Back to Journals » Medical Devices: Evidence and Research » Volume 18
Engineering a Quality Management System for Academic Research: Navigating Challenges to Comply with the New Medical Device Regulations in Europe
Authors Buist M ![]() , Ortiz-Catalan M
, Ortiz-Catalan M ![]()
Received 8 November 2024
Accepted for publication 24 February 2025
Published 4 March 2025 Volume 2025:18 Pages 137—147
DOI https://doi.org/10.2147/MDER.S448049
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Mirka Buist,1 Max Ortiz-Catalan1– 3
1Center for Bionics and Pain Research, Mölndal, Sweden; 2Prometei Pain Rehabilitation Center, Vinnytsia, Ukraine; 3Center for Complex Endoprosthetics, Osseointegration, and Bionics, Kyiv, Ukraine
Correspondence: Max Ortiz-Catalan, Prometei Pain Rehabilitation Center, Kniaziv Koriatovychiv St. 149, Vinnytsia, 21000, Ukraine, Email [email protected]
Purpose: The new Medical Device Regulations (MDR) in Europe represent unprecedented challenges for researchers in academic environments. Adherence to regulatory frameworks, like the Medical Device Directive (MDD), was mostly relevant for projects nearing commercialization. However, the MDR now extends its reach into the preliminary phases of pre-clinical research, imposing new challenges and necessitating compliance for any clinical investigation involving medical device safety or performance.
Methods: We have systematically engineered and implemented a Quality Management System (QMS) tailored to meet the distinct needs of academic institutions. Our objective was to establish a comprehensive framework that enable our research group to comply with MDR without hindering innovation and discovery. Specifically, we engineered a traditional commercial QMS aligned with ISO13485 to fulfill academic needs. We ensured the QMS focused on requirements relevant to pre-market clinical investigations and considered appropriate roles for an academic setting.
Results: We present an optimized QMS implementation to satisfy the urgent need for research institutions to align with the MDR. Notably, our efforts yielded demonstrable results, culminating in the successful approval of research projects by the Swedish Medical Product Agency (MPA). This achievement stands as a testament to the impact of our work within the regulatory landscape.
Conclusion: Here, we share the insights and challenges we encountered during the implementation of an appropriate and efficient QMS for academic research, which we believe can serve as a guiding example for other academic research groups. By presenting our QMS implementation validated by an MPA clinical investigation approval, we aim to raise awareness about the pivotal importance of MDR compliance among researchers in Europe. Our contribution seeks to provide a roadmap for fellow research groups in navigating the evolving regulatory landscape while sustaining their focus on groundbreaking research and innovation in the field of biomedical engineering.
Keywords: quality management system, QMS, medical device regulations, MDR, ISO 13485
Introduction
Biomedical engineering research is a fertile ground for innovation, where novel ideas and transformative technologies take shape to improve healthcare and patients’ quality of life.
Until recently, under the previous legislative framework guided by Medical Device Directive (MDD), researchers within the European Union (EU) enjoyed a certain degree of freedom in early stages of development. This was particularly where there were no defined intentions of commercialization. However, the introduction of the Medical Device Regulation (MDR) has significantly shifted the regulatory landscape.1 The scope of the MDR is broader than the MDD, now encompassing earlier phases of device development, including pre-clinical translational research, regardless of commercialization intent. This means that any clinical investigation assessing the safety or performance of a medical device must comply with the MDR, even if the device is not yet intended for market release. Additionally, the MDR imposes stricter requirements for documentation, risk management, and post-market surveillance compared to the MDD. These new requirements extend their reach into the earlier phases of pre-clinical translational research in a way that is unfamiliar to academic researchers, and for which academic institutions are ill-equipped to face. Any clinical investigation aimed at assessing the safety or performance of a medical device now falls under the purview of the MDR. Article 1 of the MDR states:
This Regulation lays down rules concerning the placing on the market, making available on the market or putting into service of medical devices for human use and accessories for such devices in the Union. This Regulation also applies to clinical investigations concerning such medical devices and accessories conducted in the Union.
Where “clinical investigation” is defined as “any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device”.
For research groups situated in the EU, this practically entails that an appropriate Quality Management System (QMS) is not a mere option, but a necessity—one mandated by the MDR and enforced by national Medical Product Agencies (MPAs). Unfortunately, the MDR was designed in a way that considers the development of medical devices within companies that are structured for regulatory compliance, as opposed to academic institutions in which such expertise is not only lacking but rarely financed as part of the supporting staff available to researchers. Moreover, regulatory compliance consultants are trained on industrial environments, which often renders their advice unsuitable for academic researchers. Consequently, this highlights the lack of readily available internal or external support for researchers within the EU to comply with the requirements of the MDR.
Existing QMS templates, much like the consultants, are usually focused on manufacturing and commercialization processes, which are not always relevant to academic research groups. The limitations of these templates include an overemphasis on post-market activities and a lack of flexibility to accommodate the diverse and dynamic nature of academic research projects. Therefore, there is a clear need for tailored QMS solutions that address the specific challenges faced by academic researchers, ensuring compliance without stifling innovation.
Unsurprisingly, academic institutions in the EU now face distinct compliance challenges with the MDR. Unlike commercial enterprises, academic institutions often lack dedicated regulatory affairs departments and the necessary expertise. Securing funding for regulatory compliance activities is a significant hurdle, as research grants rarely cover the administrative overheads required for QMS implementations and maintenance. Additionally, the transient nature of academic staff, such as graduate students and postdoctoral researchers, can hinder the consistency and continuity of QMS practices. Balancing the primary mission of education and the advancement of knowledge with stringent regulatory requirements is challenging for academic institutions. Furthermore, the need for a tailored QMS that fits the unique nature of academic research necessitates significant adaptation of traditional QMS templates.
In this article, we explore the critical intersection of biomedical engineering research and regulatory compliance within an academic environment. We share the experiences and insights we gained from implementing a QMS tailored to meet the unique needs of an academic research group. As we embarked on our journey to align with the MDR, we realized a significant gap in awareness among research institutions about the profound implications of these new regulations. It became evident that our experiences in implementing an appropriate QMS could serve as a guiding example for fellow research groups facing similar challenges. Many researchers are yet to realize that the MDR has a significant impact on their work, and our article aims to shed light on this critical issue. By sharing our insights, challenges, and achievements, we hope to provide a roadmap for other research groups, demonstrating how they too can adapt to the changing regulatory landscape. In doing so, we contribute to raising awareness about the importance of compliance with the MDR to be able to continue conducting groundbreaking research and innovation.
Methods
Engineering Steps
Our approach to engineering a QMS was marked by a structured and deliberate process. Recognizing the need for a tailored system that aligns with the unique context of academic institutions, we embarked on a journey that involved several key engineering steps. Figure 1 shows a flowchart with an overview of the steps in the QMS engineering process.
 |
Figure 1 Flowchart of the QMS engineering process. The process started with the evaluation of the templates based on the scope of our research, using national and international standards and requirements. Based on that, the templates were optimized and restructured to fit our specific setup. |
Template Evaluation
The initial step involved analysing templates and procedures of a “traditional” QMS aligned with ISO 13485.2 A traditional QMS is focused on the manufacturing processes and commercialization of a product,3 as this is usually tailored for commercial entities. It became evident that such templates and procedures required substantial adaptation to suit our specific situation as academic research group. This step served as the cornerstone, setting the stage for subsequent adaptations that would ensure a seamless integration of regulatory compliance within our academic framework. After analysing a stereotypical industrial QMS, we proceeded to identify which portions and sections would be required for our specific setting.
Requirements Identification
To effectively adapt a traditional QMS, we delved into a comprehensive understanding of the Medical Device Regulations (MDR). Through extensive reading and relevant courses, we strategically segmented the MDR, focusing on parts directly pertinent to pre-market clinical development and investigations. Identifying elements that were superfluous for our research objectives and isolating those that formed the essential foundation for compliance. We identified that one of the main aims of the MDR is that every medical product complies with the general safety and performance requirements (GDPR) of Annex I.4 Showing compliance to these requirements is generally achieved by referring to the section of the technical documentation that covers the listed requirement.
When it comes to Clinical Investigations, the MDR has three different pathways, Article 62, 74(1), and 82.4 In our case, article 82 is applicable as we do not intend to CE mark our devices, nor have any device on the market. Article 82 is a subset of article 62 which includes all requirements for the Clinical Investigation on the pathway to obtain a CE mark. The requirements for clinical investigations under article 82 are listed in Table 1. Besides that, article 82(2) states that member states can define additional requirements for this type of regulations. Meaning that these requirements might vary across countries within the EU.
 |
Table 1 Summary of Article 82 of the MDR |
Annex XV chapter II of the MDR4 specifies which documents shall be submitted when applying for a clinical investigation. This includes the items listed in Table 2. This is for a large part in line with the documents that the Swedish MPA requests upon submission. It is important to note that the Swedish MPA has its own interpretation of the MDR, which might differ from country to country. The methods we describe in this article are in line with the Swedish interpretation of the MDR, it might thus be possible that research institutes in other countries are required to do certain things in a slightly different manner, see Figure 1 block “National Requirements (MPA)”.
 |
Table 2 Requirements for Applying to a Clinical Investigation According to the MDR and Swedish MPA |
The MDR also requires that an “appropriate” QMS shall be in place. This is amongst other things to ensure that the required technical documentations are kept and that the production and development processes follow these procedures. The MDR does not specify when a QMS is appropriate, as this varies widely depending on the aim and setup of the organization (company or institute). It is up to the organization to adjust the QMS in such a way that it becomes appropriate.
Reduction and Restructuring of Procedures
Building on the information gathered in the previous step, we concentrated efforts on simplifying and adjusting the QMS to our needs. Elements associated with commercialization, such as manufacturer responsibilities and post-market clinical follow-up, were removed. This strategic simplification was not merely a reduction but a purposeful alignment of our QMS with the early stages of device development, in line with our research. By eliminating unnecessary complexities, we ensured that the QMS seamlessly complemented our primary focus – testing high-fidelity prototypes in clinical settings, rather than transitioning to production. Table 3 shows which Standard Operating Procedures (SOPs), we included in our QMS. It also shows a list of SOPs that are commonly found in a QMS for commercial companies, which we decided not to include in our QMS. It is important to note that this list is not exhaustive. The specific SOPs required for a QMS can vary significantly depending on the unique needs, activities, and regulatory requirements of each research group. Besides merely selecting the relevant SOPs, we also reduced the content of the relevant SOPs to match our needs. One of the SOPs that was adjusted was the Product Development Process. Figure 2 illustrates which parts of the Product Development Process were kept, and which parts were eliminated.
 |
Table 3 Chapters and SOPs That are Included and Excluded in Our QMS |
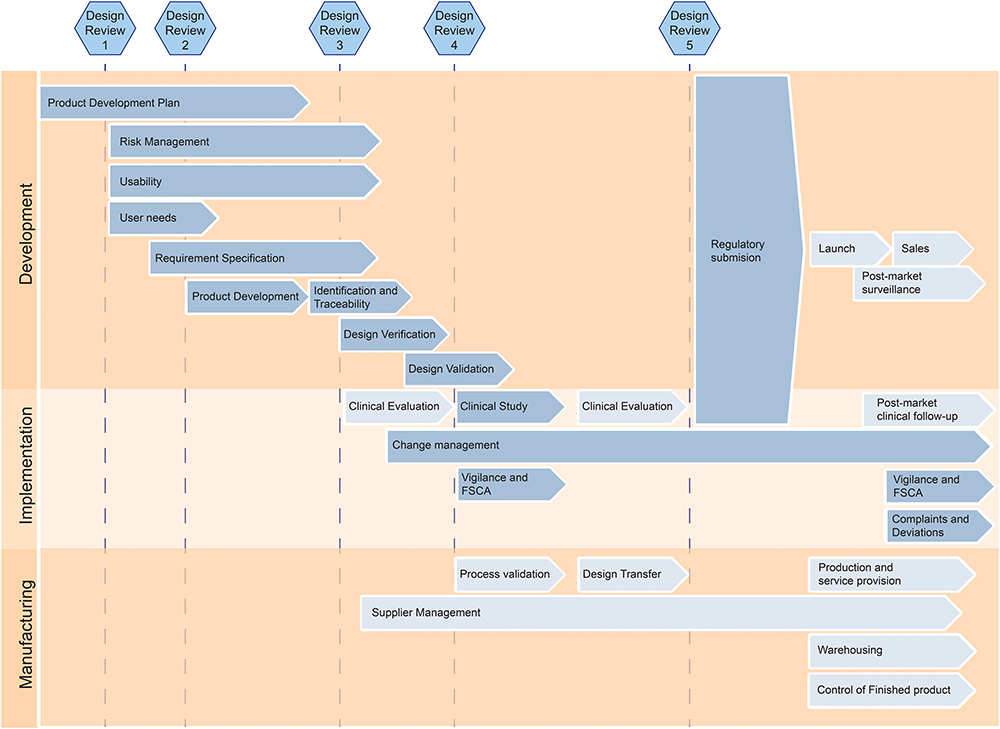 |
Figure 2 Modified Product Development Process. It highlights all relevant procedures for the development of a medical device. The arrows with a dark background indicate procedures that we included in our QMS, while the arrows with a light background indicate procedures that we deemed irrelevant for our specific academic research group. |
After the reduction of abundant elements, we also focused on optimizing the SOPs to our way of working. In Figure 2 it is apparent that the last Design Review has been removed, and in return one additional Design Review has been added in the beginning, right after establishing the product development plan. The latter works for us as a gate step for students who are working on their own project so they can start with risk management and requirement specification after creating an appropriate plan.
Role Reshaping and Redefinition
Another change from an industrial QMS was to adopt the roles of the people normally present in companies but not in academic institutions. We identified and reshaped roles such as CEO, R&D Manager, Sales and Marketing Manager, etc, to ensure they aligned with our research group’s structure. Some of these roles could be omitted as they were beyond the reach of our research projects, such as sales and marketing. Responsibilities within the remaining roles were redistributed to align more accurately with roles available in our research group at the time. In our case, these roles were Director (PI), Quality Manager, Research Coordinator, Engineering Manager, Platform Manager, and Project Supervisor.
File Structure Optimization
A functional filing system is integral to QMS effectiveness. In this step, we established a dual-part electronic file structure comprising procedures and templates on one side, and records (filled-in templates) on the other. Within our electronic QMS system (eQMS), these parts were organized into two main folders, “1. Procedures & Templates” and “2. Records”, with sub-folders corresponding to each QMS chapter. Leveraging the functionalities of the eQMS, we implemented automatic record creation from templates, streamlining the documentation process.
These engineering steps collectively ensured the adaptation of an industrial QMS to our specific research needs, laying the foundation for a system that facilitates compliance without unnecessary complexities and aligns seamlessly with our research group’s goals.
Resource and Time Allocation
Our research group consisted of approximately 20–25 members, including postdoctoral researchers, graduate and undergraduate students, and supporting staff. The group was involved in multiple projects focused on the development and pre-clinical testing of potential medical devices, ranging in complexity and classification from Class I to Class III under the MDR.
The extensive process of developing the QMS spanned various resource allocations and time commitments. A significant part involved one team member dedicating approximately 50% of full-time employment (FTE) for over a year to research and discern the QMS requirements. This effort was complemented by collaboration with a consultant (0.2 FTE) over six months to gather essential templates from the “Standard 13485” QMS database, followed by another two months spent modifying these templates to suit our research group. Subsequent efforts involved collaborative validation and reviewing of the finalized documents and templates. The QMS was eventually implemented and digitized using Microsoft SharePoint, requiring an additional one to two months of dedicated effort.
With the implementation of the QMS in Microsoft SharePoint, the first version of the QMS was finalized. Yet, ongoing maintenance and adherence to the system was, and will be, another considerable aspect of the implementation. With the size of our given research group, the support of at least one employee was necessary to undertake the Quality Assurance (QA) role at a minimum of 0.5 FTE.
The regulatory consultant that helped us with the initial phases of this project was selected after extensive meetings with nearly all medical device QMS consultants in Sweden, and even some from abroad. We explained our situation as an academic research institute, emphasizing that we were not focused on commercialization or manufacturing, but still required a QMS. Most consultants struggled to understand the differences between our needs and those of commercial institutions. However, we chose the one consultant who seemed to grasp our unique situation, despite not having prior experience with academic institutions.
Platform and Document Format Selection
All procedures and templates were written in Microsoft Word, and where appropriate Microsoft Excel. The finalized procedures were converted into PDFs to protect them against change and to allow for version control. This is important because one of the requirements from the MDR is that the files can be versioned and that changes are tracked.
The platform in which the documents are stored should be decided based on the research group’s current infrastructures and data management systems. One option is to use a paper-based filing system, or in a digital or online file manager. The highest chance of employees accepting the system, and using it in an efficient way, is when the system closely aligns with their other methods for documenting. When opting for an online file managing system, one can decide to use a one of the eQMS software options available. However, we noticed that these are often focused on commercial customers and unsuitable for research type projects. Therefore, we decided to add another step of engineering to the project, by designing an eQMS system in SharePoint.5 The integration of Microsoft Workflows (previously Power Automate) and SharePoint gave us the possibility to give the software exactly the functions that were necessary for our implementation. We implemented three automated functions in SharePoint, one to create a record from a template with a simple button click, one for a publishing request for documents in the QMS that need to be signed by the QA, and one to help us execute the onboarding procedure.
Results
Approval From Swedish Medical Product Agency
Our efforts in implementing a QMS culminated with the successful approval by the Swedish MPA for a clinical investigation with a novel medical device developed entirely within our research group. In accordance with national regulations, approval for any clinical investigation involving a medical device is a prerequisite. While future processes are anticipated to be handled through the European Database on Medical Devices (EUDAMED), the current protocol involves approval by the national authority to ensure alignment with the MDR. Our proactive approach included applying for approval of a clinical investigation at the MPA, a process necessitating submission of detailed information as outlined in Table 3.
The MPA, upon receiving our submission, raised a couple of questions, one of which focused on the presence and adequacy of our QMS. Although not explicitly mandated in the submission requirements, the MPA underscored the importance of an “appropriate” QMS. In response, we effectively demonstrated the existence and relevance of our QMS by referencing the documents within our Product Documentation. This validation through the approval process stands as a concrete testament to the efficacy of our QMS in meeting regulatory expectations.
Compliance with ISO Standards
Furthermore, our commitment to compliance extended beyond the MDR, encompassing adherence to ISO standards essential in the realm of medical device development. The QMS emerged as a valuable tool aiding conformity to additional standards such as ISO 14155,6 ISO 14971,7 and IEC 62366-1.8 Our SOPs played a pivotal role in ensuring alignment with these standards. For instance, the Risk Management SOP was written in alignment with ISO 14971, ensuring that our Risk Management Procedure follows the ISO standard. This exemplifies how our QMS became an instrumental guide in navigating various ISO standards.
Ethical Compliance
The implementation of the QMS has also helped with adherence to ethical guidelines, particularly when dealing with human subjects in clinical trials. The structured documentation and processes within the QMS help with obtaining appropriate ethical approvals. In Sweden, application to the ethical review board is now done through the Medical Product Agency (MPA) with the aim to ensure coherence and compliance. While this might vary by country, the documents generated through the QMS will undoubtedly be valuable for compiling the necessary documentation for the ethical approval process.
Functional Improvements
In addition to these regulatory milestones, our implementation of the QMS brought forth improvements in the efficiency of our work. While we present the implementation of the QMS as a necessity, forced upon us by the MDR, we embraced this implementation as an opportunity to structure our internal procedures more efficiently. The structured documentation has helped us to transfer and maintain knowledge within our research group, something that we have struggled with before as it is common in academic research groups where there is a high turnover (ie, students come and go). It has also helped us to have an efficient and consistent workflow, as well as an organized database of all project files. These enhancements underscore the tangible benefits realized through the deliberate engineering and adaptation of our QMS.
Discussion
Setting up a QMS for medical device development in a research environment faces several challenges. One of these challenges is the differences in the way of working between for-profit organizations with a focus on commercialization, and research institutes whose currency is scientific publications based on technological development and clinical validations. The roles of the people within these organizations are different, resulting in that the roles described in traditional QMS have to be modified for use in research institutes. Besides that, research groups are often on a smaller scale than for-profit organizations. This, together with the differences in organization structure, often results in less available resources for matters such as quality assurance and regulatory compliance.
Another challenge is to make the QMS adequate. Meaning that it shall be adapted in such a way that contains all the required procedures but without being a limiting factor in the way that the organization is working. Any abundant documents and procedures shall be disregarded to avoid an overly complex QMS, without cutting away anything that is required. With that, comes the challenge on deciding whether or not a certain part is required, which untimely depends on the scope of the organization. Eventually, there is no one-size-fits-all solution. A QMS shall be adapted and adjusted to specific needs.
Even though there is no one-size-fits-all solution, we believe that other research groups can still benefit from the information provided in this manuscript. Anyone who wants to pursue a clinical trial with a medical device within the EU will have to implement a QMS. And although all research groups are different, they are at least similar enough to take this as an example. One thing to take example from is to get an estimate of the time and resources required. In our case, one team member dedicated approximately 0.5 FTE for over a year to research and discern the QMS requirements, and another 2 months for the implementation and digitization of the QMS. A consultant spent 0.2 FTE over six months to gather essential templates, followed by another two months spent modifying these templates to suit our research group. Ongoing maintenance and adherence to the system require at least one employee in a Quality Assurance (QA) role at a minimum of 0.5 FTE. Financial resources were allocated primarily for consultant fees, as we used software tools already accessible to us through our educational institution.
The inclusion and exclusion of SOPs depend on the activities of each individual research group. Any relevant process should be documented. Our suggested SOPs can serve as a foundation, as most research groups likely will not need SOPs focused on post-market surveillance and other commercialization activities. The size of the QMS largely depends on the focus of the institute’s work. If an institute focuses on a specific aspect of development, only a few SOPs are needed. Conversely, if a research group or institute handles the entire process from initial prototype to commercialization, many more SOPs will be required.
Throughout our exploration of the MDR, we observed a general lack of awareness among research groups that develop and test medical devices. The scarcity of references in this manuscript reflects our limited findings on relevant articles. In our discussions with researchers from various groups, we noted low awareness of the MDR’s applicability. To our knowledge, only one academic research group in the EU currently maintains an ISO 13485 certified QMS. These observations underscore a significant knowledge gap on this subject within our peer community. Worthy of notice is that readily available consultants are lacking because of recency of the new regulations. We interviewed most regulatory consulting firms in Sweden, and several others abroad, and found the recurrent problem in that their advice was not suitable for an academic research group. Moreover, they seem often incapable to understand the difference between the familiar structure of a company versus an academic research group. While we do not have specific requirements to recommend for selecting an appropriate quality and regulatory consultant, we suggest that other researchers also engage with various consultants in their country of operation to find one who understands their unique situation. We warn caution to our fellow researchers on embarking in overly complicated QMS systems designed for large companies instead of tailored process fit to purpose.
In addition to meeting regulatory requirements, implementing a QMS is also beneficial to adhere to ethical standards. Our QMS ensures compliance with ethical guidelines by incorporating structured processes for obtaining informed consent, maintaining participant confidentiality, and ensuring participant safety and well-being throughout the study. This integration streamlines the approval process and ensures that all ethical considerations are adequately addressed, safeguarding the rights and well-being of research participants. A limitation of this study is the absence of long-term data on the QMS’s impact on research productivity, quality, and broad regulatory compliance. Despite this, publishing the findings from the initial implementation is essential to support other academic research groups in establishing their own QMS, given the challenges EU academic groups now face. In the meantime, it is important to stay informed about regulatory updates and engaging with regulatory bodies for maintaining compliance and ensuring the continued integrity and quality of research. Given the possibility that the regulations continue to evolve, and new and potentially more stringent requirements can potentially be introduced in the future. Which might require academic institutions to align elements like documentation, risk management, and post-market surveillance even further with industry standards. Eventually, we believe that making the knowledge on a QMS for medical device investigations available to fellow academic researchers should have a great impact on the quality and safety of clinical investigations within the medical device development in the EU. While the MDR can be seen as a major hurdle within the EU, it also represents an opportunity to increase the quality of our research. Implementing an appropriate QMS will be of great importance to transform this obstacle into a valuable opportunity.
Global Context
While the primary focus of this study is on the EU MDR, similar regulatory trends are emerging in other major regions, providing a broader context to the evolving regulatory landscape. In the United States, the Food and Drug Administration (FDA) has been tightening its regulations on medical devices,9 with updates that include more stringent requirements for premarket submissions and ongoing monitoring, particularly concerning cybersecurity.10 The FDA’s Quality Management System Regulation (QMSR) aims to harmonize with international standards, such as ISO 13485, to promote consistency in device regulation.9
In China, the National Medical Products Administration (NMPA) has implemented comprehensive provisions for medical device registration and filing.11 These regulations emphasize safety, effectiveness, and quality management, with a structured approach to the classification and evaluation of medical devices.12 The NMPA’s efforts align with global trends towards more rigorous oversight and standardization of medical device regulations.
By examining these regulatory frameworks, we can better understand the broader shifts towards increased scrutiny and standardization in the medical device industry. This global perspective underscores the importance of adapting to evolving regulatory environments to ensure the safety and efficacy of medical devices.
Acknowledgment
The authors would like to thank Paul Čvančara at the University of Freiburg, Germany, for the informative discussions regarding the implementation of their QMS.
Funding
This work was financed by the Promobilia Foundation (19500).
Disclosure
The authors declare that they have no competing interests.
References
1. Vollebregt E. Enriched MDR and IVDR. 2021.
2. International Organization for Standardization. ISO 13485: 2016 medical devices - quality management systems - requirements for regulatory purposes. 2016.
3. Martins EG, Pinheiro De Lima E, Gouvea da Costa SE. Developing a quality management system implementation process for a medical device manufacturer. J Manuf Technol Manag. 2015;26(7):955–979. doi:10.1108/JMTM-12-2012-0119
4. European Parliament and Council of the European Union. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. 2017.
5. Jones M. A QMS solution in SharePoint anyone can build—a step by step guide. LinkedIn Articles. 2016. Available from: https://www.linkedin.com/pulse/qms-solution-sharepoint-anyone-can-build-step-guide-melissa-jones/.
6. International Organization for Standardization. ISO 14155: 2020 clinical investigation of medical devices for human subjects – good clinical practice. 2020.
7. International Organization for Standardization. ISO 14971: 2019 medical devices - application of risk management to medical devices. 2019.
8. International Electrotechnical Commission. IEC 62366-1: 2015 medical devices – part 1: application of usability engineering to medical devices. 2015.
9. U.S. Department of Health and Human Services; Food and Drug Administration. Medical devices; quality system regulation amendments 2024-01709 (89 FR 7496). Fed Regist. 2024;82(22):7496–7525.
10. U.S. Department of Health and Human Services; Food and Drug Administration. Cybersecurity in medical devices: quality system considerations and content of premarket submissions guidance for industry and food and drug administration staff. 2023. Available from: https://www.fda.gov/media/119933/download.
11. National Medical Products Administration (NMPA). Provisions for medical device registration and filing. 2021. Available from: https://english.nmpa.gov.cn/2024-06/05/c_993242.htm.
12. National Medical Products Administration (NMPA). Rules for classification of medical devices (Decree No.15 of China Food and Drug Administration). 2015. Available from: https://english.nmpa.gov.cn/2019-10/11/c_415411.htm.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.