowest cell proliferation (Figure 6D and F, P<0.01). A TUNEL assay was used to label the apoptotic cells. The RGD-KLA/PTX-Lip-treated group exhibited the most significant levels of apoptosis (Figure 6E and G, P<0.01).
To evaluate the antiangiogenic activity of the liposomes in vivo, tumor samples were immunohistochemically stained and the MVD was calculated (Figure 7A). CD31 staining was used to identify microvessels. Very few microvessels were observed in the RGD/PTX-Lips and RGD-KLA/PTX-Lips treatment groups. Moreover, the MVD in the RGD-KLA/PTX-Lips-treated group was significantly less than in the other groups, as shown in Figure 7B (P<0.01), which confirmed the antiangiogenic effect of the cRGD-modified liposomes. Inhibiting the growth of microvessels blocked the blood supply of the tumors, which suppressed tumor growth in vivo.4,68
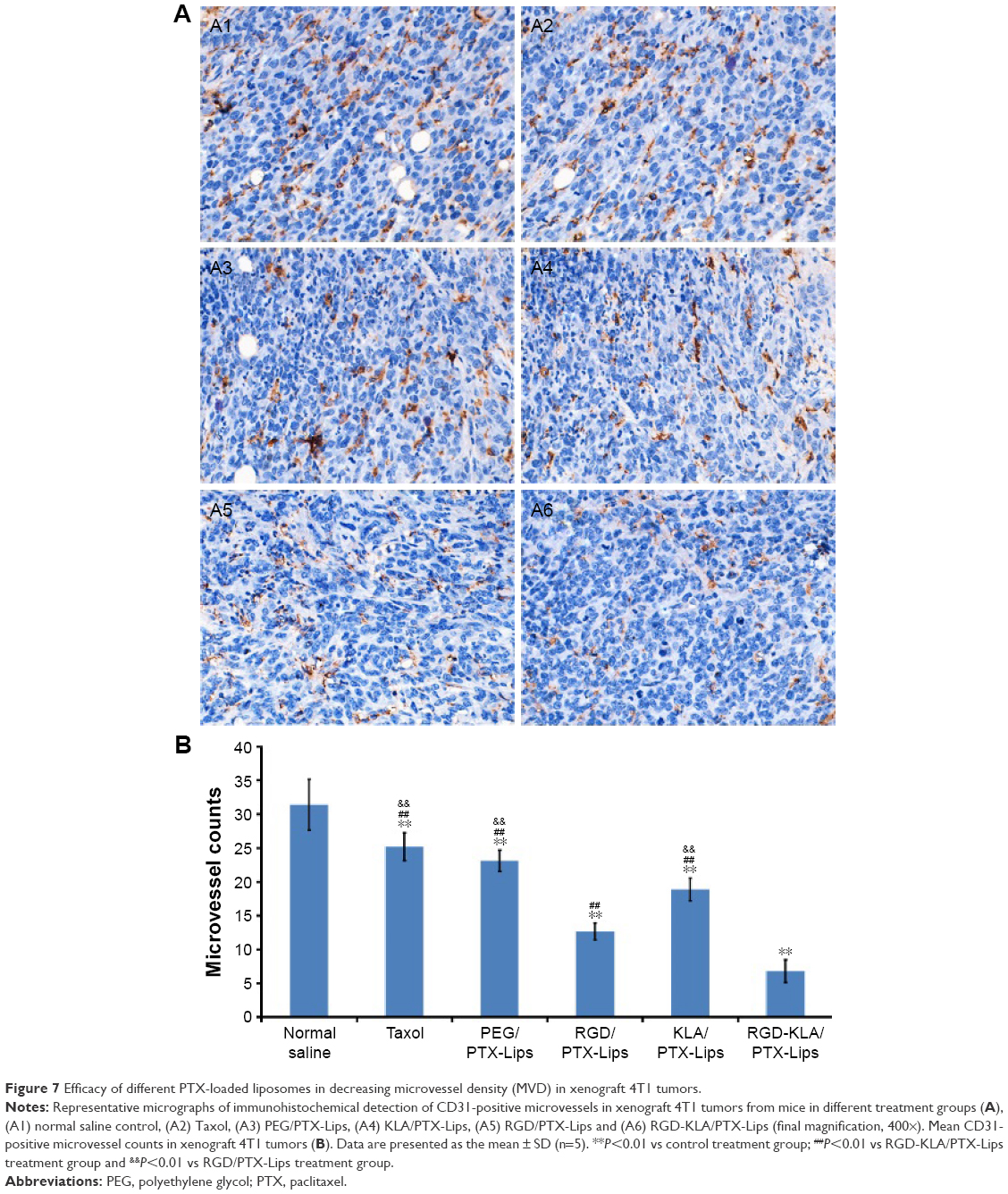 | Figure 7 Efficacy of different PTX-loaded liposomes in decreasing microvessel density (MVD) in xenograft 4T1 tumors. |
Based on the above results, the advantages of RGD-KLA/PTX-Lips may result from their passive diffusion via the EPR effect, higher accumulation in tumor vascular cells and tumor cells via receptor–ligand interactions and the activation of mitochondrial-dependent apoptosis pathways in these cells, directly killing tumor cells, decreasing the formation of blood vessels, and reducing or blocking nutrient delivery to tumors. Consequently, RGD-KLA/PTX-Lips should have an enhanced anticancer efficacy.
In vivo toxicity assays
The potential toxicity of the different formulations was assessed systematically. The body weights of all the mice were recorded and are shown in Figure 8A. No decrease in body weight was found, indicating that there was no toxicity associated with the treatments. No apparent signs of dehydration, muscle loss, locomotor impairment, anorexia or other symptoms associated with animal toxicity were observed during the treatment period. The toxicity to major organs was also investigated using H&E staining. No appreciable abnormality or noticeable organ damage was apparent after treatment with the liposomes, as shown in Figure 8B. Thus, the RGD-KLA/PTX-Lips efficiently enhanced the therapeutic index without causing side effects.
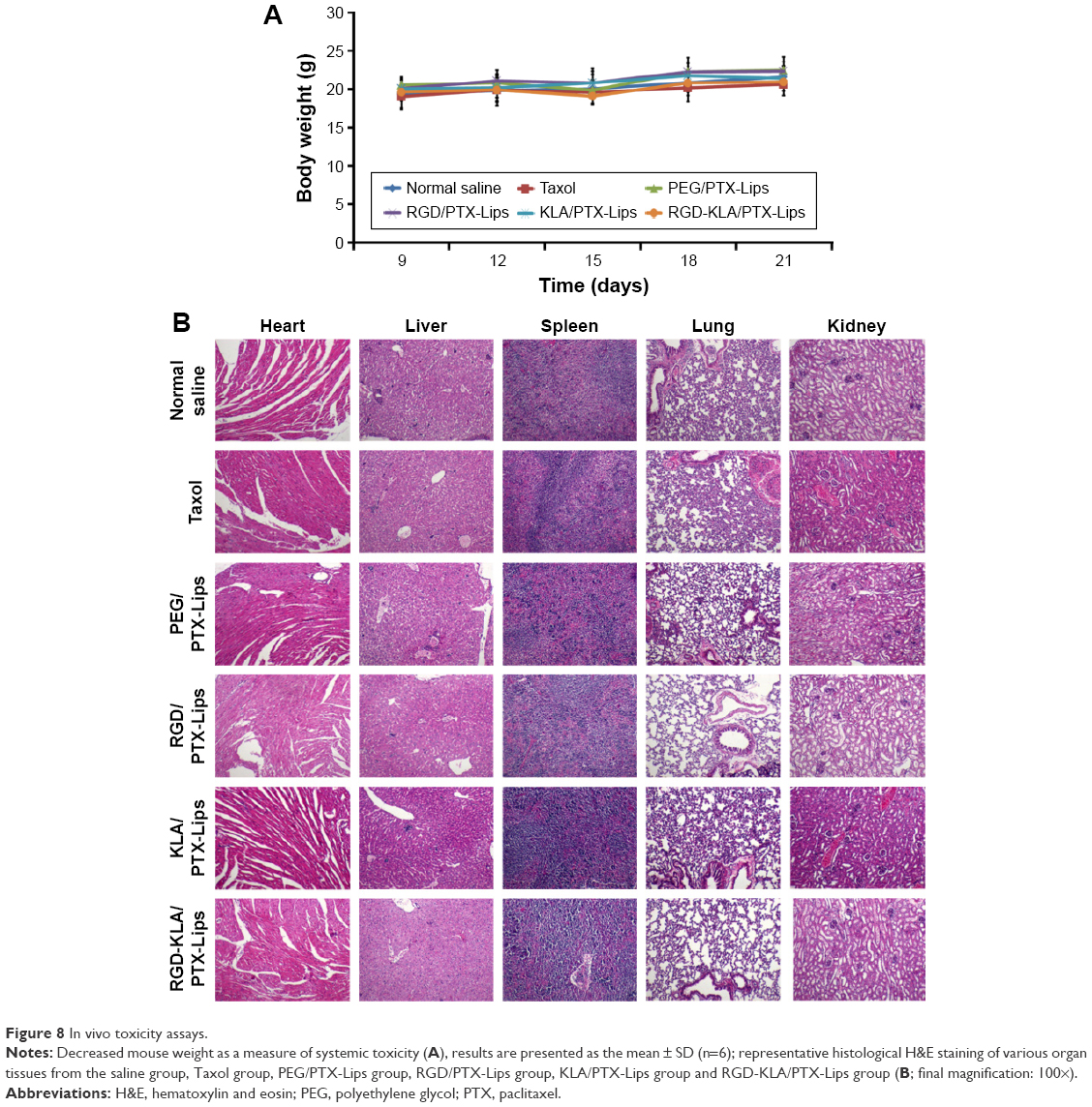 | Figure 8 In vivo toxicity assays. |
Conclusion
In the present work, a multistage tumor-targeting liposome (RGD-KLA-Lip) has been developed, in which PTX was delivered to the mitochondria of both tumor cells and tumor vascular cells to enhance anticancer activity. This PTX-loaded targeting liposome (RGD-KLA/PTX-Lips) exhibits a small size (in the range of 20–200 nm) and a slightly negative surface charge capable of achieving tumor tissue targeting through the EPR effect. RGD-PEG2000-DSPE (integrin-targeting peptide lipids) and KLA-PEG2000-DSPE (mitochondria-targeting peptide lipids) act as cell and mitochondrion-targeting moieties, respectively. The two peptides display a synergistic effect in enhancing the cellular uptake into vascular endothelial cells and tumor cells, which results in improved mitochondrial localization to facilitate mitochondria-mediated apoptosis. Consequently, superior anticancer efficacy was observed both in vitro and in vivo. Concomitantly, no systemic toxicity was found. These encouraging results demonstrate that this multistage tumor-targeting liposome is a promising entity capable of improving targeting efficiency and treatment efficacy.
Acknowledgment
The work was supported by National Natural Science Foundation of China (81371666 and 81361140343), Excellent Scholars Fund of Sichuan University (2013SCU04A09) and Sichuan University Scholarship Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
WHO. Policy and Technical Topics: Prevention and Control of Cancer–Way Forward. Geneva: WHO; 2015. | ||
Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60(15):1615–1626. | ||
Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146. | ||
Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438(7070):967–974. | ||
Folkman J. Fighting cancer by attacking its blood supply. Sci Am. 1996;275(3):150–156. | ||
Zuccari G, Milelli A, Pastorino F, et al. Tumor vascular targeted liposomal-bortezomib minimizes side effects and increases therapeutic activity in human neuroblastoma. J Control Release. 2015;211:44–52. | ||
Pastorino F, Brignole C, Di Paolo D, et al. Targeting liposomal chemotherapy via both tumor cell–specific and tumor vasculature–specific ligands potentiates therapeutic efficacy. Cancer Res. 2006;66(20):10073–10082. | ||
Xu Q, Liu Y, Su S, Li W, Chen C, Wu Y. Anti-tumor activity of paclitaxel through dual-targeting carrier of cyclic RGD and transferrin conjugated hyperbranched copolymer nanoparticles. Biomaterials. 2012;33(5):1627–1639. | ||
Fulda S. Tumor resistance to apoptosis. Int J Cancer. 2009;124(3):511–515. | ||
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. | ||
Desoize B, JarDILlier J-C. Multicellular resistance: a paradigm for clinical resistance? Crit Rev Oncol Hematol. 2000;36(2–3):193–207. | ||
Rajendran L, Knolker HJ, Simons K. Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discov. 2010;9(1):29–42. | ||
Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–464. | ||
Pathania D, Millard M, Neamati N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv Drug Deliv Rev. 2009;61(14):1250–1275. | ||
Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135(7):1161–1163. | ||
Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. | ||
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1):179–192. | ||
Wood AJ, Rowinsky EK, Donehower RC. Paclitaxel (taxol). N Engl J Med. 1995;332(15):1004–1014. | ||
André N, Braguer D, Brasseur G, et al. Paclitaxel induces release of cytochrome c from mitochondria isolated from human neuroblastoma cells. Cancer Res. 2000;60(19):5349–5353. | ||
André N, Carré M, Brasseur G, et al. Paclitaxel targets mitochondria upstream of caspase activation in intact human neuroblastoma cells. FEBS Lett. 2002;532(1):256–260. | ||
Lin B, Kolluri SK, Lin F, et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116(4):527–540. | ||
Ferlini C, Cicchillitti L, Raspaglio G, et al. Paclitaxel directly binds to Bcl-2 and functionally mimics activity of Nur77. Cancer Res. 2009;69(17):6906–6914. | ||
Mironov SL, Ivannikov MV, Johansson M. [Ca2+] i signaling between mitochondria and endoplasmic reticulum in neurons is regulated by microtubules from mitochondrial permeability transition pore to Ca2+-induced Ca2+ release. J Biol Chem. 2005;280(1):715–721. | ||
Kidd JF, Pilkington MF, Schell MJ, et al. Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J Biol Chem. 2002;277(8):6504–6510. | ||
Zhou J, Zhao W-Y, Ma X, et al. The anticancer efficacy of paclitaxel liposomes modified with mitochondrial targeting conjugate in resistant lung cancer. Biomaterials. 2013;34(14):3626–3638. | ||
Jiang L, Li L, He X, et al. Overcoming drug-resistant lung cancer by paclitaxel loaded dual-functional liposomes with mitochondria targeting and pH-response. Biomaterials. 2015;52:126–139. | ||
Yousif LF, Stewart KM, Kelley SO. Targeting mitochondria with organelle-specific compounds: strategies and applications. Chembiochem. 2009;10(12):1939–1950. | ||
Maeda H, Matsumura Y. EPR effect based drug design and clinical outlook for enhanced cancer chemotherapy. Adv Drug Deliv Rev. 2011;63(3):129–130. | ||
Nasongkla N, Shuai X, Ai H, et al. cRGD-functionalized polymer micelles for targeted doxorubicin delivery. Angew Chem Int Ed Engl. 2004;116(46):6483–6487. | ||
Guo Z, He B, Jin H, et al. Targeting efficiency of RGD-modified nanocarriers with different ligand intervals in response to integrin αvβ3 clustering. Biomaterials. 2014;35(23):6106–6117. | ||
Brooks PC, Clark R, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264(5158):569–571. | ||
Hida K, Maishi N, Sakurai Y, Hida Y, Harashima H. Heterogeneity of tumor endothelial cells and drug delivery. Adv Drug Deliv Rev. 2016;99(pt B):140–147. | ||
Law B, Quinti L, Choi Y, Weissleder R, Tung C-H. A mitochondrial targeted fusion peptide exhibits remarkable cytotoxicity. Mol Cancer Ther. 2006;5(8):1944–1949. | ||
Agemy L, Friedmann-Morvinski D, Kotamraju VR, et al. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc Natl Acad Sci U S A. 2011;108(42):17450–17455. | ||
Ellerby HM, Arap W, Ellerby LM, et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5(9):1032–1038. | ||
Biswas S, Dodwadkar NS, Deshpande PP, Torchilin VP. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J Control Release. 2012;159(3):393–402. | ||
Xiao K, Li Y, Luo J, et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32(13):3435–3446. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Yamamoto Y, Nagasaki Y, Kato Y, Sugiyama Y, Kataoka K. Long-circulating poly(ethylene glycol)–poly(d,l-lactide) block copolymer micelles with modulated surface charge. J Control Release. 2001;77(1–2):27–38. | ||
Funahashi Y, Sugi NH, Semba T, et al. Sulfonamide derivative, E7820, is a unique angiogenesis inhibitor suppressing an expression of integrin α2 subunit on endothelium. Cancer Res. 2002;62(21):6116–6123. | ||
Wei T, Liu J, Ma H, et al. Functionalized nanoscale micelles improve drug delivery for cancer therapy in vitro and in vivo. Nano Lett. 2013;13(6):2528–2534. | ||
Oehlke J, Lorenz D, Wiesner B, Bienert M. Studies on the cellular uptake of substance P and lysine-rich, KLA-derived model peptides. J Mol Recognit. 2005;18(1):50–59. | ||
Li L, Song H, Luo K, et al. Gene transfer efficacies of serum-resistant amino acids-based cationic lipids: dependence on headgroup, lipoplex stability and cellular uptake. Int J Pharm. 2011;408(1):183–190. | ||
Kibria G, Hatakeyama H, Ohga N, Hida K, Harashima H. Dual-ligand modification of PEGylated liposomes shows better cell selectivity and efficient gene delivery. J Control Release. 2011;153(2):141–148. | ||
Kluza E, van der Schaft DWJ, Hautvast PAI, et al. Synergistic targeting of αvβ3 integrin and galectin-1 with heteromultivalent paramagnetic liposomes for combined MR imaging and treatment of angiogenesis. Nano Lett. 2010;10(1):52–58. | ||
Josan JS, Vagner J, Handl HL, Sankaranarayanan R, Gillies RJ, Hruby VJ. Solid-phase synthesis of heterobivalent ligands targeted to melanocortin and cholecystokinin receptors. Int J Pept Res Ther. 2008;14(4):293–300. | ||
Mei L, Fu L, Shi K, et al. Increased tumor targeted delivery using a multistage liposome system functionalized with RGD, TAT and cleavable PEG. Int J Pharm. 2014;468(1–2):26–38. | ||
Modery-Pawlowski CL, Sen Gupta A. Heteromultivalent ligand-decoration for actively targeted nanomedicine. Biomaterials. 2014;35(9):2568–2579. | ||
Khalil IA, Kogure K, Futaki S, Harashima H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J Biol Chem. 2006;281(6):3544–3551. | ||
Nam HY, Kwon SM, Chung H, et al. Cellular uptake mechanism and intracellular fate of hydrophobically modified glycol chitosan nanoparticles. J Control Release. 2009;135(3):259–267. | ||
Lucie S, Elisabeth G, Stéphanie F, et al. Clustering and internalization of integrin αvβ3 with a tetrameric RGD-synthetic peptide. Mol Ther. 2009;17(5):837–843. | ||
Lee H, Jeong JH, Park TG. PEG grafted polylysine with fusogenic peptide for gene delivery: high transfection efficiency with low cytotoxicity. J Control Release. 2002;79(1–3):283–291. | ||
Thomas M, Klibanov AM. Non-viral gene therapy: polycation-mediated DNA delivery. Appl Microbiol Biotechnol. 2003;62(1):27–34. | ||
Yamada Y, Akita H, Kamiya H, et al. MITO-Porter: a liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim Biophys Acta. 2008;1778(2):423–432. | ||
Fu B, Long W, Zhang Y, et al. Enhanced antitumor effects of the BRBP1 compound peptide BRBP1-TAT-KLA on human brain metastatic breast cancer. Sci Rep. 2015;5:8029. | ||
Bocci G, Nicolaou K, Kerbel RS. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res. 2002;62(23):6938–6943. | ||
Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4(6):423–436. | ||
Merchan JR, Jayaram DR, Supko JG, He X, Bubley GJ, Sukhatme VP. Increased endothelial uptake of paclitaxel as a potential mechanism for its antiangiogenic effects: potentiation by Cox-2 inhibition. Int J Cancer. 2005;113(3):490–498. | ||
Jiang L, Li L, He B, Pan D, Luo K, Yi Q, Gu Z. Anti-cancer efficacy of paclitaxel loaded in pH triggered liposomes. J Biomed Nanotechnol. 2016;12(1):79–90. | ||
Milane L, Trivedi M, Singh A, Talekar M, Amiji M. Mitochondrial biology, targets, and drug delivery. J Control Release. 2015;207:40–58. | ||
Wang X-X, Li Y-B, Yao H-J, et al. The use of mitochondrial targeting resveratrol liposomes modified with a dequalinium polyethylene glycol-distearoylphosphatidyl ethanolamine conjugate to induce apoptosis in resistant lung cancer cells. Biomaterials. 2011;32(24):5673–5687. | ||
Mo R, Sun Q, Li N, Zhang C. Intracellular delivery and antitumor effects of pH-sensitive liposomes based on zwitterionic oligopeptide lipids. Biomaterials. 2013;34(11):2773–2786. | ||
Hou Z, Zhang Y, Deng K, et al. UV-emitting upconversion-based TiO2 photosensitizing nanoplatform: near-infrared light mediated in vivo photodynamic therapy via mitochondria-involved apoptosis pathway. ACS Nano. 2015;9(3):2584–2599. | ||
Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309. | ||
Murphy MP, Smith RA. Drug delivery to mitochondria: the key to mitochondrial medicine. Adv Drug Deliv Rev. 2000;41(2):235–250. | ||
Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22(8):299–306. | ||
Gasparini G, Longo R, Fanelli M, Teicher BA. Combination of antiangiogenic therapy with other anticancer therapies: results, challenges, and open questions. J Clin Oncol. 2005;23(6):1295–1311. | ||
Luo L-M, Huang Y, Zhao B-X, et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34(4):1102–1114. |
Supplementary materials
Synthesis routes of DSPE-PEG2000-RGD and DSPE-PEG2000-KLA; matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra of DSPE-PEG-RGD and DSPE-PEG-KLA; size distribution of PEG/PTX-Lips, RGD/PTX-Lips and KLA/PTX-Lips; in vitro cytotoxicity of blank RGD-KLA-Lips against NIH 3T3 fibroblast cells and in vitro cytotoxicity of Taxol, RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips at various paclitaxel (PTX) concentrations against human umbilical vascular endothelial cells (HUVECs; Figure S4A) and 4T1 cells (Figure S4B).
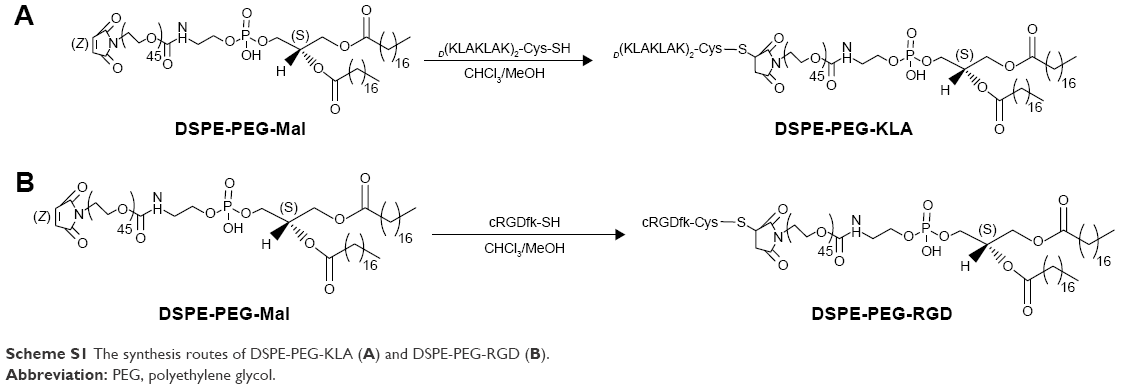 | Scheme S1 The synthesis routes of DSPE-PEG-KLA (A) and DSPE-PEG-RGD (B). |
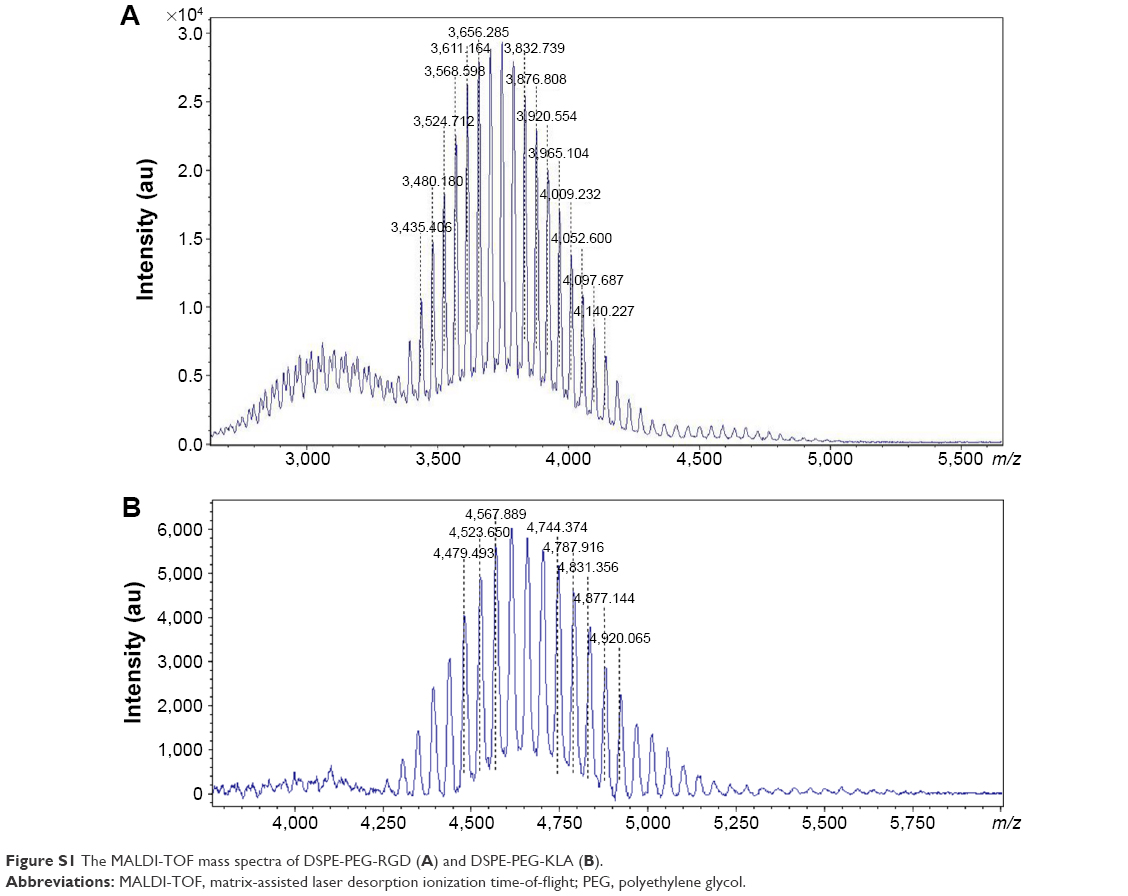 | Figure S1 The MALDI-TOF mass spectra of DSPE-PEG-RGD (A) and DSPE-PEG-KLA (B). |
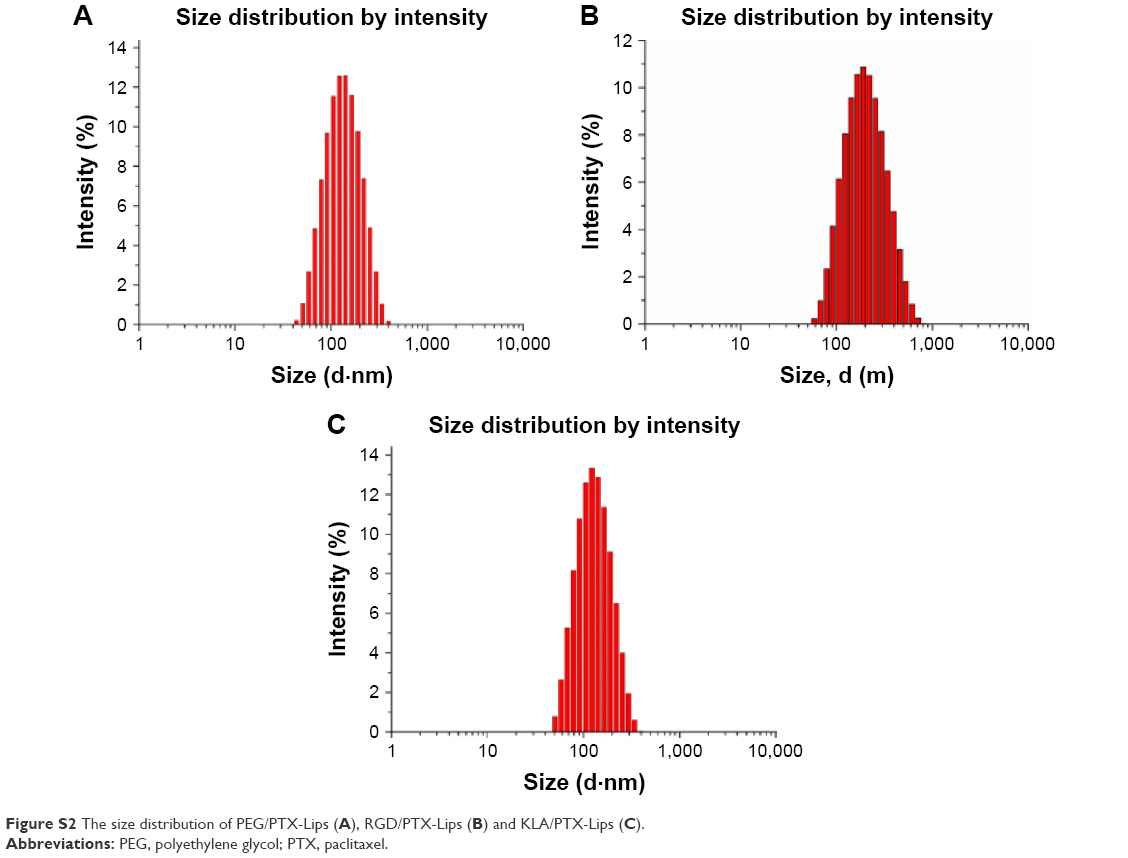 | Figure S2 The size distribution of PEG/PTX-Lips (A), RGD/PTX-Lips (B) and KLA/PTX-Lips (C). |
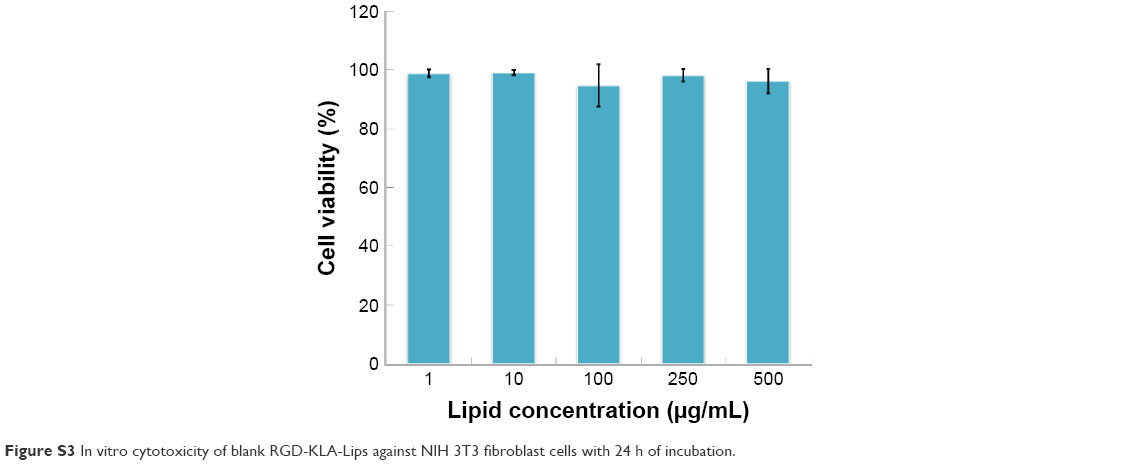 | Figure S3 In vitro cytotoxicity of blank RGD-KLA-Lips against NIH 3T3 fibroblast cells with 24 h of incubation. |
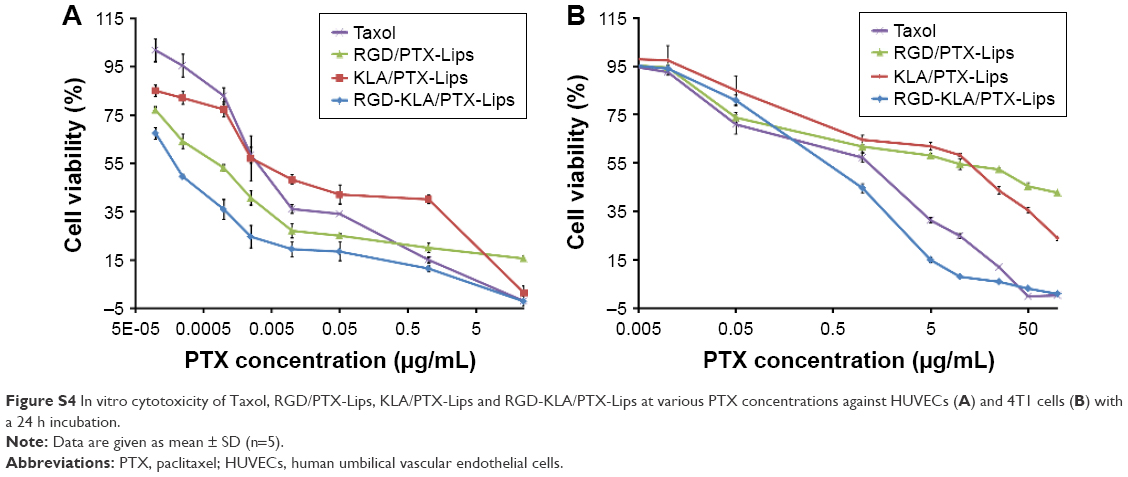 | Figure S4 In vitro cytotoxicity of Taxol, RGD/PTX-Lips, KLA/PTX-Lips and RGD-KLA/PTX-Lips at various PTX concentrations against HUVECs (A) and 4T1 cells (B) with a 24 h incubation. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.