Back to Journals » International Journal of Nanomedicine » Volume 13
Engineered exosome-mediated delivery of functionally active miR-26a and its enhanced suppression effect in HepG2 cells
Authors Liang G ![]() , Kan S, Zhu Y, Feng S, Feng W
, Kan S, Zhu Y, Feng S, Feng W ![]() , Gao S
, Gao S
Received 18 October 2017
Accepted for publication 7 December 2017
Published 30 January 2018 Volume 2018:13 Pages 585—599
DOI https://doi.org/10.2147/IJN.S154458
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Linlin Sun
Gaofeng Liang,1,2,* Shu Kan,2,* Yanliang Zhu,3 Shuying Feng,1 Wenpo Feng,1 Shegan Gao1,4
1Medical College, Henan University of Science and Technology, Luoyang, China; 2Department of Biomedical Engineering, University of California Berkeley, California, CA, USA; 3State Key laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, 4Henan Key Laboratory of Cancer Epigenetics, The First Affiliated Hospital of Henan University of Science and Technology, Luoyang, China
*These authors contributed equally to this work
Introduction: Exosomes are closed-membrane nanovesicles that are secreted by a variety of cells and exist in most body fluids. Recent studies have demonstrated the potential of exosomes as natural vehicles that target delivery of functional small RNA and chemotherapeutics to diseased cells.
Methods: In this study, we introduce a new approach for the targeted delivery of exosomes loaded with functional miR-26a to scavenger receptor class B type 1-expressing liver cancer cells. The tumor cell-targeting function of these engineered exosomes was introduced by expressing in 293T cell hosts, the gene fusion between the transmembrane protein of CD63 and a sequence from Apo-A1. The exosomes harvested from these 293T cells were loaded with miR-26a via electroporation.
Results: The engineered exosomes were shown to bind selectively to HepG2 cells via the scavenger receptor class B type 1–Apo-A1 complex and then internalized by receptor-mediated endocytosis. The release of miR-26a in exosome-treated HepG2 cells upregulated miR-26a expression and decreased the rates of cell migration and proliferation. We also presented evidence that suggest cell growth was inhibited by miR-26a-mediated decreases in the amounts of key proteins that regulate the cell cycle.
Conclusion: Our gene delivery strategy can be adapted to treat a broad spectrum of cancers by expressing proteins on the surface of miRNA-loaded exosomes that recognize specific biomarkers on the tumor cell.
Keywords: exosome, gene delivery, miR-26a, HepG2 cells
Erratum for this paper has been published.
Introduction
A major challenge in gene therapy is the development of nontoxic, molecular transport vehicles that efficiently deliver functional copies of a therapeutic gene to target cells. While viral vectors are frequently used for this purpose, their application to treat human cancers is limited by an inherent toxicity, potential infectivity, and immunogenicity.1 Alternative nonviral vector platforms have also been developed for gene targeting and delivery, examples of which include synthetic polymers, micelles, and nanoparticles.2 Although promising in animal models of human disease, these vehicles are limited by the same problems encountered by viral-derived vehicles.3 These limitations could be overcome by using host-derived, “living” gene delivery vehicles, examples of which may include engineered exosomes,4 blood platelets,5–7 and red blood cells.8 Exosomes are natural lipid membrane-enclosed vesicles that have a broad range of diameters (30–150 nm) and are synthesized and released by a variety of cells types.9 The natural internal cargo of exosomes includes specific mRNAs, miRNAs, and proteins that can be transported to remote target cells in many cases. This targeting is achieved by specific interactions of proteins on the exosomal membrane with receptor molecules on the target cell.10 We and others have also shown that the molecular cargo transported and delivered by exosomes to target cells can influence pathological and physiological processes in the target cell or tissue, including immune responses, blood coagulation, tumor growth, and tissue repair.11–15 Furthermore, the presence of specific genetic information within exosomes derived from tumor cells offers opportunities to develop simple liquid biopsy-based approaches for cancer detection or to monitor the effectiveness of a cancer treatment.16,17
Exosomes exhibit unique features that could be exploited to enhance their performance as personalized vehicles for targeted delivery of therapeutics to diseased cells and tissue. First, since they are produced in a patient, they are recognized as “self”, which increases their stability in serum. Moreover, their longer circulation compared with artificial nanovehicles will result in a significant increase in the chance of this molecule encountering with target cells, even deep-seated tumor cells.5 Also significant in this regard are studies that show exosomes traverse intact biological barriers, including the blood–brain barrier (BBB), and have an ability to deliver functional RNA and small molecule drugs to target cells.4,18–20 The intrinsic cell-targeting property of exosomes can be further enhanced by using genetic engineering techniques to introduce specific proteins to their surface, including ligands for receptors (Apo-A1) or antibodies directed against tumor biomarkers.21,22 Alvarez-Erviti et al, for example, engineered exosomes produced by dendritic cells to express the neuron-specific rabies viral glycoprotein peptide, which binds to the acetylcholine receptor expressed on neuronal cells. These exosomes were shown to cross the BBB.4 This class of exosomes is the current preferred vehicle for the delivery of small interfering RNA (siRNA) to brain cells.
Small RNAs, including siRNA and miRNA, can be used as molecular therapeutics to treat a variety of diseases, including cancer, cardiac disease, and neurodegenerative disorders.23 Most therapeutic applications of miRNA require packaging the nucleic acid in a vector or nanovehicle, example of which include adenovirus (AV), adeno-associated virus (AAV), liposomes, polycationic polymers, and organic/inorganic nanoparticles.24–29 Kota et al, for example, showed that miR-26a, a downregulated miRNA in hepatocellular carcinoma, could be delivered to liver cancer cells using AAV where it induced tumor-specific apoptosis in a mouse model of liver cancer.30 Moreover, our group developed a poly(lactic-co-glycolic acid)/polyetherimide/hyaluronan (PLGA/PEI/HA)-based vehicle as a carrier of miR-145 and further showed that PLGA/PEI/HA/miRNA complexes were delivered efficiently to tumor cells within colon carcinoma xenografts in mice where they exhibited significant antitumor effects.31
Inspired by the findings of these earlier studies, we have developed a new approach to repurpose exosomes as living vehicles that can deliver nucleic acid therapeutics to target cells. In particular, Apo-A1, the main component of high-density lipoprotein (HDL), was expressed as a fusion with the transmembrane protein CD63 to facilitate its presentation on the surface of the exosome. Apo-A1 is a known target of the scavenger receptor class B type 1 (SR-B1) receptor, which is known to mediate phospholipid transfer between HDL and the cell membrane.32 SR-B1 is highly expressed on the surface of many types of liver cancer cells, including HepG2, hepatic carcinoma, melanoma, and prostate cancer.33–35 Apo-A1-CD63 presenting exosomes produced by 293T cells were loaded with miR-26a by electroporation and shown to be internalized by HepG2 cells via SR-B1 receptor-mediated endocytosis. The exosome-mediated delivery of miR-26a to HepG2 cells was further shown to upregulate miR-26a expression, where it downregulated key cell cycle proteins that arrested the cell cycle and inhibited cell proliferation. Therefore, significance of this study is that it introduces a personalized approach to cancer treatment by engineering a natural vehicle generated from cells isolated from the patient to deliver a therapeutic miRNA to specific tumor cells.
Patients and methods
Cell culture and reagents
The human embryonic kidney 293T (HEK293T) and human liver cancer HepG2 cells were purchased from American type culture collection (ATCC, Maryland, MD, USA). Both cell lines were cultured in high glucose Dulbecco’s Modified Eagle’s Medium, supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Carlsbad, CA, USA) and penicillin (100 U/mL)/streptomycin (100 mg/mL), at 37°C in 5% CO2. β-actin, CD63, CD9, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), CCND2, CCNE2, cell division protein kinase 6 (CDK6), and anti-IgG antibodies were purchased from Abcam (Cambridge, MA, USA), and an Apo-A1 and anti-IgG antibody were purchased from R&D (Minneapolis, MN, USA). Cy5-labeled miR-26a was synthesized by Integrated DNA Technologies (Coralville, IA, USA). Other reagents were purchased from Thermo Fisher Scientific. The FBS was microvesicle depleted by 2 h ultracentrifugation at 100,000× g, followed by filtrating with a 0.22 μm steritop filter (Millipore, Billerica, MA, USA).
Construction of plasmids and preparation of exosome
The pEGFP-N2 expression system (Clontech, Mountain View, CA, USA) was used for the vector expression construct as recommended by the manufacturer. First, the sequence encoding CD63 was amplified from 293T cells using polymerase chain reaction (PCR) primers (Forward: 5′-CCCAGATCTGCCACCATGGCGGTGGAAGGAGGAATGAAATG-3′, Reverse: 5′-CCGGAATTCCATCACCTCGTAGCCACTTCTGATAC-3′). The cDNA oligos were annealed and cloned to the Bgl II/EcoR I-digested pEGFP-N2 vectors. The sequence encoding the Apo-A1-binding peptide was obtained from GE Dharmacon (Lafayette, CO, USA) and prepared with PCR primers (Forward: 5′-CTACGGGGTACCATGAAAGCTGCGGTGCTGA-3′, Reverse 5′-TGCACGGGATCCTCACTGGGTGTTGAGCTTCTTAGTG-3′) with restriction enzymatic sites (Kpn I/BamH I) directly fused into pEGFP-N2 vector. The full construct was introduced into a pEGFP-N2 vector with the Bgl II and BamH I restriction sites being located downstream of the cytomegalovirus (CMV) promoter. Each step of the vector construction was confirmed and validated by DNA sequencing and PCR and is shown collectively in Figure S1.
The Apo-A1 targeting ligand was fused to CD63 as previously described.36 The HEK 293T cells were seeded in 25-cm2 flasks (NUC; Thermo Fisher Scientific, Waltham, MA, USA). Cells at 70%–80% confluence were transfected with the Apo-CD63 plasmid using Lipofectamine 3000 (Invitrogen) as recommended by the manufacturer. The 293T cells were analyzed for green fluorescent protein (GFP) expression 48 h after the transfection. GFP-expressing 293T cells were counted and isolated using fluorescence-activated cell sorting (BD Influx, Piscatway, NJ, USA). The sorted cells were harvested and cultured in 175-cm2 flasks for 96 h. The exosomes were subsequently purified from the culture medium using an exosome isolation kit (exoEasy™; Qiagen, Hilden, Germany) as recommended by the manufacturer. The resulting pellet containing an enriched population of GFP-labeled exosomes was resuspended in 400 μL of saline.
Characterization of exosome
Exosome samples were imaged by transmission electron microscope (TEM) as follows: exosome pellets were rinsed three times with PBS for 10 min, and exosomes in the final pellet were fixed after resuspension in a droplet of 2.5% glutaraldehyde in PBS buffer at 4°C. The sample was dehydrated using increasing concentrations of alcohol. The samples were then observed under a TEM (JEM-2100; JEOL, Tokyo, Japan) at a voltage of 80 kV.
The particle size of exosome sample was measured using a Zetasizer Nano S (Malvern Instruments, Malvern, UK). The surface charge was measured by determining the zeta potential in PBS buffer.
Exosome labeling and loading
Exosomes were visualized after staining with the fluorescent probe3, 3′-dioctadecyloxacarbocyanine perchlorate (DiO), which was purchased from Invitrogen. Purified exosomes were incubated with 5 μM DiO for 15 min at 37°C and ultracentrifuged at 120,000× g for 90 min to remove unbound probe. After two wash–centrifugation cycles (PBS followed by 120,000× g centrifugation), the labeled exosomes were resuspended in PBS and used in cell studies shortly thereafter.
Exosomes with a total protein concentration of 10 μg/mL (measured by the Nanodrop instrument) were mixed with 400 nM of Cy5-labeled miR-26a in 1 mL PBS. The mixture was electrophoresed under the following condition: 400 V, 50 μF, three cycles by 30 ms pulse/2 s pause. After the loading of miR-26a, the exosome samples were diluted 10× with PBS and centrifuged at 110,000× g for 70 min to remove unbounded miR-26a. The incorporation of miR-26a into exosomes was determined by quantitative reverse transcription PCR (RT-PCR). RNA was isolated from pellets with TRIzol Reagent, as recommended by the manufacturer.
Exosome uptake
The efficiency of Apo-A1-modified exosome targeting to HepG2 cells was quantified as follows. HepG2 cells (3×105) were seeded in a 3.5-cm glass-bottom dish and incubated until they reached ~70% confluency. The cells were then washed with PBS and incubated with cell culture medium containing 108 particles/mL of exosomes labeled with DiO. The fluorescence signal of DiO in HepG2 cells was recorded in a confocal laser scanning fluorescence microscope (CLSM), and images were processed with ZEN software (CLSM; Zeiss LSM710, Oberkochen, Germany).
HepG2 cells were incubated with miR-26a-loaded exosomes for 1, 3, 6, 12, and 24 h. At each time point, the supernatant was removed and the wells were washed twice with PBS. After the final PBS wash, the preparation was fixed using 4% paraformaldehyde and incubated with the DNA stain (5 μg/mL Hoechst 33342) for 20 min. Fluorescence images were recorded with CLSM. The configurations of the confocal fluorescence filters were as follows: for DAPI (4′,6-diamidino-2-phenylindole) imaging: excitation wavelength, Haupt Farb Teiler (HFT), 405/488 nm and beam splitter pinhole diameter, 154 mm; for DiO imaging: HFT, 488/543 nm and pinhole diameter 184 mm; for Cy5 imaging: HFT, 543/633 nm and pinhole diameter, 220 mm.
Exosomes-mediated inhibition of cell migration and proliferation
The effect of miR-26a-loaded exosomes on HepG2 cell migration was quantified in vitro as follows: 3×105 HepG2 cells were seeded in six-well plates and incubated until they reached 70% confluence (~24 h). The wells were treated with either PBS, 293T cell-derived exosomes-loaded miR-26a (Exo/miR-26a), or APO-CD63 vector-engineered 293T-derived exosome-loaded miR-26a (APO-Exo/miR-26a) and incubated for 4 h at 37°C. A wound was introduced to the monolayer of attached cells by grazing the surface with a sterile pipette tip followed by gentle washing with PBS; the cells were subsequently incubated in fresh medium at 37°C. Images of cells within the scratched monolayers were recorded at 24, 48, and 96 h, respectively.
Cell proliferation was determined in a separate set of experiments using cell counting kit-8 (CCK-8) assay to detect changes in cell viability after different incubation times. HepG2 cells were seeded in 24-well plates at an initial density of 2×104 cells/well for HepG2 in 1 mL of growth medium and incubated for 24 h prior to the addition of Exo/miR-26a and APO-Exo/miR-26a at different N/P ratios. Untreated cells were taken as control with 100% viability.
RNA isolation and quantitative real-time reverse transcription PCR
Total RNA was extracted from different treated cells using the Trizol reagent (Invitrogen, Carlsbad, CA, USA), as recommended by the manufacturer. Levels of miRNA were quantified using TaqMan® miRNA assays (Applied Biosystems, Carlsbad, CA, USA). Total RNA was reverse transcribed to cDNA using a specific cDNA synthesis primer. One microliter cDNA was used for real-time PCR using qPCR master mix kit. After this reaction, the cycle threshold (CT) values were determined using the threshold setting. ThemiR-26a concentration in HepG2 cells was normalized to U6 RNA in calculations of the expression level of the miR-26a in cells. The increase in the expression level of miR-26a in exosome-transfected cells was calculated based on the U6 levels. Quantitative PCR was run on an ABI 7500 thermocycler (Applied Biosystems, Carlsbad, CA, USA), and the data were analyzed using the 2−ΔΔCt method.
Cell cycle assay
The effect of APO-Exo/miR-26a uptake on the progression of the cell cycle was analyzed using the propidium iodide (PI) method.3 HepG2 cells were seeded in a six-well plate at a density of 2×105 cells/well. After incubating for 24 h, the medium was removed and the cells were incubated in fresh medium including PBS, Exo/miR-26a, or APO-Exo/miR-26a for 48 h. The cell cycle was determined using a Propidium Iodide Kit (Invitrogen, Carlsbad, CA, USA). Briefly, after washing three times with PBS, the cells were fixed with 4 mL of 75% ethanol for 4 h at 4°C. The cells were washed twice with PBS and incubated for 30 minutes at 37°C with 100 μL of 200 μg/mL DNase-free RNaseA. Finally, 100 μL of 1 μg/mL PI was added into cell suspension and incubated for 10 minutes at room temperature. The stained HepG2 cells were then analyzed using a flow cytometer (Caliber, Becton Dickinson, San Jose, CA, USA).
Western blot analysis
Western blot analysis was performed as previously described.31 Exosome samples were lysed in sodium dodecyl sulfate loading buffer. After boiling, equal amounts (10 μg) of the proteins were electrophoresed on 12% sodium dodecyl sulfate–polyacrylamide gels and transferred to Immobilon® membranes (Millipore, Bedford, MA, USA) using wet blotting methods, and then transferred onto polyvinylidene fluoride membrane, and blocked and incubated with monoclonal antibodies APO (1:500), CD63 (1:1,000), CD9 (1:1,000), GAPDH (1:1,000), CCND2 (1:1,000), CCNE2 (1:500), CDK6 (1:500), and β-actin (1:1,000) overnight, respectively. The membrane was washed three times and incubated with horseradish peroxidase-conjugated secondary antibody (1:2,000) for 2 h at room temperature. A WesternBright™ western blotting detection kit (Advansta, Menlo Park, CA, USA) was used to visualize the bands on the membrane. The protein expression level was normalized to β-actin expression, that is, a housekeeping gene.
Statistical analysis
Data were analyzed as detailed in each section with specific values being expressed as their mean ± SD. Each measurement was repeated at least three times. Statistical analysis was carried out via the two-way analysis of variance (ANOVA) tests with values of P<0.05 being considered statistically significant.
Results
Construction and validation of pEGFP-CD63-Apo-A1
According to earlier publications, the tumor-targeting function of exosomes was introduced by expressing Apo-A1 in parent cells.37,38 The overall schematic is shown in Figure 1A. The Apo-A1 gene was cloned into the pEGFP-N2 vector downstream of the CD63 sequence. This cloning strategy led to the expression of Apo-A1 on the external face of the plasma membranes via the transmembrane domain of CD63. The transfection efficiency was determined by measuring the fluorescence signal of GFP in HepG2 cells, which was introduced downstream of CMV promoter of pEGFP-N2. The vector was verified by PCR and sequencing to ensure correct integration of the genes as shown in Figure S1. The vector was subsequently transfected in 293T cells using lipofectamine. After 48 h, untransfected cells did not generate any sensible levels of GFP fluorescence (Figure 1B), while pEGFP-CD63-Apo-A1-transfected 293T cells showed robust green fluorescence. We then sorted and isolated the cells expressing GFP by flow cytometry. The expression of GFP in the sorted cells is shown in Figure 1C. Immunofluorescence analysis of transfected cells that stained with anti-Apo-A1 antibody showed Apo-A1 was expressed in transfected 293T cells (Figure S2).
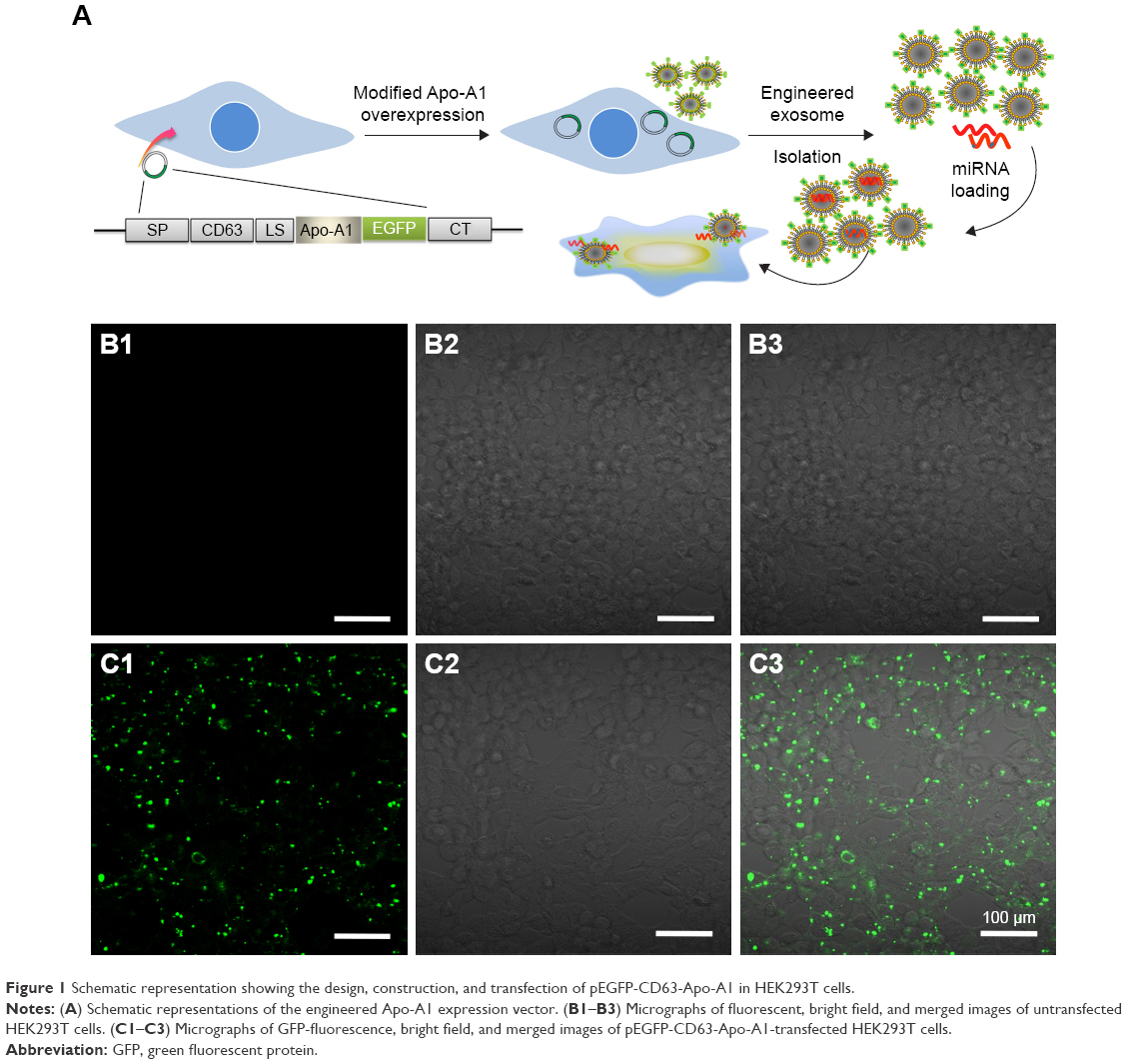 | Figure 1 Schematic representation showing the design, construction, and transfection of pEGFP-CD63-Apo-A1 in HEK293T cells. |
Characterization of Apo-A1 exosomes
The plasmid encoding the gene fusion between CD63 and Apo-A1 was transfected in HEK293T cells for 96 h prior to harvesting exosomes. Exosomes were isolated separately from culture supernatants of untransfected and Apo-A1-transfected HEK 293T cells using the exoEasy Kit. The purified exosomes were characterized using TEM and dynamic light scattering (DLS). TEM photographs of exosomal preparations (Figure 2A) revealed a number of expected morphological characteristics, including an average diameter of 120±9.7 nm and a closed membrane with a surface potential of −16±2.2 mV. It should be noted that there were some modest differences in the topography of the miR-26a-loaded exosomes, including slightly larger average diameter and surface potential, that is, 135±11.3 nm (Figure 2B) and −21±3.1 mV, respectively. Regarding these variations, we speculate that it is a consequence of electroporating miR-26a in exosomes, as all other procedures used to produce exosomes were identical. The average diameter of exosomes recorded using DLS was consistent with those determined from an analysis of exosomes using TEM images (Figure 2C and D). It was noted that there is a common phenomenon of two peaks (about 40 and 120 nm) in the exoEasy Kit-purified exosomes.
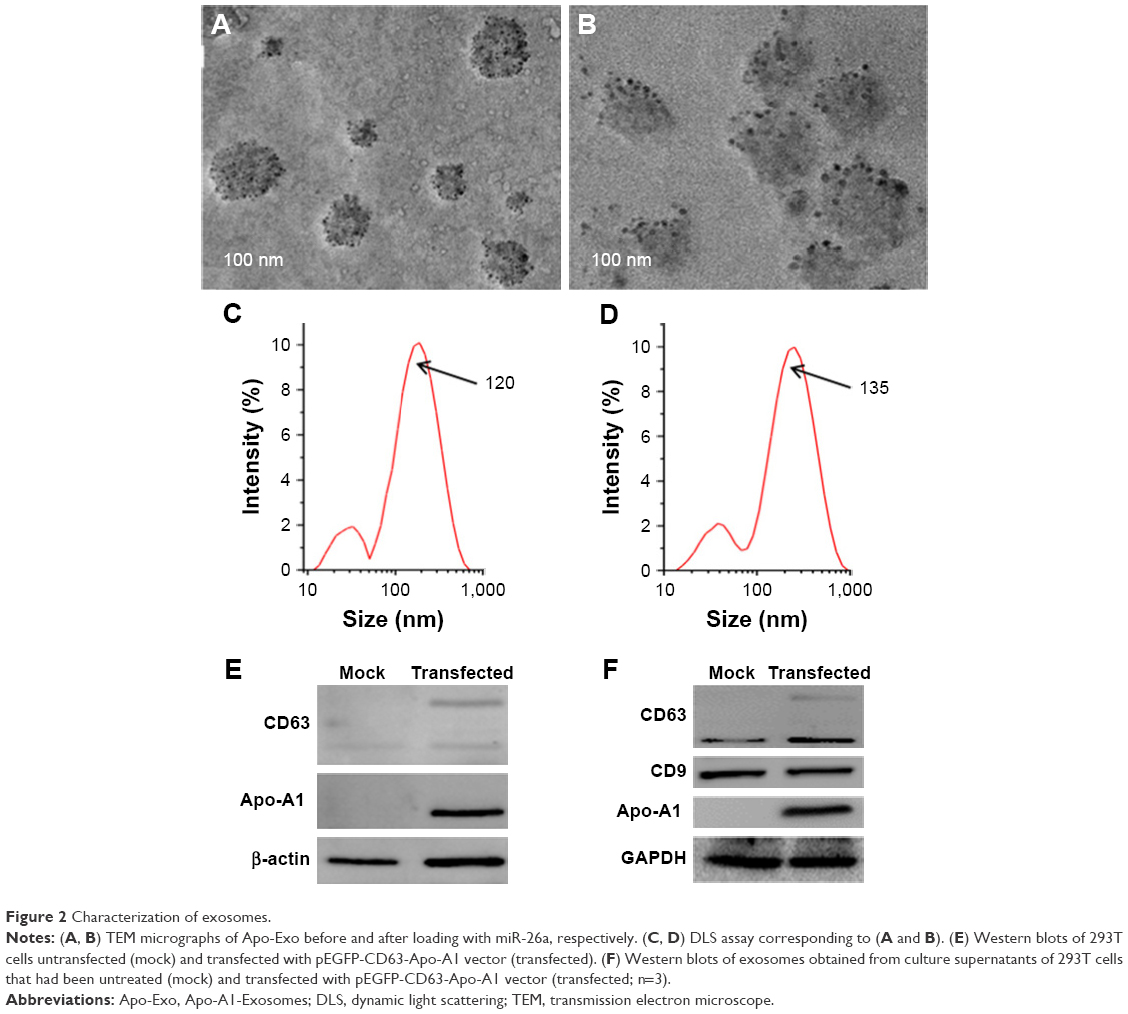 | Figure 2 Characterization of exosomes. |
Furthermore, CD63 and CD9 in the membrane of exosomes were confirmed using western blot analysis of exosomes isolated from transfected 293T cells (Figure 2F). As expected, we determined Apo-A1 was expressed only in pEGFP-CD63-Apo-A1-transfected exosomes. On the other hand, CD63 and CD9 (exosome markers) were expressed in both transfected and untransfected exosomes. In addition, the two bands in western blot analysis correspond to endogenous CD63 (26 kDa), and the larger engineered fusion protein (CD63-Apo-A1, 54 kDa) was detected in exosomes isolated from transfected 293T cells (Figure 2E and F). In addition, consistent with previous reports, GAPDH was also detected in exosomes (Figure 2F).39 These results suggest that the isolated sample is exactly the exosome.
Quantification of miR-26 loading into exosome
Next, we investigated the efficiency of electroporation-mediated loading of miRNA in exosomes expressing Apo-A1.40 The amount of miR-26a loaded into purified exosomes was evaluated using a qRT-PCR assay. A calibration curve was established for the qRT-PCR assay using known amounts of pure miR-26a in a set of serially diluted samples. The Pearson correlation coefficient for the standard curve was determined as R2>0.99 (Figure S3A). The linear range of the CT value was from 17.28 to 30.11, which corresponds to miR-26a levels of 10 amol to 100 fmol (Figure S3B). The amount of miR-26a loaded into exosomes as determined from this standard curve is 0.12 pmol/μg. The results demonstrated that electroporation of exosomes results in an efficient loading of miR-26a.
Uptake of exosome by HepG2 cells
Next, we determined the specificity and efficiency of exosome uptake by HepG2 cells. HepG2 cells were treated with either 200 μg/mL of control exosomes (no Apo-A1; Exo-control) or test Apo-A1-Exosomes (Apo-Exo) at 37°C for 12 h. Both exosome preparations were labeled with the fluorescent probe DiO. CLSM was used to monitor the fluorescence intensity of DiO-labeled exosomes in HepG2 cells. The fluorescence intensity of HepG2 cells treated with DiO-labeled Apo-A1-modified exosomes (Apo-Exo-A1) was higher than that in cells incubated with DiO-labeled control exosomes (Exo-control). Figure 3A shows that cells in the control sample emitted a very low green fluorescence signal. Figure 3B indicates that the Apo-Exo is more easily uptake by HepG2 cells. In separate control studies, the green fluorescence cannot be detected in non-treated HepG2 cells, or in HepG2 cells incubated with the unlabeled Exo-control (Figure S4). Similary, Apo-Exo shows the similar results as HepG2 cells in SMMC-7721 cells that is known as the expression of SR-B1 on the cell’s surface (Figure S5).
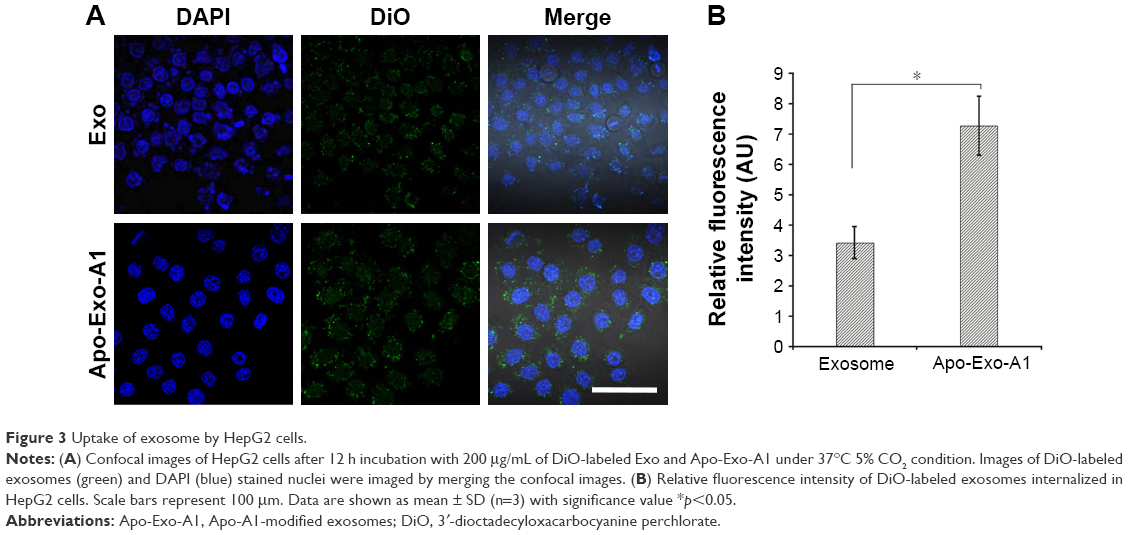 | Figure 3 Uptake of exosome by HepG2 cells. |
Next, we investigated the dynamics of the uptake of miR-26a-loaded Apo-Exo in HepG2 cells. Differential interference contrast and confocal fluorescence images of HepG2 cells were recorded at 1, 3, 6, 12, and 24 h after incubating 200 μg/mL of Apo-Exo-A1 loaded with Cy5-labeled miR-26a. CLSM fluorescence images of cells incubated with APO-Exo/miR-26a (red) with DAPI (blue), along with the merged images (DAPI/Cy5) from this study are shown in Figure 4. Analysis of the images show the intensity of red fluorescence (Cy5-miR-26a) in HepG2 cells increased up to the 6 h time point and thereafter decreased for 24 h; its intensity of red fluorescence was about two-thirds of that of the 6-h time point. The decrease in Cy5 fluorescence may result from processing of internalized APO-Exo/miR-26a by HepG2 cells, and/or loss of the Cy5 molecules. Interestingly, Cy5-labeled miR-26a was found to localize primarily to the cytoplasm of HepG2 cells, although we cannot exclude the possibility that lower levels of the probe exist in the nucleus (Figure 4).
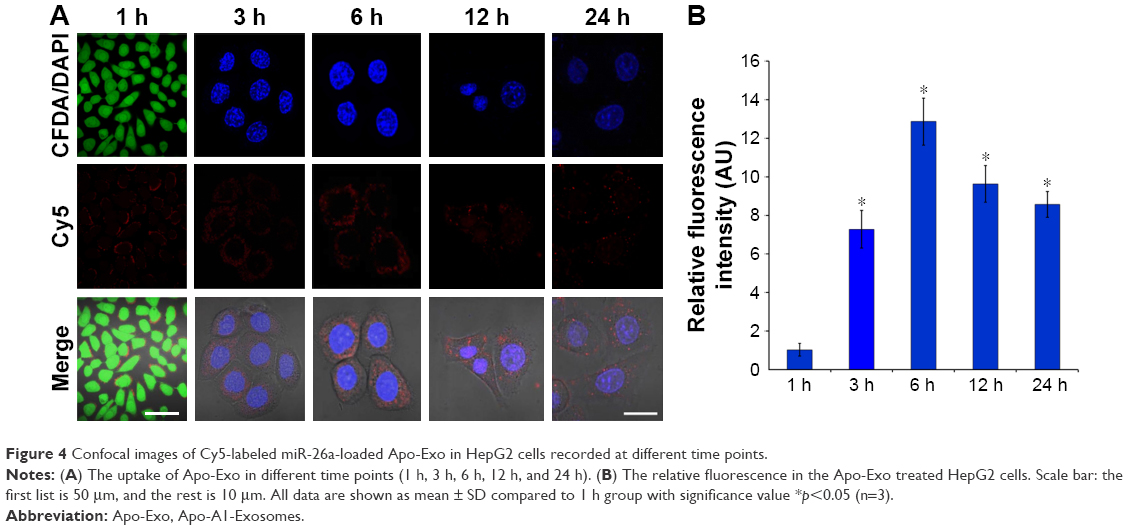 | Figure 4 Confocal images of Cy5-labeled miR-26a-loaded Apo-Exo in HepG2 cells recorded at different time points. |
The effects of miR-26a delivered by Apo-Exo on HepG2 cells
Next, we investigated the effects of the uptake of miR-26a-Apo-Exo by HepG2 cells on their wound-healing response. The migratory properties of HepG2 incubated with Apo-Exo/miR-26a were monitored over 4 days within a simple wound-healing assay. HepG2 cells were first incubated for 6 h with each of the following: Exo/miR-26a (exosome control) or Apo-Exo/miR-26a (test) and an untreated control. Confluent monolayers of cells were scratched with a sterile tip, washed twice with PBS, and cultured for a further 24 or 72 h. Bright field microscopy was used to measure the distance that the cells migrated into the wound. After 72 h, the wound-like gaps in the untreated and Exo/miR-26a-treated cells had healed almost completely (Figure 5A), indicating that the HepG2 cell migration was normal in the control samples. The results from the quantitative analysis (Figure 5B) indicated a statistically significant reduction in the wound closures in the Apo-Exo/miR-26a-treated cells compared with the untreated and the negative control cells, respectively.
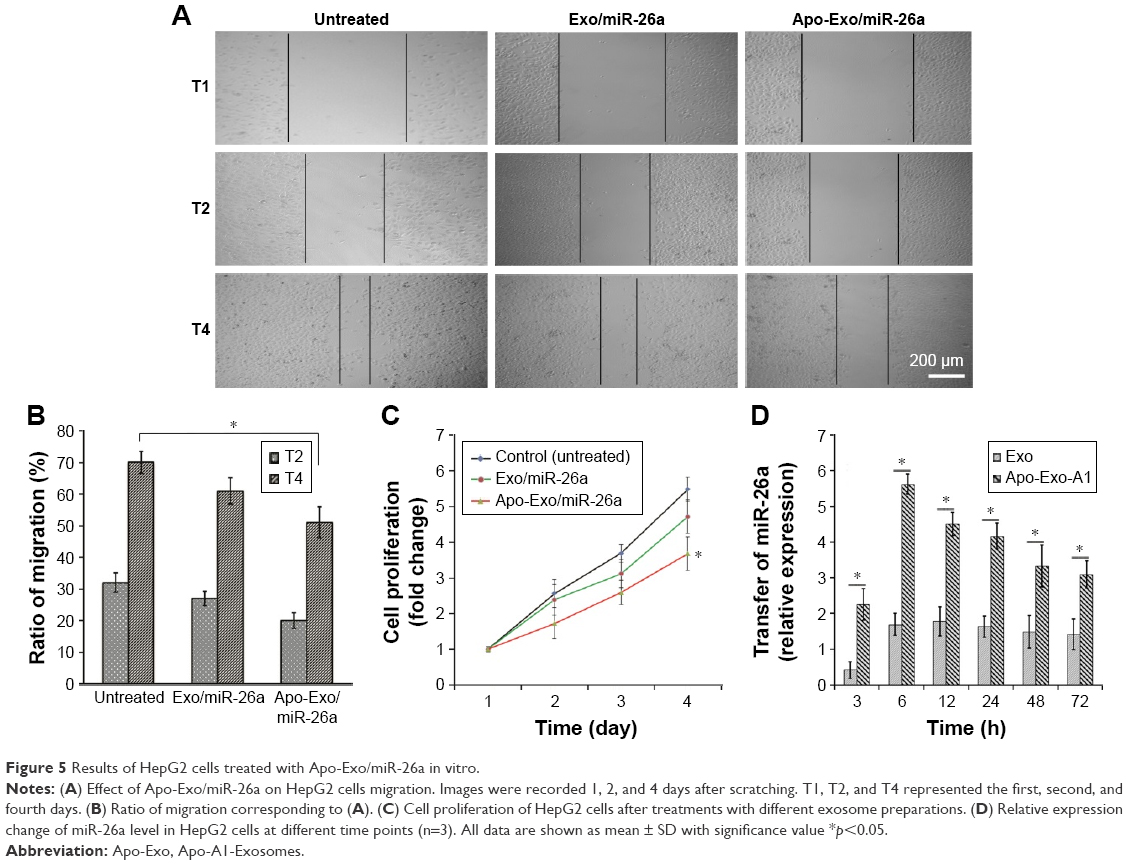 | Figure 5 Results of HepG2 cells treated with Apo-Exo/miR-26a in vitro. |
Next, we assessed the rate of cell proliferation for HepG2 cells treated with miR-26a-loaded Apo-A1 exosomes. The CCK-8 assay was used to quantify cell proliferation. Quantitative analysis of these data showed a statistically significant reduction in the rate of cell proliferation in Apo-Exo/miR-26a-treated cells compared with untreated cells (Figure 5C). In summary, these studies show that the internalization of miR-26a in HepG2 cells is accompanied by specific changes in cell physiology, including cell migration and proliferation.
Furthermore, we explored the mechanism underlying the inhibition of HepG2 cell migration and proliferation following internalization of exosomes loaded with miR-26a. qRT-PCR was used to quantify the expression level of miR-26a in HepG2 treated with miR-26a-loaded exosomes. The results showed a time-dependent elevation of miR-26a (upregulation) in HepG2 cells that had been incubated with Apo-Exo-loaded miR-26. Interestingly, unmodified exosomes loaded with miR-26a were also found to be taken up by HepG2 cells, albeit at much lower levels. The low levels of miR-26a delivered by control exosomes may result from nonspecific uptake of exosomes by HepG2 cells, or else because the exosomes generated from 293T cell contain a background of surface proteins that are recognized by receptors on HepG2 cells. A closer inspection of data recorded at 3, 6, 12, 24, 48, and 72 h showed the levels of miR-26a in cells treated with Apo-Exo/miR-26a were indeed significantly higher than those of control exosomes (Figure 5D).
miR-26a delivered by Apo-Exo inhibited cell cycle and protein expression
The uptake of Apo-Exo loaded with miR-26a by HepG2 cells inhibited the progression of the cell cycle by arresting cells at the G1 phase, as shown in Figure 6A–C. The HepG2 cell cycle was unaffected in the two control cell groups (untreated and incubated with Exo/miR-26a). Figure 6D shows that an enhanced expression of miR-26a in HepG2 cells correlates with an increase in the probability of their arrest at G1 compared with untreated cells, or those incubated with control exosomes (Exo/miR-26a). This finding is consistent with earlier reports on the effects of miR-26a.41 Correspondingly, our study shows that the upregulation of miR-26a expression in HepG2 cells inhibits the rates of proliferation and migration.
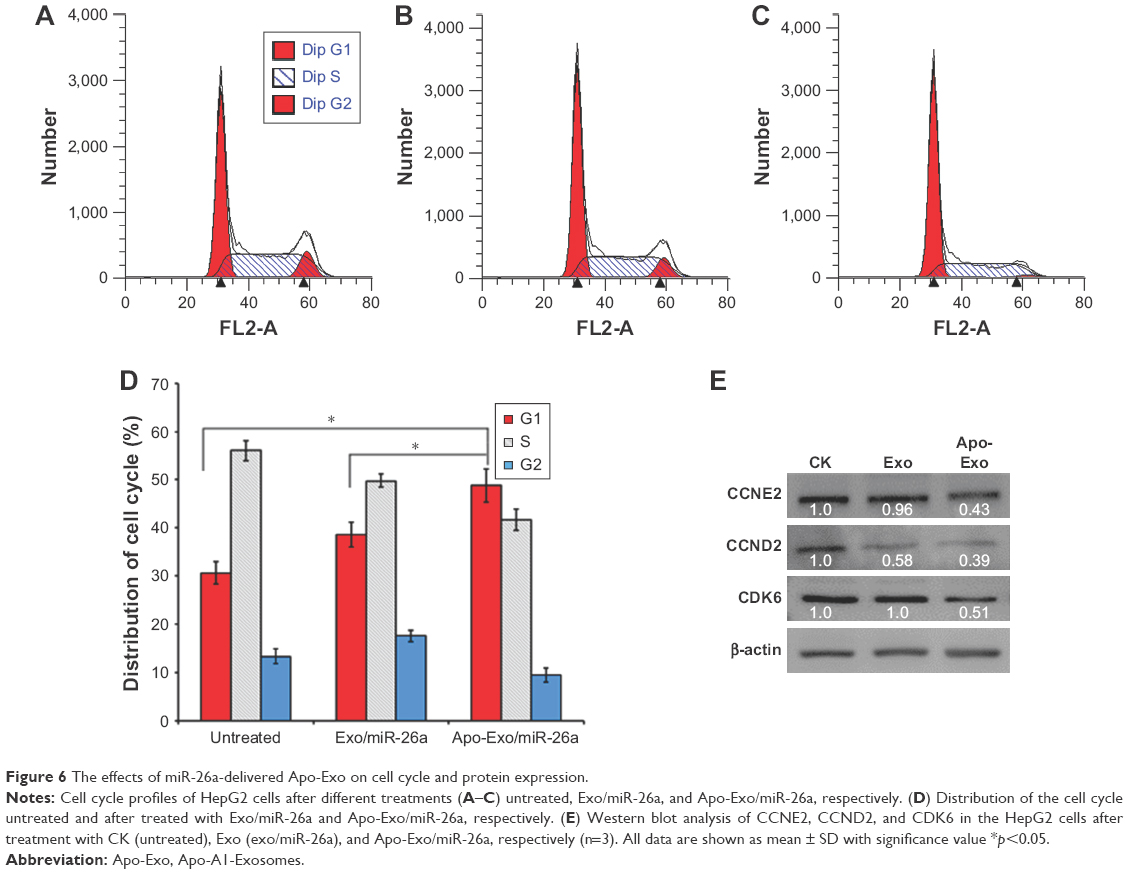 | Figure 6 The effects of miR-26a-delivered Apo-Exo on cell cycle and protein expression. |
miRNAs are attractive candidates for tumor therapy, because they can be designed to target specific gene or even multiple genes, and they can exert their effects by targeting defined molecules that regulate critical cellular processes.42 To further investigate the mechanism of miR-26a-mediated inhibition of cell proliferation, we quantified the expression levels of key cell cycle checkpoint proteins, including cyclin D2 (CCND2), cyclin E2 (CCNE2), and CDK6. These proteins have been predicted and identified as the targets of miR-26a, and they play important roles in regulating the passage through G1/S.30 As shown in Figure 6E, the uptake of Apo-Exo/miR-26a by HepG2 cells results in a significant downregulation of the expression levels of CCNE2 and CDK6. In contrast, the amounts of these proteins are similar in the two control groups (untreated cells and control exosomes). This finding suggested that the miR-26a delivered by Apo-Exo arrests the cell cycle by reducing the amount of key protein regulators of the G1/S transition (CCNE2, CDK6, and CCND2). Taken together, we have shown that Apo-A1-engineered exosomes are preferentially endocytosed by HepG2 cells where their cargo of miR-26a triggers cell cycle arrest and results in reductions in the rates of cell proliferation and migration.
Discussion
Exosomes are natural delivery vehicles that exchange macromolecular cargo and information between remote cells in the body.43 The sealed membrane of the exosome protects its internal cargo of nucleic acids and proteins from the destructive effects of circulating nucleases and proteases. Significantly, proteins on the external membrane of the exosome may bind to target receptors on target cells where they are internalized by receptor-mediated endocytosis.44 Exosomes possess a far longer period of circulation in the body compared with artificial nanoparticles, which increases the chance of encountering with rare, remote, and deep-seated target cells.5 These favorable features are driving efforts to engineer exosomes for the targeted delivery of exogenous nucleic acids and therapeutic drugs to diseased cells,4,45 especially tumor cells.14,46
In the present study, we introduced a novel class of engineered exosomes that are shown to deliver a therapeutic miRNA cargo to target cells. In particular, human 293T cells were engineered, using standard molecular biology approaches, to prepare exosomes with a targeting Apo-A1 sequence, a component of HDLs. After purification, these exosomes were loaded with a cargo of fluorescently labeled miR-26a by electroporation and characterized using a variety of physical, biochemical, cell biological, and high-resolution fluorescence and electron microscope imaging techniques. Apo-A1-modified exosomes were shown to bind specifically to HepG2 cells via their SR-B1 receptor. SR-B1 is abundant on the surface of liver cancer cells and tumor-associated vascular endothelium, which is believed to facilitate vascular and tissue penetration of antitumor agents.47 Apo-A1-presenting exosomes were shown to be efficiently endocytosed by HepG2 cells. Subsequently, miR-26a released into the cytoplasm of these HepG2 cells resulted in its upregulation and led to an enhanced suppression in the rates of cell migration and proliferation. Further studies showed that the elevated miR-26a downregulated three key cell cycle control proteins, the consequence of which includes an increase in the arrest of the cell cycle at the G1/S checkpoint and inhibition of cell proliferation.30 In addition, the efficiency of our engineered exosomes in targeting and suppressing HepG2 cells arises from several enhanced and optimized features of the engineered exosomes. These features include correct presentation of Apo-A1 on the outer membrane of the exosome, high-efficiency loading of fluorescent miR-26a, the selective targeting and uptake of Apo-A1 exosomes by cancer cells that overexpress SR-B1, and finally, the release of functional miR-26a into targeted cells. Furthermore, we showed that miR-26a delivered to HepG2 cells was upregulated in the host cell, which enhanced its ability to suppress the levels of key protein regulators of the cell cycle. Interestingly, we note that the delivery of therapeutic cargo to late endosomal/lysosomal compartments by exosomes could also be exploited to deliver drugs specifically to intracellular-resident microbial pathogens.44
Over the past decade, thousands of researchers have contributed to the development of exosome-based vehicles for targeted delivery of therapeutics to diseased, including neurodegenerative diseases4 and cancer.48 Collectively, these studies have identified advantages of exosomes, over viruses and artificial nanoparticles, for the targeted delivery of therapeutics to diseased cells. However, the potential of engineered exosomes for clinical therapies is limited by their very challenging manufacture, and the need to conduct a comprehensive molecular and functional characterization on each preparation prior to a treatment, both of which are expensive and labor-intensive.49 These challenges could be overcome by developing robust and automated devices that can carry out operations associated with the production and purification of disease-targeted exosomes from genetically engineered 293T cells (or stem cell or CAR-T cells). The engineered exosomes can eventually be made in the form of injectable for therapeutic drugs, including miRA and protein-based therapeutics. Efforts to automate the preparation of tumor-targeted exosomes for use in small animal models of human cancer are under way in our laboratories.
Conclusion
We have engineered exosomes as living vehicles for tumor-targeted delivery and uptake of a small RNA therapeutic to a model cell of human liver cancer. Our results showed that miR-26a was delivered with high efficiency to target cells where it was further released, and it led to the downregulation of key regulatory proteins of the cell cycle. The development of a simple approach to engineer human cell-derived exosomes for the targeted delivery of specific and functional therapeutic miRNA species to tumor cells could be further exploited for personalized medicine and gene therapy. In summary, our study demonstrates the potential of using engineered exosomes as living vehicles for the targeted delivery of functional small RNAs for the effective treatment of cancer.
Acknowledgments
The National Natural Science Foundation of China (U1404824), Young Backbone Teacher Project of Henan Provincial Universities (2015GGJS-049), and The China Scholarship Council (201508410057) financially supported this work.
Disclosure
The authors report no conflicts of interest in this work.
References
Niidome T, Huang L. Gene therapy progress and prospects: nonviral vectors. Gene Ther. 2002;9(24):1647–1652. | ||
Lee SJ, Kim MJ, Kwon IC, Roberts TM. Delivery strategies and potential targets for siRNA in major cancer types. Adv Drug Deliv Rev. 2016;104:2–15. | ||
Liang G, Li Y, Feng W, et al. Polyethyleneimine-coated quantum dots for miRNA delivery and its enhanced suppression in HepG2 cells. Int J Nanomedicine. 2016;11:6079–6088. | ||
Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29(4):341–345. | ||
Dai L, Gu N, Chen BA, Marriott G. Human platelets repurposed as vehicles for in vivo imaging of myeloma xenotransplants. Oncotarget. 2016;7(16):21076–21090. | ||
Marriott G. Engineering platelets for tumour targeting. Aging (Albany NY). 2016;8(8):1572–1573. | ||
Li J, Sharkey CC, Wun B, Liesveld JL, King MR. Genetic engineering of platelets to neutralize circulating tumor cells. J Control Release. 2016;228:38–47. | ||
Shi J, Kundrat L, Pishesha N, et al. Engineered red blood cells as carriers for systemic delivery of a wide array of functional probes. Proc Natl Acad Sci U S A. 2014;111(28):10131–10136. | ||
Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci. 2000;113:3365–3374. | ||
Fuhrmann G, Herrmann IK, Stevens MM. Cell-derived vesicles for drug therapy and diagnostics: opportunities and challenges. Nano Today. 2015;10(3):397–409. | ||
Eckard SC, Rice GI, Fabre A, et al. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat Immunol. 2014;15(9):839–845. | ||
Gidlof O, van der Brug M, Ohman J, et al. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood. 2013;121(9):3908–3917. | ||
Karantalis V, Hare JM. Use of mesenchymal stem cells for therapy of cardiac disease. Circ Res. 2015;116(8):1413–1430. | ||
Powers S, Pollack RE. Inducing stable reversion to achieve cancer control. Nat Rev Cancer. 2016;16(4):266–270. | ||
Tian T, Zhu YL, Zhou YY, et al. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J Biol Chem. 2014;289(32):22258–22267. | ||
Ghai V, Wang K. Recent progress toward the use of circulating microRNAs as clinical biomarkers. Arch Toxicol. 2016;90(12):2959–2978. | ||
Lee C, Carney RP, Hazari S, et al. 3D plasmonic nanobowl platform for the study of exosomes in solution. Nanoscale. 2015;7(20):9290–9297. | ||
Tian Y, Li S, Song J, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35(7):2383–2390. | ||
Kalra H, Adda CG, Liem M, et al. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics. 2013;13(22):3354–3364. | ||
Rana S, Malinowska K, Zoeller M. Exosomal tumor microRNA modulates premetastatic organ cells. Neoplasia. 2013;15(3):281–295. | ||
Didiot MC, Hall LM, Coles AH, et al. Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol Ther. 2016;24(10):1836–1847. | ||
Morishita M, Takahashi Y, Matsumoto A, Nishikawa M, Takakura Y. Exosome-based tumor antigens-adjuvant co-delivery utilizing genetically engineered tumor cell-derived exosomes with immunostimulatory CpG DNA. Biomaterials. 2016;111:55–65. | ||
O’Connor RM, Gururajan A, Dinan TG, Kenny PJ, Cryan JF. All roads lead to the miRNome: miRNAs Have a central role in the molecular pathophysiology of psychiatric disorders. Trends Pharmacol Sci. 2016;37(12):1029–1044. | ||
Kortylewski M, Nechaev S. How to train your dragon: targeted delivery of MicroRNA to cancer cells in vivo. Mol Ther. 2014;22(6):1070–1071. | ||
Rincon MY, VandenDriessche T, Chuah MK. Gene therapy for cardiovascular disease: advances in vector development, targeting, and delivery for clinical translation. Cardiovasc Res. 2015;108(1):4–20. | ||
Hatakeyama H, Murata M, Sato Y, et al. The systemic administration of an anti-miRNA oligonucleotide encapsulated pH-sensitive liposome results in reduced level of hepatic microRNA-122 in mice. J Control Release. 2014;173(1):43–50. | ||
Scomparin A, Polyak D, Krivitsky A, Satchi-Fainaro R. Achieving successful delivery of oligonucleotides – From physico-chemical characterization to in vivo evaluation. Biotechnol Adv. 2015;33(6 Pt 3):1294–1309. | ||
Kheirolomoom A, Kim CW, Seo JW, et al. Multifunctional nanoparticles facilitate molecular targeting and miRNA delivery to inhibit atherosclerosis in ApoE(−/−) mice. Acs Nano. 2015;9(9):8885–8897. | ||
Chen F, Huang P, Zhu YJ, Wu J, Zhang CL, Cui DX. The photoluminescence, drug delivery and imaging properties of multifunctional Eu3+/Gd3+ dual-doped hydroxyapatite nanorods. Biomaterials. 2011;32(34):9031–9039. | ||
Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137(6):1005–1017. | ||
Liang G, Zhu Y, Jing A, et al. Cationic microRNA-delivering nanocarriers for efficient treatment of colon carcinoma in xenograft model. Gene Ther. 2016;23(12):829–838. | ||
Urban S, Zieseniss S, Werder M, Hauser H, Budzinski R, Engelmann B. Scavenger receptor BI transfers major lipoprotein-associated phospholipids into the cells. J Biol Chem. 2000;275(43):33409–33415. | ||
Yamamoto S, Fukuhara T, Ono C, et al. Lipoprotein receptors redundantly participate in entry of hepatitis C virus. PLoS Pathog. 2016;12(5):e1005610. | ||
Kharaziha P, Chioureas D, Rutishauser D, et al. Molecular profiling of prostate cancer derived exosomes may reveal a predictive signature for response to docetaxel. Oncotarget. 2015;6(25):21740–21754. | ||
Lazar I, Clement E, Ducoux-Petit M, et al. Proteome characterization of melanoma exosomes reveals a specific signature for metastatic cell lines. Pigment Cell Melanoma Res. 2015;28(4):464–475. | ||
Yim N, Ryu SW, Choi K, et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat Commun. 2016;7:12277. | ||
Hung ME, Leonard JN. Stabilization of exosome-targeting peptides via engineered glycosylation. J Biol Chem. 2015;290(13):8166–8172. | ||
Mo ZC, Ren K, Liu X, Tang ZL, Yi GH. A high-density lipoprotein-mediated drug delivery system. Adv Drug Deliv Rev. 2016;106(Pt A):132–147. | ||
Kanada M, Bachmann MH, Hardy JW, et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc Natl Acad Sci U S A. 2015;112(12):E1433–E1442. | ||
Lunavat TR, Jang SC, Nilsson L, et al. RNAi delivery by exosome-mimetic nanovesicles − Implications for targeting c-Myc in cancer. Biomaterials. 2016;102:231–238. | ||
Chen L, Zheng J, Zhang Y, et al. Tumor-specific expression of MicroRNA-26a suppresses human hepatocellular carcinoma growth via cyclin-dependent and -independent pathways. Mol Ther. 2011;19(8):1521–1528. | ||
Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release. 2014;194:238–256. | ||
Tkach M, Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–1232. | ||
Malhotra H, Sheokand N, Kumar S, et al. Exosomes: tunable nano vehicles for macromolecular delivery of transferrin and lactoferrin to specific intracellular compartment. J Biomed Nanotechnol. 2016;12(5):1101–1114. | ||
Choi JS, Yoon HI, Lee KS, et al. Exosomes from differentiating human skeletal muscle cells trigger myogenesis of stem cells and provide biochemical cues for skeletal muscle regeneration. J Control Release. 2016;222:107–115. | ||
Tian Y, Li S, Song J, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35(7):2383–2390. | ||
Martinez LO, Jacquet S, Esteve JP, et al. Ectopic beta-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature. 2003;421(6918):75–79. | ||
Tominaga N, Yoshioka Y, Ochiya T. A novel platform for cancer therapy using extracellular vesicles. Adv Drug Deliv Rev. 2015;95:50–55. | ||
Jo W, Kim J, Yoon J, et al. Large-scale generation of cell-derived nanovesicles. Nanoscale. 2014;6(20):12056–12064. |
Supplementary materials
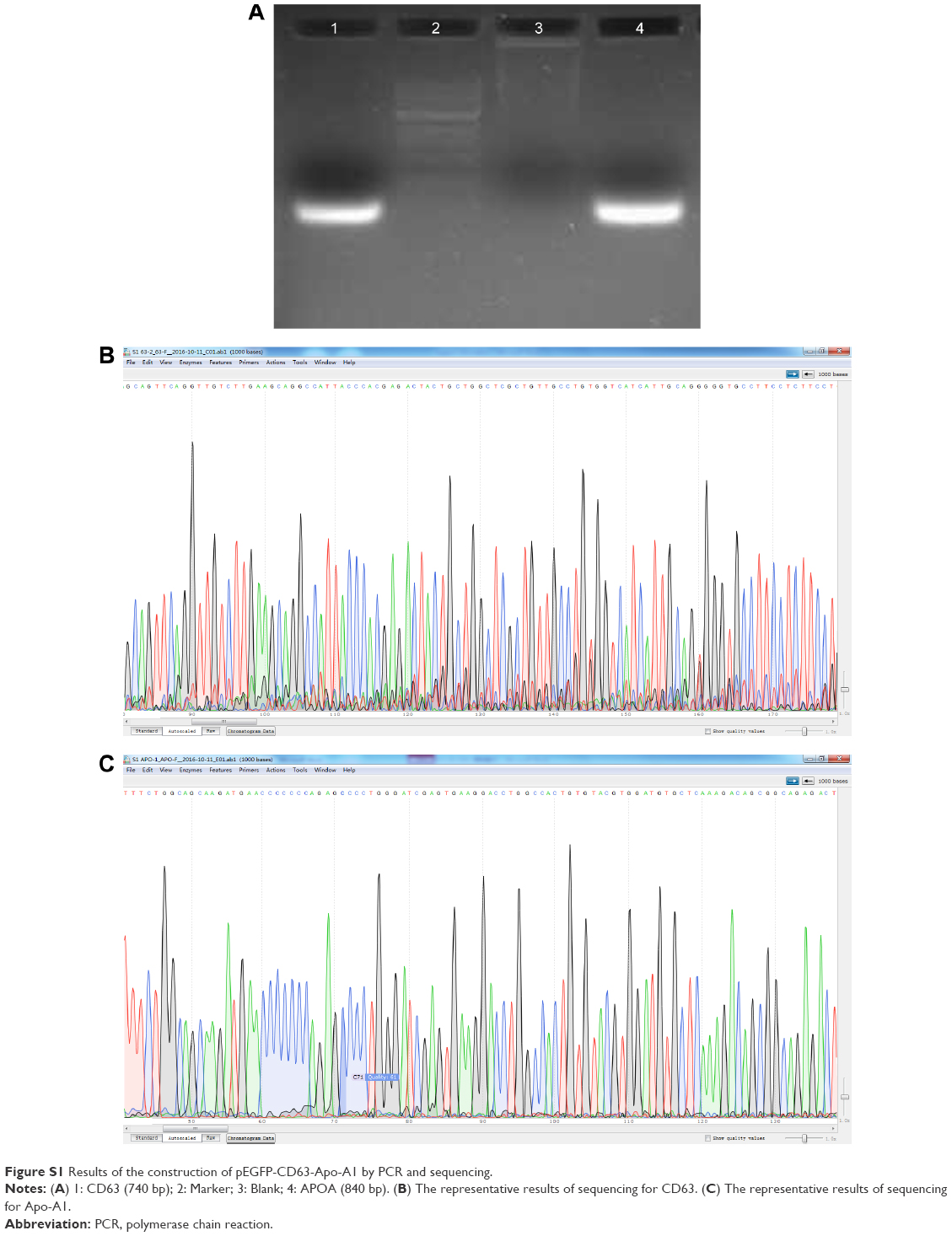 | Figure S1 Results of the construction of pEGFP-CD63-Apo-A1 by PCR and sequencing. |
 | Figure S2 Immunofluorescence analysis for Apo-A1 expression. |
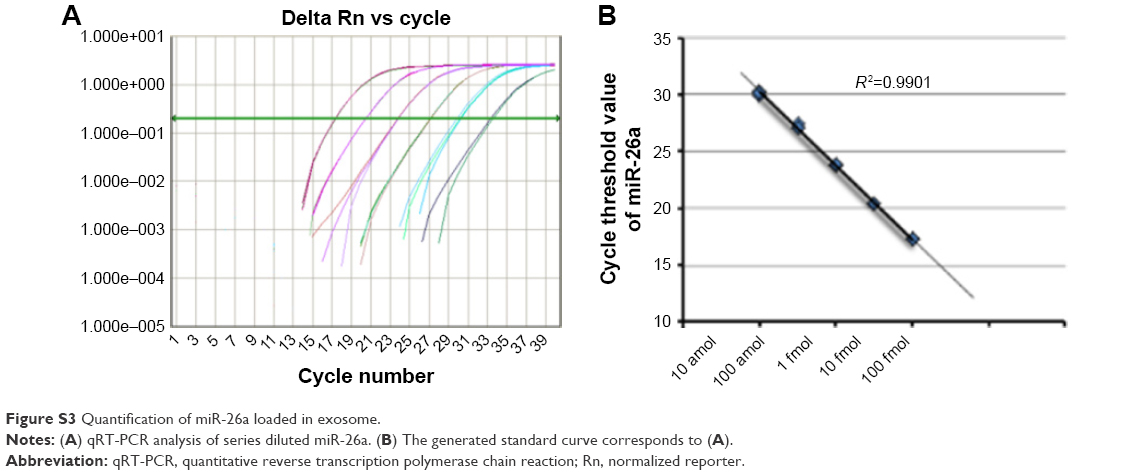 | Figure S3 Quantification of miR-26a loaded in exosome. |
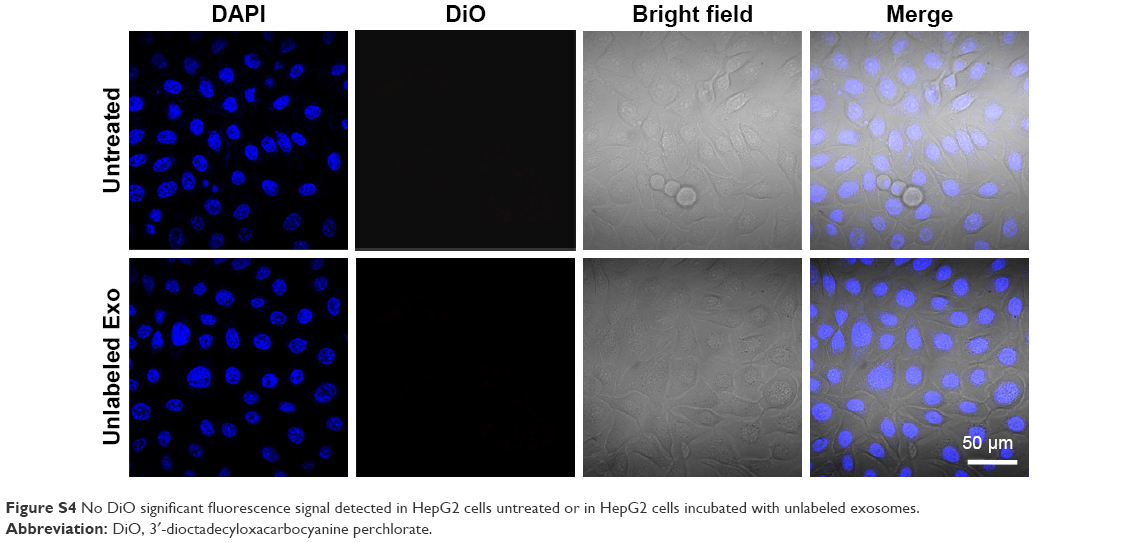 | Figure S4 No DiO significant fluorescence signal detected in HepG2 cells untreated or in HepG2 cells incubated with unlabeled exosomes. |
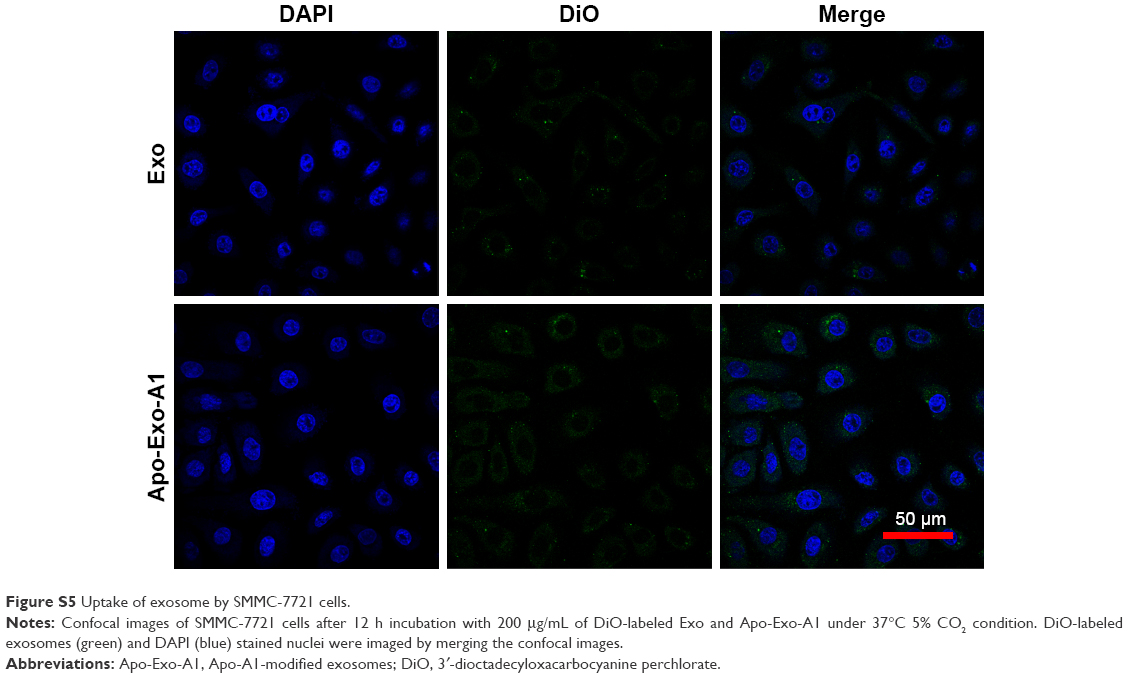 | Figure S5 Uptake of exosome by SMMC-7721 cells. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.