Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 6
Enantiomer-selective pharmacokinetics, oral bioavailability, and sex effects of various alpha-lipoic acid dosage forms
Authors Hermann R ![]() , Mungo J, Cnota PJ, Ziegler D
, Mungo J, Cnota PJ, Ziegler D
Received 22 July 2014
Accepted for publication 30 September 2014
Published 28 November 2014 Volume 2014:6 Pages 195—204
DOI https://doi.org/10.2147/CPAA.S71574
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Robert Hermann,1 Julius Mungo,2 Peter Jürgen Cnota,2 Dan Ziegler3
1Clinical Research Appliance (cr appliance), Gelnhausen, Germany; 2MEDA Pharma GmbH & Co KG, Bad Homburg, Germany; 3Institute for Clinical Diabetology, German Diabetes Center, Heinrich Heine University, Düsseldorf, Germany
Abstract: The present study aimed to examine the enantiomer-selective pharmacokinetics (PK), relative bioavailability (Frel), and sex effects of various oral dosage forms of racemic alpha-lipoic acid (ALA). In an open-label, randomized, four-period, four-sequence crossover study, 24 healthy adult subjects (12 males and 12 females) received single doses of 600 mg of ALA in fasted state at four different occasions as follows: three 200 mg tablets (T 200); two 300 mg tablets (T 300); one 600 mg tablet (T 600); and a racemic ALA solution (OS). All tablet formulations (Thioctacid HR) were considered test treatments, while the OS (Thioctacid, 600 T) served as the reference treatment. Serial blood samples were collected over 8 hours postdose to quantify R-(+)- and S-(–)-ALA enantiomer plasma concentrations for the PK evaluation. The maximum observed plasma concentration (Cmax) and total exposure (area under the curve [AUC]0–t) were compared between treatments by analysis of variance. Weight-normalized Cmax and the AUC data of male and female study subjects were applied to examine the presence of sex effects. All treatments displayed rapid absorption of both enantiomers with median time to maximum concentration (tmax) values ranging from 0.33–0.5 hours. The Frel of all tablet formulations was comparable, with R-(+)-enantiomer Cmax test/reference ratios ranging from 36% (T 600) to 43% (T 200), and R-(+)-enantiomer AUC test/reference ratios ranging from 64% (T 600) to 79% (T 300), indicating a favorable Frel of all tablet formulations, especially in terms of the total extent of absorption (AUC). An examination of weight-normalized female/male Cmax and AUC sex ratios for both ALA enantiomers indicated the absence of a significant sex effect for Cmax, as well as 20%–26% and 25%–32% higher R-(+)- and S-(–)-ALA enantiomer AUC outcomes in females when compared to males. The observed modest sex effect was comparable for both ALA enantiomers and across all formulations, and it did not appear to require a dose adjustment in clinical practice.
Keywords: alpha-lipoic acid, thioctic acid, enantiomers, sex effect, formulation effect, bioavailability
Introduction
Alpha-lipoic acid (ALA) (synonymously also referred to as thioctic acid) is a well-established treatment for neuropathic symptoms and deficits in type 2 diabetic patients with distal symmetric polyneuropathy (DSP). Oral treatment with 600 mg of ALA (Thioctacid® 600 HR; MEDA Pharma GmbH & Co KG, Bad Homburg vor der Höhe, Germany) once daily for 5 weeks improved positive sensory symptoms and neuropathic deficits in diabetic patients with DSP (SYDNEY 2 trial).1 In addition, the long-term efficacy and safety of once-daily 600 mg ALA (Thioctacid 600 HR) over 4 years was established by the NATHAN 1 trial2 in patients with mild-to-moderate diabetic distal symmetric sensorimotor polyneuropathy.
ALA exists in the form of two enantiomers: R-(+)-lipoic acid (R-(+)-ALA); and S-(–)-lipoic acid (S-(–)-ALA). The R-(+)-enantiomer is the naturally occurring form of ALA and acts as an essential cofactor of the mitochondrial pyruvate dehydrogenase multienzyme complex.3,4 The neuroprotective properties of ALA in diabetes are considered to result from a variety of mechanisms, including protection against exaggerated oxidative stress, restoration of glutathione levels, improvement of insulin-stimulated glucose disposal, enhancement of nerve blood flow, and protection against protein glycation.5
The enantiomer-selective human pharmacokinetics (PK) of intravenously administered and various oral dosage forms of racemic ALA have been extensively studied by our group in healthy adult subjects and special populations.6–8 Also, food- and drug-interaction studies were accomplished.9,10 In addition, ALA plasma kinetics, metabolism, and urinary excretion were studied in healthy adult subjects, patients with severe renal impairment, and those with end-stage kidney disease requiring hemodialysis using a nonenantiomer-selective high-performance liquid chromatography assay with electrochemical detection.11–13
All of these PK studies employing oral solid ALA dosage forms were conducted with historical products from the same manufacturer (Thioctacid film-coated tablets; ASTA Medica AG, Frankfurt, Germany), which are no longer available on the market, while the pivotal clinical efficacy studies were conducted with a successor formulation (Thioctacid 600 HR tablets) with improved biopharmaceutical performance characteristics with substantially reduced intersubject and interoccasion variability in terms of the rate and extent of absorption.1,2,14
Since the PK data of the current ALA formulation have not been published as yet in detail, it is one aim of this paper to present the respective enantiomer-selective PK and bioavailability data of the currently available 200 mg, 300 mg, and 600 mg ALA tablet formulations. These data have been established by a comparative bioavailability study conducted by our group at a single site in Frankfurt, Germany, from August–September 1996.
As the effects of the intrinsic factors, sex and body weight (BW), on the disposition of ALA enantiomers have not been formally studied as yet, this paper also evaluates the effects of these covariates on the bioavailability and PK of ALA enantiomers.
Methods
Ethical conduct of the study
The study protocol, subject information, and informed consent were reviewed and approved by the Ethics Committee of the General Medical Council of Hesse, Germany, and submitted to the competent authorities (Federal Institute for Drugs and Medical Devices [German BfArM]). The study was conducted in accordance with the principles of the International Conference on Harmonisation’s good clinical practice guidelines and the revised Declaration of Helsinki of the World Medical Assembly, which was current at that time. All subjects gave their written informed consent after having received ample information about the product characteristics, the study plan and procedures, as well as the overall scope, meaning, and consequences of the trial.
Study design
This was an open-label, randomized, four-period, and four-sequence crossover trial evaluating the enantiomer-selective PK and relative oral bioavailability of 200 mg, 300 mg, and 600 mg of racemic ALA immediate-release tablets (Thioctacid HR film-coated tablets; MEDA Pharma GmbH & Co KG) in comparison to an orally administered racemic ALA solution (OS) (Thioctacid 600 T, 24 mL ampoule containing 600 mg of ALA; MEDA Pharma GmbH & Co KG). All treatments were administered after overnight fasts of at least 10 hours with oral 600 mg ALA single doses, as follows: T 200 =3×200 mg tablets (Thioctacid 200 HR; batch number 9602-046/02); T 300 =2×300 mg tablets (Thioctacid 300 HR; batch number 9602-087/02); T 600 =1×600 mg tablet (Thioctacid 600 HR; batch number 6902-030/02); and OS =1×600 mg orally administered solution (Thioctacid 600 T, 24 mL ampoule; batch number 025003). The tablet treatments were administered with 200 mL of tap water. The OS treatment (24 mL) was metered with a syringe, transferred into a water glass, and diluted with 76 mL of water to a total volume of 100 mL. Following the administration of the OS, the water glass was again filled with another 100 mL of water to be taken by the subjects. After administration of the treatments, the subjects continued fasting for another 4 hours. At each treatment period, subjects were hospitalized for about 28 hours – ie, from late afternoon on the pretreatment days (ie, day –1) until 12 hours postdose on treatment days (day 1). During hospitalization, subjects received standardized meals and beverages. Between treatments, wash-out periods of at least 6 days were observed. A final end-of-study visit was accomplished 1–10 days after the conclusion of the last study period.
Subjects
Male and female healthy adult subjects of any ethnic origin underwent a thorough evaluation of their medical and surgical history, a full physical examination including vital signs, electrocardiogram (ECG), evaluation of biochemistry and hematology, hepatitis and HIV serology, and an illicit drug urine screen. Subjects were required to be in good physical and mental health with an uneventful medical history, normal clinical blood and urine laboratory tests, vital signs (blood pressure and pulse rate), ECG findings without relevant abnormalities, aged 18–45 years (inclusive), have a BW within ±20% of predicted normal (ie, Broca Index: 0.8–1.2), as well as be willing and able to provide written informed consent.
Subjects were not eligible for enrollment if they had any clinically relevant abnormal physical findings that could interfere with the objectives of the trial; if they demonstrated legal incapacity or limited legal capacity; an anticipated lack of compliance with the study requirements; a history or current evidence of allergy/idiosyncrasy (ascertained or presumptive) to the active drug substance and/or any of the excipients, or a history of anaphylaxis to drugs or foods; any clinical chemical, hematological, or other laboratory parameters that were clinically relevant outside the normal range; any clinically significant cardiovascular, renal, and hepatic disease, or any significant respiratory, endocrine, ophthalmologic, neurologic, or psychiatric illness; any surgical or medical condition that could interfere with the absorption, metabolism, or excretion of drugs; any chronic or clinically relevant acute infections; any history or evidence of substance abuse or addiction; or were pregnant or breastfeeding.
Subjects were also excluded if they required or used any prescription or over-the-counter medication within 2 weeks of study initiation; reported the intake of microsomal enzyme inhibitors/inducers within 2 months before trial participation, including hormonal contraceptives; smoked more than 15 cigarettes or equivalent per day; reported excessive consumption of caffeine-containing/xanthine-containing drinks or foods; tested positive for serum b-human chorionic gonadotropin (pregnancy test), hepatitis B surface antigen, antihepatitis B core antibody, antihepatitis C virus, or HIV antibodies (anti-HIV 1 and 2); donated blood (≥400 mL) within 2 months prior to dosing; or participated in clinical trials with investigational medicinal products within 2 months prior to the administration of the study medication. If deemed suitable, subjects were enrolled and randomized into the study.
Blood sampling, sample processing, and bioanalytical methods
For the quantification of ALA R-(+)- and S-(–)-enantiomer plasma concentrations, a total of 16 serial blood samples were collected predose and at 10 minutes, 20 minutes, 30 minutes, and 40 minutes, as well as at 1 hour, 1.5 hours, 2 hours, 2.5 hours, 3 hours, 3.5 hours, 4 hours, 5 hours, 6 hours, 7 hours, and 8 hours postdose. After blood withdrawal (7.5 mL, NH4-Monovette®; Sarstedt AG & Co, Nümbrecht, Germany), the samples were centrifuged at 4°C for 10 minutes at 3,400× g. The resulting supernatant of the samples was transferred into polypropylene plastic tubes and frozen in the upright position. Samples were stored in a freezer under continuous temperature control at ≤−20°C until analysis.
Plasma concentrations of ALA R-(+)- and S-(–)- enantiomers were determined using enantiomer-selective, reversed-phase, high-performance liquid chromatography with fluorescence detection, as described previously.15 The lower limit of quantification was 15 ng/mL for each ALA enantiomer, while the lower limit of detection was 2 ng/mL for each enantiomer.
PK and statistical analyses
The drug concentration–time data for each subject were analyzed by standard noncompartmental PK methods. The PK parameters were calculated using the Microsoft Excel-based PK-evaluation software FUNCALC (Collection of functions for noncompartmental Excel-based pharmacokinetic evaluations. Version 1997, ASTA Medica AG, Frankfurt).
The following PK parameters were taken directly from the data or were calculated according to standard procedures and algorithms: maximum observed plasma concentration (Cmax); time to maximum observed plasma concentration (tmax); total exposure (area under the plasma concentration–time curve from time zero to the time of the last quantifiable measurable concentration [AUC0–t]); terminal plasma disposition half-life (t1/2); and terminal rate constant determined from the terminal slope of the log-transformed plasma concentration curve using linear regression on the terminal data points of the curve (»z). It should be noted that (»z) could not be determined in all subjects and at all occasions with sufficient confidence, as ALA enantiomer concentrations often declined steeply in a nonlog-linear fashion, thereby resulting in insufficient data points in the terminal log-linear part of the concentration–time curves. For this reason, no AUC data extrapolated to infinity (AUC0–8) were calculated.
The primary PK parameters for the assessment of formulation, sex, and BW effects on the PK of ALA R-(+)- and S-(–)-enantiomers were Cmax and AUC0–t. For the assessment of the relative bioavailability (Frel) of the different ALA tablet strengths (ie, 200 mg, 300 mg, and 600 mg Thioctacid HR tablets), treatments with the orally administered OS was considered the reference treatment.
The statistical analyses of all PK variables were performed using SAS software (version 9.2; SAS Institute Inc., Cary, NC, USA) (mixed-models procedure) and referred to the PK population, which comprised all randomized subjects without major protocol deviations with potential relevance for the PK analyses. Analysis of variance (ANOVA) was applied to the log-transformed PK parameters with treatment, sequence, and period as a fixed effect and subjects within the sequence as a random effect. The effects of formulation, sex, and BW on the exposure of ALA R-(+)- and S-(–)-enantiomers were determined based on the log-transformed maximum (Cmax) and total exposure (AUC0–t) data with two-sided 90% confidence intervals (CI) of the geometric means. The 90% CIs of the ratio of least squares ([test] T/[reference] R) for the log-transformed values of PK parameters were applied to assess relative bioavailability, while the ANOVA-derived 95% CI are given for the primary PK parameter estimates (ie, Cmax and AUC). All other PK parameters of ALA R-(+)- and S-(–)-enantiomers were subject to descriptive analysis. For the purpose of the sex effect analysis, the originally obtained individual AUC and Cmax data were normalized to the BW of a 75 kg subject, according to the following equation:
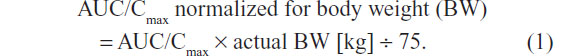
The sample size of the study was calculated for the purpose of product comparison (ie, relative bioavailability in terms of AUC outcomes), not for the purpose of the sex effect analysis, as this was done retrospectively in an exploratory fashion. Sample size calculation was based on a preceding PK study conducted with a total number (N) =16 subjects using a replicate design and the 200 mg tablet formulation, in which the AUC point estimates and 90% CI were about 96% (84%–110%) for both enantiomers (Meda Pharma GmbH & Co KG, unpublished data, 1995). Based on this, it was concluded that for N=24 subjects and an alpha =0.10, a precision factor of 1.112 (ie, $±11%) around the geometric mean AUC outcomes could be expected. Therefore, the chosen sample size of N=24 was considered adequate and sufficient to address the primary study objective.
Safety and tolerability assessments
Safety and tolerability assessments included monitoring of systolic/diastolic blood pressure, pulse rate (each study period up to 8 hours postdose), ECG, safety laboratory (screening and end-of-study examinations), and the subjects’ well-being in terms of the nature, severity, and incidence of adverse event (AEs) throughout the entire study period.
Results
Subject demographics and disposition
A total of 24 healthy male and female adult subjects (12 males, 12 females), aged 24–43 years, mean ± standard deviation (SD) of 31.8±6.8 years, with a mean ± SD BW of 70.0±10.2 kg, and a mean ± SD Broca Index of 1.11±0.13, were enrolled and randomized to treatment. The mean ± SD age and BW of the female participants was 32.6±3.5 years and 65.8±7.6 kg, respectively, while the mean ± SD age and BW of the male participants was 31.0±5.3 years and 74.3±10.9 kg, respectively. All subjects completed the study according to protocol and were eligible for PK evaluation.
PK and relative bioavailability
Predose plasma concentrations of both ALA enantiomers were below the limit of quantitation in all periods and in all subjects – a finding that is consistent with earlier reports using the same bioanalytical assay, and which indicates adequate wash-out intervals between study treatments.
The mean plasma concentration–time profiles of both ALA enantiomers after the administration of different dosage forms of 600 mg racemic ALA single doses are displayed in linear scale in Figure 1A and B, respectively. Plasma concentration–time profiles indicate a distinctly higher exposure to the R-(+)-enantiomer as compared to the S-(–)- form, and higher exposure levels for the OS treatment as compared to the tablet formulations.
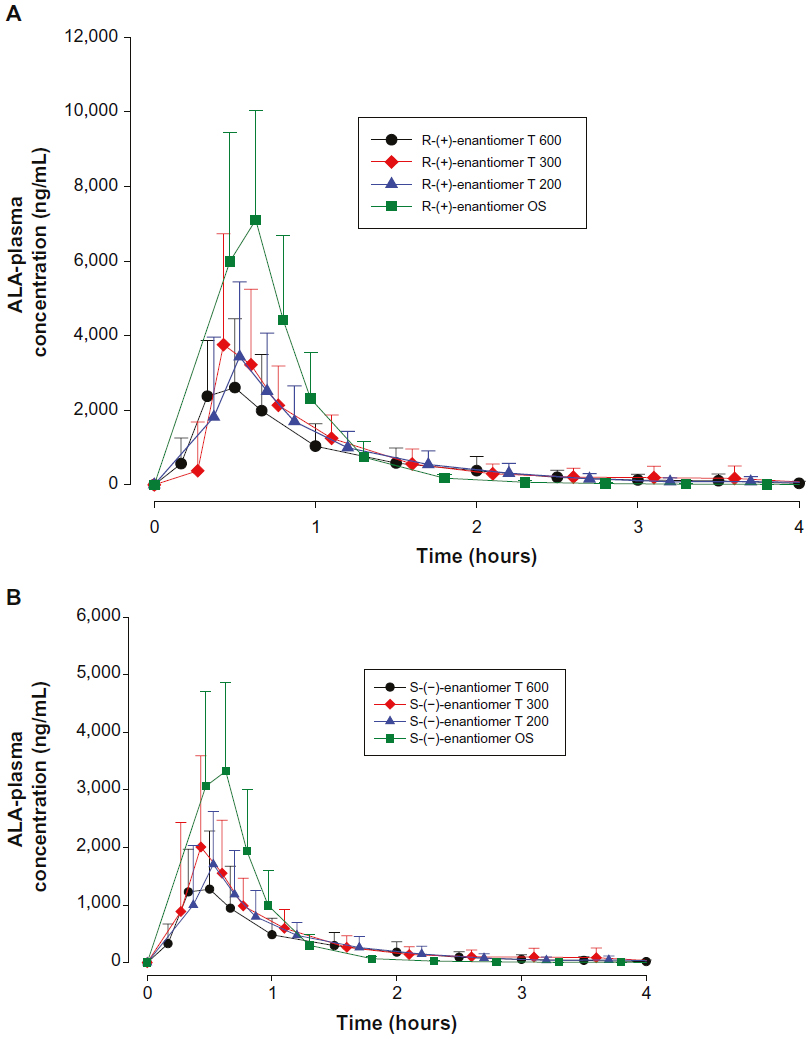 | Figure 1 ALA plasma concentration–time curves by treatment. |
All treatments displayed similar rapid absorption of both enantiomers with median times to observed peak plasma concentrations (tmax) observed at 0.33 hours with the OS (range:0.17–0.50 hours) and the T 200 tablet (range: 0.17–1.0 hours) formulations, and at 0.42/0.33 hours (R-(+)/S-(−)- enantiomers; range: 0.33–3.5 hrs) and 0.5 hours (range: 0.33–1.0 hours) observed with the T 300 and T 600 tablet formulations, respectively.
The geometric means of primary PK parameters (ie, Cmax and AUC0–t) along with 95% CI of both ALA enantiomers are summarized for all treatments in Tables 1 and 2, respectively. As expected, the highest Cmax and AUC values for both ALA enantiomers were observed with the OS, while the respective exposure data of all tablet formulations examined were generally well comparable.
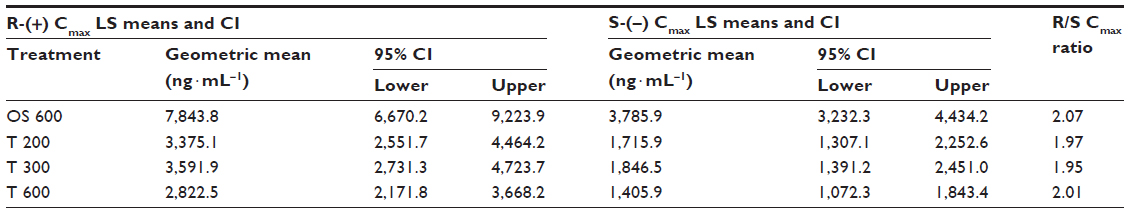 | Table 1 ANOVA-derived maximum observed ALA enantiomer geometric LS mean plasma concentrations (Cmax) and the corresponding 95% CI by treatment |
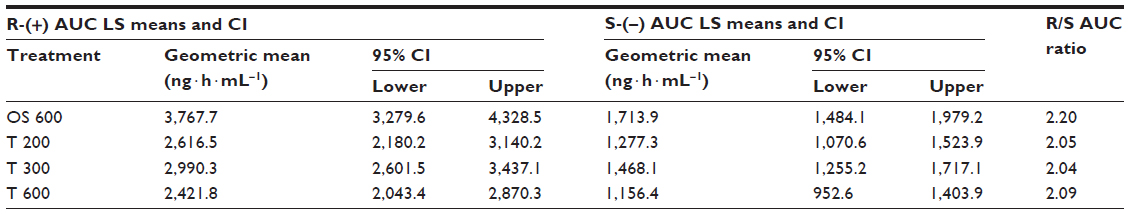 | Table 2 ANOVA-derived ALA enantiomer geometric LS mean total exposure data (AUC0–t) and the corresponding 95% CI by treatment |
The geometric mean Cmax and AUC R/S enantiomer ratios indicated a twofold superior bioavailability of the R-(+)-enantiomer in terms of rate and the extent of absorption, as compared to the S-(–)-enantiomer, respectively (Tables 1 and 2). Notably the R/S ratio is highly consistent for both Cmax and AUC data (ie, it is formulation independent).
The bioavailability data for the maximum (Cmax) and total (AUC0–t) exposure outcomes of all tablet formulations relative to the OS treatment (ie, test [T]/reference [R] treatment ratios) are summarized in Tables 3 and 4, respectively. The relative bioavailability of all solid oral dosage forms is generally well comparable, with R-(+)-enantiomer Cmax treatment ratios ranging between 36% (T 600) to 43% (T 200), and R-(+)-enantiomer AUC treatment ratios ranging between 64% (T 600) and 79% (T 300), thereby indicating favorable relative bioavailability of all tablet formulations, especially in terms of total exposure (ie, AUC).
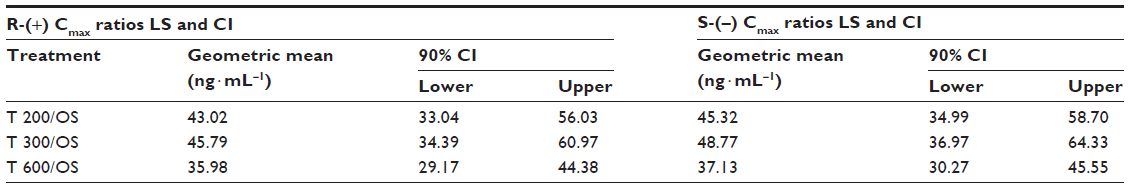 | Table 3 Test/reference geometric LS mean maximum exposure (Cmax) treatment ratios and 90% CI of R-(+)- and S-(−)-ALA enantiomers by treatment |
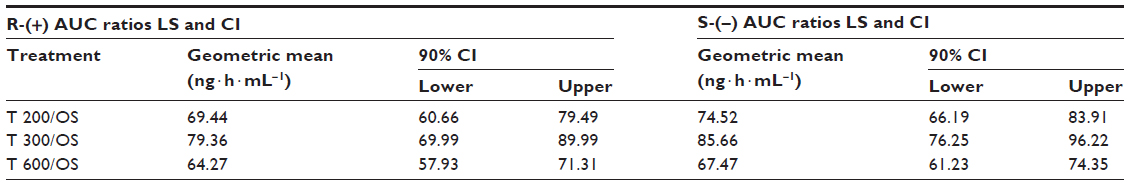 | Table 4 Test/reference geometric LS mean total exposure (AUC0–t) treatment ratios and 90% CI of R-(+)- and S-(−)-ALA enantiomers by treatment. |
While the R-(+)- and S-(–)-enantiomer T/R ratios were similar for the Cmax data (Table 3), the relative bioavailability of the tablet formulations tended to be modestly higher for the S-(–)-enantiomer (as compared to the R-(+)-form), when the AUC data are considered (Table 4).
Sex effects
Figure 2 indicates, as exemplified by the 600 mg tablet formulation data, that at corresponding times, both R-(+)- and S-(–)-ALA enantiomer plasma concentrations were consistently higher in females than in males. This finding was generally applicable and formulation independent.
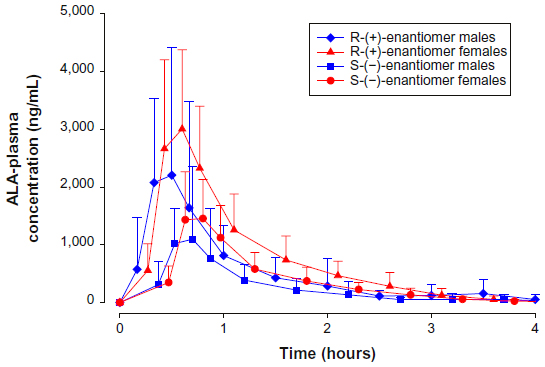 | Figure 2 ALA plasma concentration–time curves by sex. |
Geometric means and 95% CIs of primary PK parameters of both ALA enantiomers are summarized by sex in Tables 5 and 6 (Cmax data of R-(+)- and S-(–)-ALA), and Tables 7 and 8 (AUC data of R-(+)- and S-(–)-ALA), respectively.
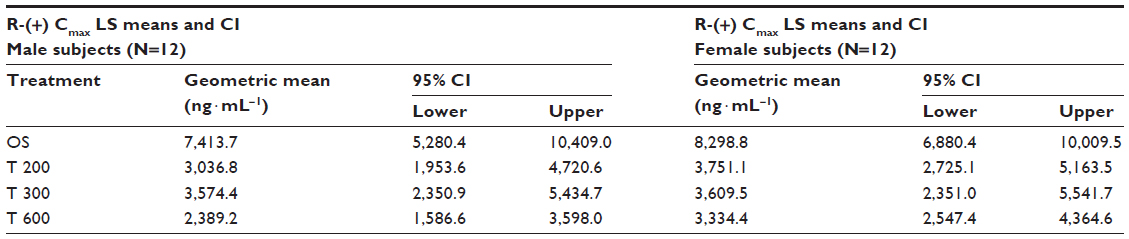 | Table 5 ANOVA-derived maximum observed R-(+)-ALA enantiomer geometric LS mean plasma concentrations (Cmax) and the corresponding 95% CI by sex (12 males/12 females) and treatment |
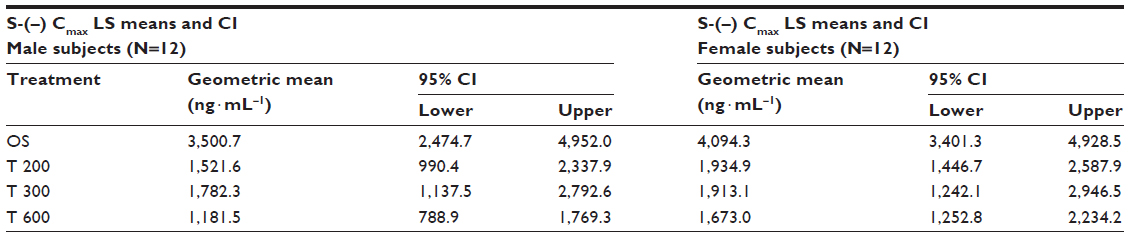 | Table 6 ANOVA-derived maximum observed S-(-)-ALA enantiomer geometric LS mean plasma concentrations (Cmax) and the corresponding 95% CI by sex (12 males/12 females) and treatment |
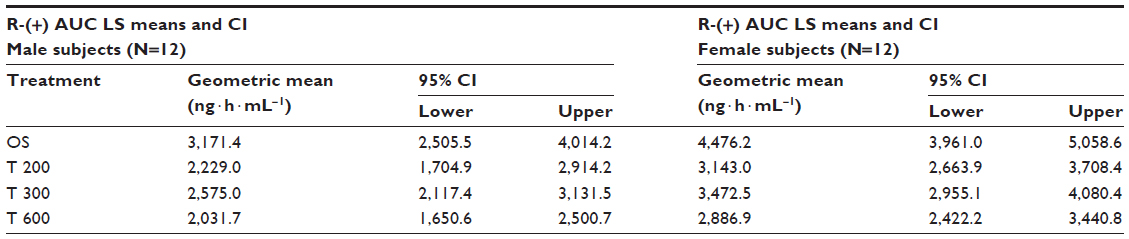 | Table 7 ANOVA-derived R-(+)-ALA enantiomer geometric LS mean total exposure data (AUC0–t) and the corresponding 95% CI by sex (12 males/12 females) and treatment |
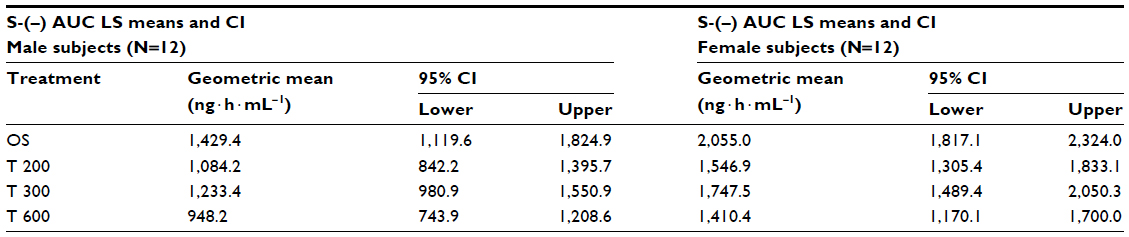 | Table 8 ANOVA-derived S-(–)-ALA enantiomer geometric LS mean total exposure data (AUC0–t) and the corresponding 95% CI by sex (12 males/12 females) and treatment |
The female/male exposure ratios for the maximum (Cmax) and total (AUC0–t) exposure outcomes of all formulations are given in Tables 9 (Cmax data) and 10 (AUC data), respectively. For Cmax, only a modest, nonsignificant trend toward higher maximum exposure levels in female subjects appears notable, albeit with quantitatively different outcomes observed for the different treatments (Table 9). When data from all treatments are considered together, the female/male Cmax sex ratio amounts to 110.47% (CI: 89.94–135.68) for R-(+)-ALA and to 115.33% (CI: 92.48–143.83 for S-(–)-ALA. Since the 90% CIs for both ALA enantiomers include unity, these numerical differences are not considered indicative of statistically significant differences.
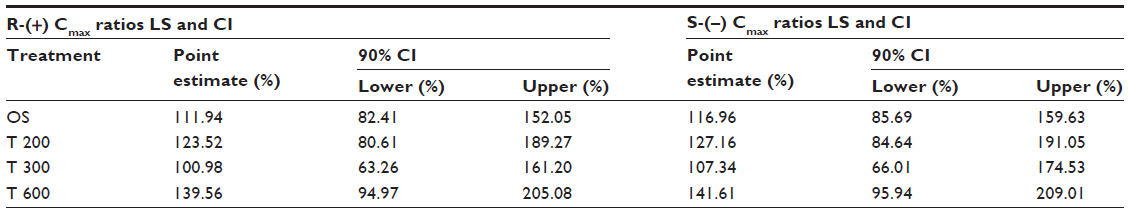 | Table 9 Female/male geometric LS mean maximum exposure (Cmax) ratios and the 90% CI of R-(+)- and S-(−)-ALA enantiomers by treatment |
The overall outcome was much more distinct and consistent for the total exposure data, indicating a significant and formulation-independent sex effect in terms of $40% higher AUC values in females as compared to males. The observed sex effect in terms of total exposure was quantitatively similar for all formulations and for both ALA enantiomers (Table 10).
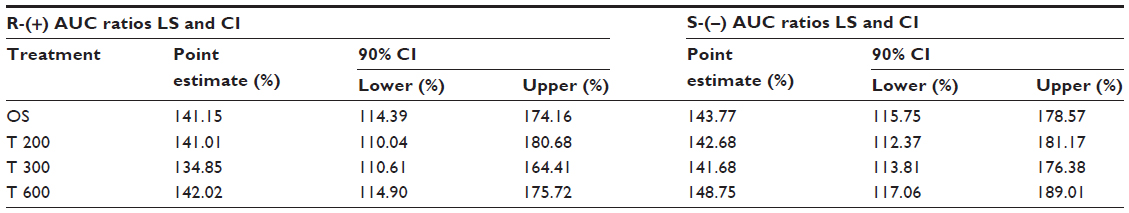 | Table 10 Female/male geometric LS mean total exposure (AUC0–t) ratios and the 90% CI of R-(+)- and S-(−)-ALA enantiomers by treatment |
The BW-normalized AUC data are displayed in Table 11. (The BW-normalized analysis of Cmax data is not shown, as the original nonnormalized data did not show a significant sex effect.) The BW-normalized data indicated that the differences in BW may not solely explain the observed sex differences in total R-(+)- and S-(–)-ALA enantiomer exposure. BW-normalized point estimates indicate 20%–26% and a 25%–32% higher R-(+)- and S-(–)-ALA enantiomer exposure in females as compared to males, respectively. The observed BW-normalized sex effect was well comparable across all formulations, although the sex difference remained statistically significant only for the OS and 600 mg tablet treatments when the range of the BW-normalized 90% CI is considered (Table 11).
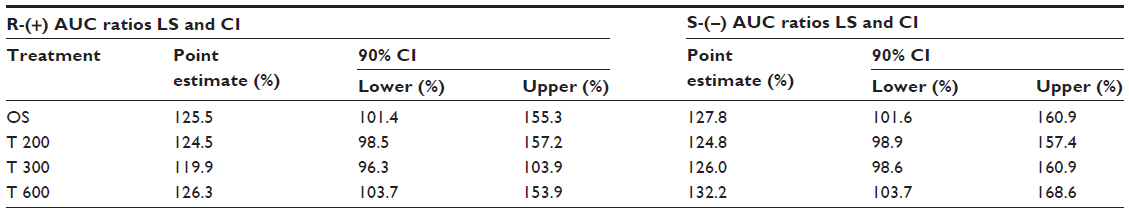 | Table 11 Body weight-normalized sex effect analysis |
Safety and tolerability
There were no serious or unexpected adverse events (AEs) noted throughout the study, and there were no premature study discontinuations. The repeated administration of racemic 600 mg ALA single doses at four different occasions was generally well tolerated. A total of seven out of 24 subjects reported at least one AE. In total, N=25 AEs were reported throughout the study with the following frequencies by treatment: OS, four of 24; T 200, four of 24; T 300, eight of 24; and T 600, nine of 24. The most frequently reported single AE was headache (N=16), followed by fatigue (N=2). These were also the only AEs that were reported more than once. All AEs were of mild intensity, except headache in one subject after treatment with T 300, which was rated as being of “severe” intensity. All AEs were of a transient nature and fully recovered spontaneously without sequelae.
Discussion
The present PK/bioavailability study conducted in healthy male and female subjects formed the scientific and regulatory basis for the development, registration, and marketing of racemic ALA tablet formulations with improved biopharmaceutical performance characteristics (ie, Thioctacid HR 200 mg, 300 mg, and 600 mg tablets) as compared to the historical forerunner products (Thioctacid 50 mg and 200 mg tablets).
The publication of these data fills an important gap in the published evidence on the human PK and bioavailability characteristics of currently marketed racemic ALA formulations, as the entire available evidence in the public domain on the enantioselective PK of racemic ALA has been established with the historical forerunner products, which have been replaced by the currently available formulations because of their nonoptimal absorption characteristics.14 In contrast, however, the best clinical outcome data have been generated with the current 600 mg ALA product, thereby resulting in missing information on the exposure–response characteristics of racemic ALA (ie, exposure levels required to confer clinical efficacy) in the treatment of diabetic patients with DSP.1,2
Across study comparisons of PK and bioavailability performance characteristics of the current products with those of the historical tablet formulation (ie, a comparison of the 200 mg tablet formulations, dose-normalized to 600 mg) using a well-comparable study design and population, as well as the same bioanalytical assay15 (as published by our group6), indicates two- to threefold improvements in the rate of absorption (ie, tmax), as well as about twofold improvements in the maximum (ie, Cmax) and total exposure (ie, AUC) characteristics of both ALA enantiomers, which were achieved with the currently available ALA successor products.
The presented study results of the approximately twofold higher bioavailability of the natural R-(+)-enantiomer, as compared to the S-(–)- form, essentially confirmed the existing evidence from prior studies employing the same enantiomer-selective ALA assay.6–10 Notably, the present data also indicated that the different PK and bioavailability characteristics of the ALA enantiomers are similar for all of the formulations investigated (ie, they were formulation independent). One mechanistic background might be the existence of active transporter-mediated intestinal uptake of the natural R-(+)- form, which might not be available (or similarly effective) for the S-(–)-enantiomer. However, this hypothesis needs to be examined in dedicated studies.
A sex effect in terms of a higher maximum, total exposure, and the oral bioavailability of racemic ALA in female individuals has been suggested previously based on a pooled analysis of a number of clinical PK studies.16 However, this information is available in abstract form only, and it neither provided methodological details nor precise quantitative outcomes. Hence, it is not known, as yet, whether the described sex effect was solely based on the BW differences between the male and female subjects, whether it may have another contributing mechanistic background, and whether the effect size would be of clinical meaning in terms of dosing requirements. We aimed, therefore, to take advantage of the sex-balanced population and the reasonable sample size of the present study by applying a dedicated sex analysis on the original and the BW-normalized maximum, as well as on the total ALA enantiomer exposure data. Essentially, the analysis confirmed the existence of a significant and robust sex effect (ie, on average, about 40% increased AUC outcomes in female subjects as compared to males) for the total ALA exposure (both enantiomers; Table 10), while modestly increased maximum exposure outcomes in the group of female subjects did not achieve statistical significance (Table 9). Further analysis of BW-normalized AUC data indicated a remaining 20%–26% and 25%–32% higher R-(+)- and S-(–)-ALA enantiomer exposure in females as compared to males, which could not be explained by sex differences in BW and is, therefore, likely to have another sex-specific mechanistic background. In this respect, a comparison with PK data from earlier studies employing the intravenous administration of racemic ALA (data on file; MEDA Pharma GmbH & Co KG) indicates that there are no sex-specific differences, neither with regard to the total ALA enantiomer clearance, nor in the apparent volume of distribution. In light of this evidence, the observed exposure differences between females and males may be explained mechanistically only by a higher relative bioavailability of orally administered ALA in women, putatively by a less-pronounced first-pass effect. In principle, this effect can mechanistically either be based upon varying capacities/activities of presystemic (ie, intestinal) or hepatic enzymes, or it may be based on differences with regard to the abundance/activity of intestinal uptake or efflux transporters. However, dedicated studies are required to shed further light on the mechanistic basis of the observed sex effect.
Notably, the sex effect observed in the present study was quantitatively well comparable for all formulations examined (ie, it was formulation independent). The overall effect size of $20%–30% in terms of a higher total exposure in female subjects does not appear to be of clinical relevance in therapeutic practice, in view of the well-established favorable safety profile and the wide therapeutic index of oral ALA treatment. Hence, any dose adjustment considerations based on the intrinsic sex factor alone do not appear warranted.
Disclosure
The study was sponsored by ASTA Medica AG, Frankfurt, a predecessor company of MEDA Pharma GmbH & Co KG, 61352 Bad Homburg, Germany. The authors report no other conflicts of interest in this work.
References
Ziegler D, Ametov A, Barinov A, et al. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care. 2006;29(11):2365–2370. | |
Ziegler D, Low PA, Litchy WJ, et al. Efficacy and safety of antioxidant treatment with α-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care. 2011;34(9):2054–2060. | |
Packer L, Witt EH, Tritschler HJ. alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med. 1995;19(2):227–250. | |
Biewenga G, Vriesmann MF, Haenen GRMM, Bast A. The identification of a new metabolite of lipoic acid in man: 3-ketolipoic acid. In: Biewenga G. Lipoic Acid – A Pharmacochemical Study [thesis]. Amsterdam, the Netherlands: Vrije Universiteit; 1997:137–152. | |
Rochette L, Ghibu S, Richard C, Zeller M, Cottin Y, Vergely C. Direct and indirect antioxidant properties of α-lipoic acid and therapeutic potential. Mol Nutr Food Res. 2013;57(1):114–125. | |
Hermann R, Niebch G, Borbe HO, et al. Enantioselective pharmacokinetics and bioavailability of different racemic α-lipoic acid formulations in healthy volunteers. Eur J Pharm Sci. 1996;4:167–174. | |
Hermann R, Wildgrube HJ, Ruus P, Niebch G, Nowak H, Gleiter CH. Gastric emptying in patients with insulin dependent diabetes mellitus and bioavailability of thioctic acid-enantiomers. Eur J Pharm Sci. 1998;6(1):27–37. | |
Breithaupt-Grögler K, Niebch G, Schneider E, et al. Dose-proportionality of oral thioctic acid – coincidence of assessments via pooled plasma and individual data. Eur J Pharm Sci. 1999;8(1):57–65. | |
Gleiter CH, Schug BS, Hermann R, Elze M, Blume HH, Gundert-Remy U. Influence of food intake on the bioavailability of thioctic acid enantiomers. Eur J Clin Pharmacol. 1996;50(6):513–514. | |
Gleiter CH, Schreeb KH, Freudenthaler S, et al. Lack of interaction between thioctic acid, glibenclamide and acarbose. Br J Clin Pharmacol. 1999;48(6):819–825. | |
Teichert J, Preiss R. High-performance liquid chromatographic assay for alpha-lipoic acid and five of its metabolites in human plasma and urine. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;769(2):269–281. | |
Teichert J, Hermann R, Ruus P, Preiss R. Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers. J Clin Pharmacol. 2003;43(11):1257–1267. | |
Teichert J, Tuemmers T, Achenbach H, et al. Pharmacokinetics of alpha-lipoic acid in subjects with severe kidney damage and end-stage renal disease. J Clin Pharmacol. 2005;45(3):313–328. | |
Böhm S (Editor). What can oral thioctic therapy accomplish? Steadier plasma levels of thioctic acid with the help of a new oral formulation. MMW Special. Münch med Wschr. 1999;141(Suppl):1–5. | |
Niebch G, Büchele B, Blome J, et al. Enantioselective high-performance liquid chromatography assay of (+)R- and (−)S-alpha-lipoic acid in human plasma. Chirality. 1997;9(1):32–36. | |
Schug BS, Hermann R, Blume HH, Elze M. Gender effect on the pharmacokinetics of thioctic acid: An evaluation of several bioavailability studies. Naunyn–Schmiedebergs Archives of Pharmacology. 1998; 357(Suppl 4):R4–R4. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.