Back to Archived Journals » Cell Health and Cytoskeleton » Volume 8
Emerging perspectives on laminopathies
Authors Lattanzi G, Benedetti S, D'Apice MR, Maggi L, Carboni N, Scarano E, Politano L
Received 30 September 2015
Accepted for publication 13 January 2016
Published 7 April 2016 Volume 2016:8 Pages 25—35
DOI https://doi.org/10.2147/CHC.S59507
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Denis Wirtz
Giovanna Lattanzi,1,2 Sara Benedetti,3 Maria Rosaria D’Apice,4 Lorenzo Maggi,5 Nicola Carboni,6 Emanuela Scarano,7 Luisa Politano8
1National Research Council of Italy, Institute for Molecular Genetics (CNR-IGM), Unit of Bologna, 2Rizzoli Orthopedic Institute, Laboratory of Musculoskeletal Cell Biology, Bologna, 3Laboratory of Clinical Molecular Biology and Cytogenetics, San Raffaele Scientific Institute, Milan, 4Fondazione Policlinico Tor Vergata, Rome, 5Neuromuscular Diseases and Neuroimmunology Unit, IRCCS Neurological Institute C Besta, Milan, 6Division of Neurology, Hospital San Francesco, Nuoro, 7Pediatric Endocrinology and Rare Diseases Unit, Department of Pediatrics, S Orsola-Malpighi University Hospital, University of Bologna, Bologna, 8Department of Experimental Medicine, Cardiomyology and Medical Genetics, Second University of Naples, Naples, Italy
Abstract: Laminopathies are a group of inherited disorders caused by mutations in the lamin A/C gene, and can affect diverse organs or tissues, or can be systemic, causing premature aging. In the present review, we report on the composition and structure of the nuclear lamina and the role of lamins in nuclear mechanics and their involvement in human diseases, and provide some examples of laminopathies and current therapeutic approaches.
Keywords: lamin A/C, emerin, laminopathies, Emery–Dreifuss muscular dystrophy, Hutchinson–Gilford progeria
Introduction
In eukaryotes, chromosomes are confined to the nucleus, whose perimeter is defined by the nuclear envelope (NE), composed of a double membrane: the outer nuclear membrane, continuous with the endoplasmic reticulum and containing many proteins; and the inner nuclear membrane, containing a distinct set of proteins, some of which interact with chromatin. Both membranes are fused at sites of nuclear pore complexes, macromolecular structures allowing selective passage of proteins, RNA, and other molecules to and from the nucleus.1 In most eukaryotes, underlying the inner nuclear membrane is a network of proteins made of lamins and lamin-associated proteins. Lamins are ubiquitous nuclear intermediate-filament proteins that form a scaffold, termed nuclear lamina, at the nuclear periphery. A small fraction of lamins also localize throughout the nucleoplasm. This network provides nuclear stability, helps connect the nucleus to the cytoskeleton, and contributes to modulating chromatin organization, gene regulation, genome stability, and cellular differentiation.2,3
Since the discovery that Emery–Dreifuss muscular dystrophy (EDMD) was caused by mutations in the EMD gene encoding a nuclear protein,4 considerable interest has been focused on the NE in the last 15 years, following the realization that several human diseases are linked to defects in genes encoding nuclear-specific proteins. These diseases are now comprehensively described as laminopathies. Recent findings suggest that lamin mutations decrease nuclear stability, increase nuclear fragility, and disturb mechanotransduction signaling, possibly explaining the specific defects of many laminopathies.3,5 Moreover, lamins have been recently involved in stress response.6
In this review, we focus on the role of lamins in nuclear mechanics and their involvement in human diseases, and report some examples of laminopathies and current therapeutic prospects.
The nuclear envelope
A- and B-type lamins are located in the nuclear lamina, beneath the inner nuclear membrane, where they form a meshwork of intermediate filaments interacting with chromatin on the nucleoplasmic side and with nuclear membrane and cytoskeleton linkers on the cytoplasmic side. A-type lamins include the four splicing products of the LMNA gene: lamin A and C, the most abundant isoforms, lamin C2, and lamin AΔ10.7 B-type lamins are lamin B1, encoded by the LMNB1 gene, and lamin B2 and B3, alternative splicing products of the LMNB2 gene.7 Although lamins are considered ubiquitous proteins, it has been demonstrated that diverse expression levels and different isoforms can be found in different cell types and organs. For instance, lamin A/C is expressed in monocytes upon stimulus, and lamin A is downregulated in brain by microRNA-9-mediated silencing.8,9 Both A- and B-type lamins are transcribed as precursor proteins (prelamins) that undergo farnesylation at their C-terminal CaaX box, subsequent cleavage, and carboxymethylation. B-type lamins remain permanently farnesylated, while mature lamin A is devoid of the farnesylated residue.10 The farnesylation state avoids formation of mixed networks of A- and B-type lamins and influences their interaction with NE constituents, chromatin-associated proteins, and transcription factors.10–14 Moreover, lamins undergo phosphorylation and SUMOylation, further regulating their own intermolecular interactions. The best-recognized binding partner of lamin A/C at the NE is emerin, while the best-known partner of B-type lamins is the lamin B receptor (LBR).7 Both emerin and LBR mediate lamin interplay with chromatin, either through interaction with BAF15 and other LEM-domain proteins,16 such as LAP2α, or by direct recruitment of chromatin domains.16,17 The lamin–chromatin interface has been getting better defined at the molecular level in recent years, and dynamic lamina-associated chromatin domains known as LADs have been identified.17 Regulation of chromatin in response to diverse cellular and extracellular stimuli appears to be the key function of lamins under physiological conditions. More recently, the role of lamin in physiological processes has been elucidated. Indeed, lamins are required for muscle cell differentiation, nuclear and cellular migration, stem cell homeostasis, and even formation of immunological synapses. In all these contexts, lamins are involved in stress response and aging mechanisms through their role as sensors and chromatin regulators. Lamins and mostly prelamin A act as sensors of stress to modify chromatin arrangement in a way that facilitates repair of damaged DNA. In this context, the reported euchromatization of nuclei induced by prelamin A accumulation not only makes accessible DNA sites able to be repaired but favors recruitment of DNA-repair factors, such as 53BP1, into the nucleus.18 In addition, lamins play key roles in cellular differentiation. Lamin A, prelamin A, and lamin B1 anchor transcription factors at the nuclear periphery, thus regulating their nuclear import. This is particularly relevant for Oct1 and SREBP1, two transcription factors regulating adipogenesis, a process impaired in most lamin-linked diseases. Moreover, lamin A and prelamin A regulate nuclear positioning through functional interaction with SUN1 and nesprins, key constituents of the LINC complex connecting the nucleoskeleton to the cytoskeleton. The relevance of the latter function is highlighted by the effects elicited in muscle by either lamin A/C or SUN1 mutations,19 which lead to myonuclear clustering and aberrant distribution of myonuclear domains.
Finally, lamins regulate cellular signaling, at least to control their own expression levels. This occurs through modulation of TGFβ2 levels, which in turn affects AKT1 activity required for lamin A degradation.20,21 A general dysregulation of TGFβ signaling has been reported in muscular laminopathies, hinting at a major role of lamin A in this cellular pathway in muscle cells.21
Molecular basis of and pathways in nuclear lamin dysfunction
Alterations of the NE have been associated with several disorders, including autosomal-dominant forms of EDMD, dilated cardiomyopathy (DCM) with conduction-system defects, limb-girdle MD 1B (LGMD1B) with atrioventricular conduction disturbances, Dunnigan-type familial partial lipodystrophy, mandibuloacral dysplasia (MAD), autosomal-recessive (AR) forms of axonal Charcot–Marie–Tooth (ARCMT2, CMT2B), and Hutchinson-Gilford Progeria Syndrome. To date, more than 450 LMNA mutations have been reported in locus-specific databases (http://www.umd.be/LMNA/, http://www.dmd.nl).
Besides lamins, mutations in other genes coding for NE proteins (Table 1) have been implicated in neuromuscular disorders: 1) emerin, encoded by STA/EMD associated with X-linked EDMD; 2) SYNE1 and SYNE2, coding for nesprin 1 and 2, which interact with the nucleoplasm through SUN proteins and lamins, associated with MD and cardiomyopathy;22 3) TMPO, coding for the inner nuclear membrane integral protein LAP2α in DCM;23 4) TOR1AIP1, coding for LAP1B in MD and DCM;24 and 5) TMEM43, coding for LUMA, another nuclear inner-membrane protein, in EDMD-like patients.25
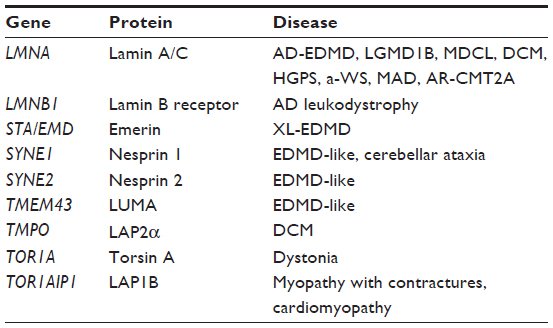 | Table 1 Laminopathies and related genes |
In contrast, loss of function of the LMNB1 gene has not been reported in any disease, while its duplication has been associated with an autosomal-dominant form of leukodystrophy, a progressive neurological disorder.26 The TOR1A gene has been implicated in dystonia.27
Different, probably not mutually exclusive, pathogenic hypotheses have been proposed. The “mechanical stress” hypothesis (the oldest) posits that mutations in lamin A/C or proteins participating in the LINC complex would lead to more fragile nuclei and weaken nucleus anchorage to the cytoskeleton. This follows reduced nuclear stability, especially in tissues subjected to constant mechanical stress, such as muscles.28 Indeed, recent discoveries have focused on key interactions between A-type lamins and stress-response pathways.29 Recently, it has been reported that LMNA-mutated cells present severe defects in their capacity to sense the stiffness of the environment and respond to mechanical stress by inappropriate activation of the YAP transcription factor.30 The altered gene-expression hypothesis is based on the ability of NE proteins, in particular LEM proteins (LAP2, emerin, MAN1), to tether repressive chromatin at the nuclear periphery by binding to BAF DNA-binding protein and to transcription factors. Analysis of N195K LMNA knock-in mice displaying cardiac defects revealed alterations of Ht1b/Sp4 transcription factor and connexin 40 and 43 gap-junction proteins, suggesting a pathogenetic mechanism based on disruption of intercellular junctions, similar to other cardiomyopathies, such as arrhythmogenic right-ventricle dysplasia.31 The alteration of signaling pathway hypothesis is based on an abnormal activation of MAP kinases ERK1/2 and JNK, and has been reported in H222P LMNA knock-in and emerin-mutant mice.32,33 H222P LMNA knock-in mice also activate the AKT pathway, providing possible therapeutic targets based on inhibitors of these signaling pathways, like rapamycin and temsirolimus.34 Similarly, disruption of TGFβ signaling has been reported in LAP2α mutants through alteration of SMAD2 and SMAD3 transcription factors.35
Mutations in the LMNA and in ZMPSTE24 genes, the latter encoding a posttranslational processing enzyme of prelamin A, can cause progeroid laminopathies, a group of disorders with different age of onset, grade of severity and prognosis, and prematurely aged appearance. Prevalent mutations determine altered prelamin A processing, with consequent accumulation of immature nuclear forms, causing alterations in nuclear structure and functional activities.36 Affected cells losing integrity of the nuclear lamina show irregular NE shape, chromatin disorganization, cellular senescence, apoptotic death, changes in epigenetic regulation and gene expression, telomere shortening, genomic instability, and delay of DNA-damage repair.37
Special emphasis has been put on chromatin organization and DNA-repair defects contributing to the pathophysiology of progeroid diseases. Lamins regulate heterochromatin organization through interaction with diverse chromatin-binding factors and proteins of the nuclear membrane. Fibroblasts from progeroid patients or mouse models of progeria exhibit elevated basal levels of γH2AX (a phosphorylated histone marker of unrepaired DNA damage), persistent activation of the DNA-damage response-checkpoint kinases, and higher sensitivity to agents causing double-strand breaks.38,39 The recent availability of high-throughput sequencing technologies has made it feasible to uncover mutations in genes producing other nuclear proteins interacting directly or indirectly with A-type lamins in progeroid syndromes. Patients with Néstor–Guillermo progeria syndrome, a chronic progeria with early onset but slow clinical course, and overlapping clinical features of Hutchinson–Gilford progeria syndrome (HGPS) and MAD had the same homozygous mutation – c.34G>A (p.Ala12Thr) – in the BANF1 gene, encoding BAF conserved DNA-binding protein involved in higher-order chromatin structure and nuclear assembly.40,41 A morphological analysis of mutant fibroblasts revealed profound nuclear abnormalities, including blebs and aberrations previously described in other laminopathies, and significant differences in the subcellular distribution of emerin compared to control cells.41 Recently, several patients have been described with mandibular hypoplasia, deafness, progeroid features, and associated progressive lipodystrophy (MDPL) syndrome, characterized by those who share a recurrent in-frame deletion (c.1812_1814delCTC, p.Ser605del) of a single codon or a missense mutation (c.1519C>T, p.Arg507Cys) in the POLD1 gene, affecting the active polymerase site or exonuclease domain, respectively.42 The DNA polymerase-δ is responsible for DNA synthesis of the lagging strand during DNA replication and cooperation with DNA helicase encoded by the RECQL2 gene to maintain genome stability. Mutations in this gene cause Werner syndrome (WS), an AR premature aging disorder characterized by early onset of aging-associated diseases, chromosomal instability, and cancer predisposition.44 A subset of WS patients are known as atypical cases, 20% of them having missense mutations in the amino-terminal globular domain or in the heptad repeats of the lamin-rod domain.45
Mechanisms and pathogenesis of some examples of laminopathies
LMNA-related myopathies
LMNA-related myopathies (LMs) represent a more consistent subgroup of diseases, due to mutations in the LMNA gene. Three main different phenotypes have been reported, based on distribution of muscle weakness or age at onset: LGMD1B, EDMD2, and a form of congenital MD (congenital, LMNA-related [MDCL]). However, all three may be associated with the same mutation and present in the same family;46,47 in addition, considerable clinical overlap exists among these three phenotypes, suggesting they should be considered a continuum in the clinical spectrum of LMs. Indeed, the heart is involved in all three entities, with similar features.47 Interestingly, cardiomyopathy may precede onset of muscle weakness.
Creatine kinase is usually normal or mildly elevated. Muscle histological findings are usually not specific, including myopathic and sometimes dystrophic features, making muscle biopsy unnecessary for diagnosis in patients with typical clinical features. Muscle magnetic resonance imaging may be helpful in differential diagnosis for other myopathies, eg, LM associated with predominant fatty infiltration of medial gastrocnemius and vasti muscles with relative sparing of the rectus femoris.48 EDMD2 was the first described myopathy clinically characterized by the triad of 1) early ankle, elbow, and spine contractures, 2) scapulohumeroperoneal muscle weakness and wasting, particularly in early stages, and 3) heart involvement presenting in adult life as conduction-system defects associated with a high frequency of sudden cardiac death and DCM.49–55 Muscle weakness usually presents within the beginning of second decade, sometimes preceded by contractures, which may cause difficulties with posture and gait.47,53 Compared to EDMD2, the X-linked form (EDMD1), due to mutations in the STA1 gene, shows humeroperoneal distribution of weakness, contractures as the presenting symptom, less frequent loss of ambulation, and lower heart risk of sustained ventricular tachyarrhythmia and DCM.51 Few cases of X-linked EDMD have been associated with mutations in the FHL1 gene, differing from EDMD1 and EDMD2 for the presence of hypertrophic cardiomyopathy.56
LGMD1B differs from EDMD2 in the distribution of muscle weakness and wasting, being characterized by predominant proximal muscle involvement. However, in later stages of the disease, pelvic muscle weakness may be marked, as in EDMD2 patients. Furthermore, age at onset is later than EDMD2, usually in the third or fourth decade. Of note, tendon retractions, initially considered absent or late in disease course,53 have been recently observed in about two-thirds of LGMD1B patients, sometimes in early disease stages, although elbow contractures appear to be more specific for EDMD2.47 LGMD1B was the most frequent muscle phenotype in a large cohort of Italian LM patients.47 The study confirmed that the natural history of LM appears mainly marked by heart involvement and related complications, while only a minority of patients lost the ability to walk or needed assisted ventilation. Therefore, cardiologic follow-up is strongly recommended in these patients.
More recently, a form of MDCL has been described in patients with congenital onset or presentation within the first 2 years of life.57,58 In particular, two phenotypes have been observed: a severe congenital form with minimal or absent motor development, and a milder and more frequent myopathy characterized by prominent axial weakness, defined as “dropped head syndrome”, after normal acquisition of head control, with preservation of walking ability. MDCL patients may progress both to EDMD2 or LGMD1B.47,58 Respiratory failure is very frequent, whereas cardiac involvement is less common, and when present it is characterized by the presence of arrhythmias. The central nervous system is not involved, except for a single case report of a girl with dropped head syndrome and focal white-matter changes.59 No clear genotype–phenotype correlation has been demonstrated to date. However, missense mutations seem to be more frequent in EDMD2 and MDCL, and frameshifts more frequent in LGMD1B.47,60 In EDMD2 and LGMD1B, mutations are confined mostly to Ig-like (exons 7–10) and coil 2B (exon 6) regions, respectively, whereas in MDCL they are confined to the N-terminal and the first part of the rod domains (exons 1, 4, and 5).47,60 Environmental factors and additional possible genetic modifiers have been postulated to explain the phenotypic variability.61–63
LMNA-related cardiomyopathies
The first cases presenting with selective cardiac diseases associated with mutations affecting the LMNA gene were reported in 1999.64 Very often, the first signs of cardiac disease proved to be silent; electrocardiography findings consisted in cardiac rate and rhythm abnormalities, followed by sinus-node alterations or progressive atrioventricular blocks. A number of subjects developed atrial fibrillation or flutter. Later on, several study patients developed DCM ranging in severity from mild to severe, leading to heart failure and requiring in some cases heart transplantation.
The frequency of LMNA mutations associated with the different cardiac and skeletal muscle phenotypes was studied by Vytopil et al65 in 166 patients subdivided into four groups: EDMD, isolated heart disease, isolated MD, and idiopathic “hyperCKemia”. Mutation analysis demonstrated that the presence of heart involvement and age of onset were distinguishing features separating patients with EDMD and LMNA mutations from those that were LMNA/STA-negative, while distribution of myopathy was not a reliable diagnostic criterion. The frequency of LMNA mutations in isolated heart disease was 3%–5%, similar to that of other identified genes, underscoring the high heterogeneity of the cardiac phenotype and a role of LMNA in this group of disorders.
Both intra- and interfamilial variability has been reported, with cases showing a rapid and negative prognosis caused by sudden cardiac death or quick deterioration of cardiac status, and familial cases with milder disease course characterized by low prevalence of atrial fibrillation and DCM and no evidence of sudden death.66 Cardiac compromise does not significantly differ between patients with the neuromuscular phenotype and patients with selective cardiac disease.66 It usually manifests first with electrocardiography findings of low P-wave and prolonged PR interval, with a narrow QRS complex. The majority of them developed dysrhythmias after the age of 30 years, and many underwent pacemaker implantation; heart failure was relatively frequent after the age of 50 years, and occurred less commonly than dysrhythmias.
Recently, Nigro et al demonstrated that P-wave dispersion, an independent risk factor for the development of atrial fibrillation, was significantly increased in subjects carrying LMNA-gene mutations, despite preserved systolic and diastolic cardiac function.67
Interestingly, sudden cardiac death was the modality of exit most frequently observed even in implanted subjects, indicating that they are at high risk of sudden death and that pacemakers are unable to protect them against this dramatic event.55 Sudden cardiac death is attributed to the development of life-threatening arrhythmias that may occur in the presence of normal left ventricular systolic function. Heterogeneity of ventricular repolarization, stated by the electrocardiographic parameters QTc dispersion and JTc dispersion, is considered to provide an electrophysiological substrate for malignant arrhythmias and sudden death.68 It was observed that compared to the healthy controls, LMNA-mutated patients presented increased values of QTc dispersion and JTc dispersion.68 In a retrospective longitudinal study on a large number of families whose index case was affected by cardiolaminopathy, a high rate of major cardiac events was reported, including malignant ventricular arrhythmias and heart failure, usually occurring in patients already affected by DCM.47 Furthermore, competitive sport and being a carrier of a nonmissense LMNA-gene mutation are considered factors predicting sudden cardiac death. Malignant ventricular arrhythmias tend to occur in subjects carrying at least two of the following variables: mutations other than missense on the LMNA gene, ejection fraction <45% at first clinical evaluation, male sex, and unsustained ventricular tachycardia.69 These findings help in understanding the natural course of cardiolaminopathies, and provide clinicians with important suggestions on how to prevent sudden cardiac death.
Systemic laminopathies
Systemic laminopathies refer to a group of progeroid syndromes that includes HGPS, restrictive dermopathy (RD), MAD, and atypical progeroid syndrome (APS). HGPS (OMIM 176670) is a rare dominant genetic disorder characterized by accelerated aging caused by de novo mutations in LMNA gene, first described by Jonathan Hutchinson in 1886 and Hasting Gilford in 1897. The estimated incidence is one per 4–8 million live births. A total of 132 cases of HGPS have been reported, with 40 known cases worldwide.
Mutated LMNA encodes for an aberrant protein called progerin, present in high concentrations in cells and causing distortion of the nuclear membrane, altered function of chromatin, and reduced life expectancy.70 More than 50% of patients share the G608G mutation in exon 11, but further variants have been also reported.71,72 Some patients have shown AR mutations in LMNA72,73 or in ZMPSTE24, with atypical features overlapping MAD or RD.74 HGPS affects growth, skeleton, body fat, skin, hair, and the cardiovascular system. Children with classical HGPS typically appear normal at birth, but usually present a profound failure to thrive and a sclerodermatous skin, with loss of subcutaneous fat over the trunk in the first year of life. The phenotype is characterized by “plucked-bird” appearance, stunted growth, prominent scalp veins, distinctive facial appearance (frontal bossing, protruding ears with absent lobes, a glyphic nose, prominent eyes, thin lips, and micrognathia), short clavicles, generalized joint stiffness, and osteolysis involving distal phalanges and clavicles. These changes result in an aged appearance. Intelligence is normal. Growth in weight is more disturbed than growth in height, and growth delay starts prenatally. Secondary sexual characteristics fail to develop. Lipodystrophy is generalized, except for intra-abdominal fat. Cardiovascular problems are extremely variable: stroke and coronary dysfunction are very frequent. Decreased high-density lipoprotein levels are also characteristic of this syndrome. Most patients die at 6–20 years of age (average life span 13 years) of complications of severe atherosclerosis or cardiac/cerebrovascular diseases.75–78
RD (OMIM 275210) is a rare AR lethal multiple congenital syndrome characterized by severely taut skin restricting intrauterine growth and movements and causing akinesia or hypokinesia. Newborns die in the first week of life due to respiratory insufficiency.79 Patients show tight skin, joint contractures, rocker-bottom feet, and characteristic facial dysmorphisms (micrognathia, mouth fixed in an “O” position, small pinched nose, lack of eyebrows and eyelashes). The disorder is due to mutations in the ZMPSTE24 or LMNA gene. ZMPSTE24 mutations abolish normal functioning of a zinc metalloproteinase responsible for the correct processing and maturation of lamin A, while LMNA mutations produce a truncated precursor to lamin A.80 RD is regarded as a more severe form of HGPS, with a possible correlation between the cellular levels of progerin and prelamin A and the severity of the symptoms.81
MAD (OMIM 248370) is a rare AR disorder characterized by postnatal growth retardation, craniofacial features, and skeletal and cutaneous manifestations.82 Patients show mandibular hypoplasia, progressive osteolysis of terminal phalanges and clavicles, and cutaneous changes (mottled hyperpigmentation, skin atrophy). Some patients may show marked premature aging features, including bird-like faces, high-pitched voice, alopecia, and nail dysplasia, and less commonly hypogonadism and delayed puberty, sensorineural deafness, cutaneous calcinosis, and scleroderma. Lipodystrophy, hyperlipidemia, and metabolic complications associated with insulin resistance are typical of this disorder. Appearance is normal at birth, but signs progress with age.83–85 The most common genetic defects are homozygous missense mutations in the LMNA86 or ZMPSTE24 genes.87 MADA, caused by mutations in LMNA gene, is characterized by partial loss of fat from extremities, with normal or excessive deposition in the face and neck.86 Mandibuloacral dysplasia type B (MADB), caused by mutations in the ZMPSTE24 gene, is characterized by generalized loss of subcutaneous fat, affecting the face as well as extremities.87 The majority of Mandibuloacral dysplasia type A (MADA) patients are homozygotes for the p.R527H mutation.86 MADB patients develop clinical manifestations earlier in life, are premature at birth, and can develop focal segmental glomerulosclerosis and calcified skin nodules during adulthood. Differential diagnosis may be difficult, especially in infancy, because of common clinical features and poor evidence of lipodystrophy in childhood.
The features of MAD show significant clinical overlap with HGPS and APS. Lipodystrophy and pinched nose are more prominent in HGPS than in MAD, while acroosteolysis and clavicular and mandibular hypoplasia are less prominent in APS. The similarity confirms that these forms represent a clinical spectrum of related disorders.88 Patients with MADA have a significantly longer life expectancy than those with HGPS or RD.
APS is a progeroid syndrome involving heterozygous LMNA-gene mutations, with heterogeneous phenotypes.89 Patients show clinical signs of HGPS, MAD, or atypical WS: short stature, beaked nose, premature graying, partial alopecia, high-pitched voice, skin atrophy over the hands and feet, diabetes, generalized lipodystrophy, skin pigmentation, and mandibular hypoplasia. No clear genotype–phenotype correlation has been shown. The same mutations (R133L, R644C) could be associated with extreme phenotypic diversity similar to that observed in myolaminopathies.88,90 Many of the mutations are de novo. In contrast to patients with MAD or HGPS, APS patients generally have no acroosteolysis or only mild changes of the terminal phalanges, slight evidence of loss of scalp hair, and a slightly delayed onset of manifestations. Partial or generalized lipodystrophy is a common sign. A large number of patients show cardiac valve involvement and metabolic diseases. Phenotypic heterogeneity could be explained by ethnic differences, environmental factors, and various roles of the nuclear lamina.90
Prospects for therapeutic intervention
Presently, there is no cure for laminopathies; treatment can only be symptomatic and performed by a multidisciplinary medical team. The strategic use of established medications might delay the cardiovascular complications in certain laminopathies. However, different therapeutic strategies are developing91 as pharmacological and gene approaches for both systemic and nonsystemic laminopathies.
Pharmacological approach
Systemic laminopathies
Hutchinson-Gilford progeria
A possible therapeutic approach for systemic laminopathies caused by farnesylated prelamin A accumulation is the degradation of prelamin A or progerin, expected to improve the disease phenotype.92 Prelamin A elicits toxic effects in cells, leading to chromatin damage and cellular senescence, ultimately causing accelerated skin aging.92
Rapamycin, an antibiotic belonging to the class of macrolides, is able to inhibit progerin, dramatically and selectively decreasing protein levels through a mechanism involving autophagic degradation. Progeria cells treated by rapamycin show lower levels of progerin and wild-type prelamin A, suggesting the drug can serve as a therapeutic tool to eliminate progerin, avoid farnesylated prelamin A accumulation, and restore chromatin dynamics.92,93
Quite promising results have been found with the use of farnesyltransferase inhibitors (FTIs). Initially developed as a target drug for an oncogenic RAS gene, FTIs have been shown to block the enzyme responsible for the farnesylation step on prelamin A in HGPS. In transgenic mice expressing progerin in the skin, administration of FTIs or a combination of pravastatin and zolendronate rescue nuclear morphological abnormalities in keratinocytes.94
Both prelamin A and its truncated form progerin/LAΔ50 undergo alternative prenylation by geranylgeranyl transferase in the setting of farnesyltransferase inhibition, which could explain the low efficiency of FTIs in ameliorating the phenotypes of progeroid mouse models.94 A combination of statins and aminobisphosphonates efficiently inhibits both farnesylation and geranylgeranylation of progerin and prelamin A, and markedly improves the aging-like phenotypes of ZMPSTE24-deficient mice. Likewise, the longevity of these mice is substantially extended.95 These findings opened a new therapeutic approach for human progeroid syndromes associated with NE abnormalities.
In clinical studies, inhibition of farnesylation, though not curing the pathological cause, averts buildup of progerin on the nuclear rim and prevents further disease progress.91,96 Frequency of clinical strokes, headaches, and other complications were greatly reduced.96 Gordon et al recently stated that there is evidence that survival may be improved by FTIs,97 although they are clearly not curative, many features of the disease persisting despite treatment. The inhibition of isoprenylcysteine methylation, causing accumulation of progerin, could be a further therapeutic approach for HGPS, being able to restore cellular processes in both transgenic mice and human cells.98
Nonsystemic laminopathies
LMNA dilated cardiomyopathy
A defective autophagy in hearts of LMNAH222P/H222P mice has been observed, caused by hyperactivation of the AKT–mTOR pathway.35,99,100 Reducing mTOR activity by pharmacological intervention ameliorated autophagy in correlation with improved heart function.100 Furthermore, in vivo administration of the rapamycin analog temsirolimus prevents deterioration of cardiac function. These findings provide a rationale for a novel treatment of LMNA cardiomyopathy and implicate defective autophagy as a pathogenic mechanism of cardiolaminopathy.99
Mandibuloacral dysplasia
Rapamycin seems to be a potential treatment for MAD.101 In fact, treatment with rapamycin of MAD cells, which feature very low levels of the nicotine adenine dinucleotide (NAD)-dependent sirtuin SIRT1 in the nuclear matrix, restores SIRT1 localization and chromatin-marker distribution, elicits release of the transcription factor Oct1, and determines shortening of the prolonged S-phase.101
Gene therapy
The general approach for gene therapy in laminopathies will depend on the type of mutation and disease phenotype. Treatment of simple AR disorders would “only” require the expression of the wild-type allele in the diseased tissue, while treatment of dominant negative-type laminopathies presents considerably more difficulties. The main difficulty seems to be the development of strategies neutralizing the mutated protein without affecting the normal allele expression. The most promising methods (the least dangerous and relatively fast to develop) would be to design small peptides or other molecules specifically interacting with mutated lamin protein only, and making them inert for interactions with other proteins.91
Systemic laminopathies
The gene therapy for HGPS is based on RNA interference (RNAi), in which mutant LMNA messenger RNAs (mRNAs) are selectively destroyed. In RNAi, the 21–23 mers of double-stranded RNAs introduced to cells hybridize to target mRNA and recruit machineries to destroy the target mRNAs. Double-stranded RNA can be delivered to cells utilizing expression of short-hairpin RNA.102 In HGPS fibroblasts and lymphocytes, Scaffidi and Misteli103 introduced morpholino oligonucleotides targeted to the activated cryptic splicing site, and observed that mutant proteins were reduced to ~25% compared to untransfected cells, and cellular disease phenotypes, including nuclear morphology and replicative life spans, were partially restored.103 Alternatively, synthetic oligonucleotides with long half-lives can be employed.91,104 Both approaches were shown to effectively restore various cellular and nuclear phenotypes. However, though these show potential clinical applications, systemic delivery of short-hairpin RNA or synthetic oligonucleotides remains a major hurdle for any gene therapy.
Progerin has been shown to accumulate in patient-derived vascular cells from skin,105 while pathological studies of HGPS mice suggest that progressive loss of vascular smooth-muscle cells may be the cause of fatal cardiac complications.105 The simple reduction of the toxic mutant protein using RNAi might become a potential treatment for selected LMNA phenotypes in which accumulation of the toxic mutant lamin is the predominant mechanism of disease.
Of particular interest in the field is a study by Osorio et al.106 Using a genetically modified mouse strain carrying HGPS mutation, they developed an antisense morpholino-based therapy that prevented pathogenic LMNA splicing, markedly reducing the accumulation of progerin and its associated nuclear defects, and resulted in a marked amelioration of the progeroid phenotype and life span. These data support the effectiveness of antisense oligonucleotide-based therapies for treating human diseases of accelerated aging.104,107
Nonsystemic laminopathies
Some promise for an efficient gene therapy was given by the discovery that human hematopoietic stem cells (HSCs) or mesenchymal stem cells can differentiate into different cell types.105,108 Among others, they can differentiate into myoblasts and subsequently into myotubes. HSCs have been widely used for the treatment of a large variety of human disorders.91,109,110 Stem cells can be delivered to damaged tissue for regeneration, or must be stimulated to differentiate into muscle satellite cells or mature cells in vitro and then delivered to patients. The major drawback of ex vivo stem cell therapy with adult muscle stem cells in this approach is the low migration potential of muscle satellite cells. The proposed gene therapy for muscle laminopathies may also be based on the isolation of HSCs from the patient, transformation with the wild allele of the lamin gene, and cotransfection with the gene(s) of transcription factors, responsible for differentiation into “primary” myoblasts (eg, Shh, Pax7, Myo-D, Sca-1), and infusion/microinjection back into patients.110,111 Infused cells should differentiate into primary myoblasts, and as such should be directed very efficiently to the muscle cells, where they can repopulate and fuse with existing myotubes or form new ones.
Exon skipping to restore a reading frame or switch protein isoforms is under clinical trial in some genetic diseases. The hypothesis is that by removing an in-frame exon containing a pathogenic mutation, phenotypes could also improve, as recently demonstrated by Scharner et al.112 However, provided the high pathogenic potential of any LMNA mutation, gene therapy must be regarded with several caveats.
Conclusion
Mutations in genes encoding nuclear lamins, particularly LMNA encoding the A type lamins, cause a range of phenotypically different diseases. The phenotypes and genetic abnormalities of these disorders have been described. A significant amount of current research is aimed at deciphering pathogenic mechanisms, and some has connected mutations in LMNA to posttranslational protein modifications and alterations in cell-signaling pathways.
In this review, we attempted to summarize the current state of knowledge on laminopathies and to describe the current prospects for future treatment, addressing the different aspects of this complex group of diseases.
Acknowledgments
All authors are members of the Italian Network for Laminopathies (http://www.igm.cnr.it/1/laminopatie). The support of Telethon-Unione Italiana Lotta alla Distrofia Muscolare (UILDM) (grants GUP 11001C and GUP 11002L to LP and 5‰- 2012 Rizzoli Orthopedic Institute to GL) is acknowledged. We thank patients, families, and patient/family associations (Associazione Italiana Progeria Sammy Basso, Associazione Italiana Distrofia di Emery-Dreifuss, and Alessandra Proietti Onlus) for their collaboration and constant support.
Disclosure
The authors report no conflicts of interest in this work.
References
Hoelz A, Debler EW, Blobel G. The structure of the nuclear pore complex. Annu Rev Biochem. 2011;80:613–643. | |
Khadija SG, Chen F, Hadden T, Commissaris RL, Kowluru A. Biology and regulatory roles of nuclear lamins in cellular function and dysfunction. Recent Pat Endocr Metab Immune Drug Discov. 2015;9:111–120. | |
Davidson PM, Lammerding J. Broken nuclei – lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014;24:247–256. | |
Bione S, Maestrini E, Rivella S, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. | |
Camozzi D, Capanni C, Cenni V, et al. Diverse lamin-dependent mechanisms interact to control chromatin dynamics: focus on laminopathies. Nucleus. 2014;5:427–440. | |
Houben F, Ramaekers FC, Snoeckx LH, Broers JL. Role of nuclear lamina-cytoskeleton interactions in the maintenance of cellular strength. Biochim Biophys Acta. 2007;1773:675–686. | |
Maraldi NM, Capanni C, Cenni V, Fini M, Lattanzi G. Laminopathies and lamin-associated signaling pathways. J Cell Biochem. 2011;112:979–992. | |
Zini N, Avnet S, Ghisu S, et al. Effects of prelamin A processing inhibitors on the differentiation and activity of human osteoclasts. J Cell Biochem. 2008;105:34–40. | |
Jung HJ, Coffinier C, Choe Y, et al. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A. 2012;109:E423–E431. | |
Lattanzi G. Prelamin A-mediated nuclear envelope dynamics in normal and laminopathic cells. Biochem Soc Trans. 2011;39:1698–1704. | |
Capanni C, Cenni V, Haraguchi T, et al. Lamin A precursor induces barrier-to-autointegration factor nuclear localization. Cell Cycle. 2010;9:2600–2610. | |
Jung HJ, Nobumori C, Goulbourne CN, et al. Farnesylation of lamin B1 is important for retention of nuclear chromatin during neuronal migration. Proc Natl Acad Sci U S A. 2013;110:E1923–E1932. | |
Lattanzi G, Columbaro M, Mattioli E, et al. Pre-lamin A processing is linked to heterochromatin organization. J Cell Biochem. 2007;102:1149–1159. | |
Infante A, Gago A, de Eguino GR, et al. Prelamin A accumulation and stress conditions induce impaired Oct-1 activity and autophagy in prematurely aged human mesenchymal stem cell. Aging (Albany NY). 2014;6:264–280. | |
Kind J, van Steensel B. Stochastic genome-nuclear lamina interactions: modulating roles of lamin A and BAF. Nucleus. 2014;5:124–130. | |
Wagner N, Krohne G. LEM-domain proteins: new insights into lamin-interacting proteins. Int Rev Cytol. 2007;261:1–46. | |
Guelen L, Pagie L, Brasset E, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. | |
Lattanzi G, Ortolani M, Columbaro M, et al. Lamins are rapamycin targets that impact human longevity: a study in centenarians. J Cell Sci. 2014;127:147–157. | |
Meinke P, Mattioli E, Haque F, et al. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet. 2014;10:e1004605. | |
Evangelisti C, Bernasconi P, Cavalcante P, et al. Modulation of TGFβ 2 levels by lamin A in U2-OS osteoblast-like cells: understanding the osteolytic process triggered by altered lamins. Oncotarget. 2015;6:7424–7437. | |
Chatzifrangkeskou M, Bonne G, Muchir A. Nuclear envelope and striated muscle diseases. Curr Opin Cell Biol. 2014;32:1–6. | |
Zhang X, Xu R, Zhu B, et al. Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development. 2007;134:901–908. | |
Taylor MR, Slavov D, Gajewski A, et al. Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat. 2005;26:566–574. | |
Kayman-Kurekci G, Talim B, Korkusuz P, et al. Mutation in TOR1AIP1 encoding LAP1B in a form of muscular dystrophy: a novel gene related to nuclear envelopathies. Neuromuscul Disord. 2014;24:624–633. | |
Liang WC, Mitsuhashi H, Keduka E, et al. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol. 2011;69:1005–1013. | |
Padiath QS, Saigoh K, Schiffmann R, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114–1123. | |
Sharma N, Franco RA Jr, Kuster J, et al. Genetic evidence for an association of the TOR1A locus with segmental/focal dystonia. Mov Disord. 2010;25:2183–2187. | |
Isermann P, Lammerding J. Nuclear mechanics and mechanotransduction in health and disease. Curr Biol. 2013;23:R1113–R1121. | |
Swift J, Ivanovska IL, Buxboim A, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. | |
Bertrand AT, Ziaei S, Ehret C, et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J Cell Sci. 2014;127:2873–2884. | |
Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet. 2005;14:2167–2180. | |
Muchir A, Pavlidis P, Bonne G, Hayashi YK, Worman HJ. Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum Mol Genet. 2007;16:1884–1895. | |
Muchir A, Pavlidis P, Decostre V, et al. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest 2007;117:1282–1293. | |
Ramos FJ, Chen SC, Garelick MG, et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012;4:144ra103. | |
Cohen TV, Gnocchi VF, Cohen JE, et al. Defective skeletal muscle growth in lamin A/C-deficient mice is rescued by loss of Lap2α. Hum Mol Genet. 2013;22:2852–2869. | |
Novelli G, D’Apice MR. Protein farnesylation and disease. J Inherit Metab Dis. 2012;35:917–926. | |
Liu B, Wang J, Chan KM, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11:780–785. | |
Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci. 2006;119:4644–4649. | |
di Masi A, D’Apice MR, Ricordy R, Tanzarella C, Novelli G. The R527H mutation in LMNA gene causes an increased sensitivity to ionizing radiation. Cell Cycle. 2008;7:2030–2037. | |
Cabanillas R, Cadinanos J, Villameytide JA, et al. Néstor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A. 2011;155A:2617–2625. | |
Puente XS, Quesada V, Osorio FG, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. 2011;88:650–656. | |
Weedon MN, Ellard S, Prindle MJ, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45:947–950. | |
Pelosini C, Martinelli S, Ceccarini G, et al. Identification of a novel mutation in the polymerase delta 1 (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism. 2014;63:1385–1389. | |
Goto M, Imamura O, Kuromitsu J, et al. Analysis of helicase gene mutations in Japanese Werner’s syndrome patients. Hum Genet. 1997;99:191–193. | |
Chen L, Lee L, Kudlow BA, et al. LMNA mutations in atypical Werner’s syndrome. Lancet. 2003;9;362(9382):440–445. | |
Mercuri E, Poppe M, Quinlivan R, et al. Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: from congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch Neurol. 2004;61:690–694. | |
Maggi L, D’Amico A, Pini A, et al. LMNA-associated myopathies: the Italian experience in a large cohort of patients. Neurology. 2014;83:1634–1644. | |
Mercuri E, Clements E, Offiah A, et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol. 2010;67:201–208. | |
Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–288. | |
Emery AE. Emery-Dreifuss muscular dystrophy: a 40-year retrospective. Neuromuscul Disord. 2000;10:228–232. | |
Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354:209–210. | |
Muchir, A, Bonne G, van der Kooi AJ, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. 2000;9:1453–1459. | |
Bonne G, Mercuri E, Muchir A, et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48:170–180. | |
Boriani G, Gallina M, Merlini L, et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: a long-term longitudinal study. Stroke. 2003;34:901–908. | |
Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52:1250–1260. | |
Gueneau L, Bertrand AT, Jis JP, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2009;85:338–353. | |
Quijano-Roy S, Mbieleu B, Bönnemann CG, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. 2008;64:177–186. | |
Menezes MP, Waddell LB, Evesson FJ, et al. Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy. Neurology. 2012;78:1258–1263. | |
Hattori A, Komaki H, Kawatani M, et al. A novel mutation in the LMNA gene causes congenital muscular dystrophy with dropped head and brain involvement. Neuromuscul Disord. 2012;22:149–151. | |
Benedetti S, Menditto I, Degano M, et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology. 2007;69:1285–1292. | |
Muntoni F, Bonne G, Goldfarb LG, et al. Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain. 2006;129:1260–1268. | |
Granger B, Gueneau L, Drouin-Garraud V, et al. Modifier locus of the skeletal muscle involvement in Emery-Dreifuss muscular dystrophy. Hum Genet. 2011;129:149–159. | |
Arimura T, Onoue K, Takahashi-Tanaka Y. Nuclear accumulation of androgen receptor in gender difference of dilated cardiomyopathy due to lamin A/C mutations. Cardiovasc Res. 2013;99:382–394. | |
Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. | |
Vytopil M, Benedetti S, Ricci E, et al. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet. 2003;40:e132. | |
van Berlo JH, de Voogt WG, van der Kooi AJ, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl). 2005;83:79–83. | |
Nigro G, Russo V, Rago A, et al. Regional and transmural dispersion of repolarisation in patients with Emery-Dreifuss muscular dystrophy. Kardiol Pol. 2012;70:1154–1159. | |
Russo V, Rago A, Politano L, et al. Increased dispersion of ventricular repolarization in Emery Dreifuss muscular dystrophy patients. Med Sci Monit. 2012;18:CR643–CR647. | |
van Rijsingen IA, Nannenberg EA, Arbustini E, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013;15:376–384. | |
De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. | |
Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. | |
Plasilova M, Chattopadhyay C, Pal P, et al. Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome. J Med Genet. 2004;41:609–614. | |
Madej-Pilarczyk A, Rosinskan-Borkowska D, Rekawek J, et al. Progeroid syndrome with scleroderma like skin changes associated with homozygous R435C LMNA mutation. Am J Med Genet A. 2009; 149A:2387–2392. | |
Shackletton S, Smallwood DT, Clayton P, et al. Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. J Med Genet. 2005;42:e36. | |
DeBusk FL. The Hutchinson-Gilford progeria syndrome: report of 4 cases and review of the literature. J Pediatr. 1972;80:697–724. | |
Badame AJ. Progeria. Arch Dermatol. 1989;125:540–544. | |
Gordon LB, McCarten KM, Giobbie-Hurder A, et al. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120:824–833. | |
Merideth MA, Gordon LB, Clauss S, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008; 358:592–604. | |
Loucks C, Parboosingh JS, Chong JX, et al. A shared founder mutation underlies restrictive dermopathy in old colony (Dutch-German) Mennonite and Hutterite patients in North America. Am J Med Genet A. 2012;158A:1229–1232. | |
Morais P, Magina S, Riberio MC, et al. Restrictive dermopathy – a lethal congenital laminopathy: case report and review of the literature. Eur J Pediatr. 2009;168:1007–1012. | |
McKenna T, Rosengardten Y, Viceconte N, Baek JH, Grochova D, Eriksson M. Embryonic expression of the common progeroid lamin A splice mutation arrests postnatal skin development. Aging Cell. 2014;13:292–302. | |
Yang LW, Radebaugh JF, Rubin P, Sensenbrenner JA, Fiorelliu G, McKusick VA. New syndrome manifested by mandibular hypoplasia, acroosteolysis, stiff joints and cutaneous atrophy (mandibuloacral dysplasia) in two unrelated boys. Birth Defects Org Artic Ser. 1971;7:291–297. | |
Hoeffel JC, Mainard L, Chastagner P, Hoeffel CC. Mandibulo-acral dysplasia. Skeletal Radiol. 2000;29:668–671. | |
Simha V, Agarwal AK, Oral EA, Fryns JP, Garg A. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia associated lipodystrophy. J Clin Endocrinol Metab. 2003;88:2821–2824. | |
Cefle A, Cefle K. A case of mandibuloacral dysplasia presenting with features of scleroderma. Int J Clin Pract. 2004;58:635–638. | |
Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA encoding lamin A/C. Am J Hum Genet. 2002;71:426–431. | |
Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. | |
Jacob KN, Garg A. Laminopathies: multisystem dystrophy syndromes. Mol Genet Metab. 2006;87:289–302. | |
Garg A, Subramanyam L, Agarwal AK, et al. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab. 2009;94:4971–4983. | |
Rankin J, Auer-Grumbach M, Bagg W, et al. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am J Med Genet A. 2008;146A:1530–1542. | |
Zaremba-Czogalla M, Dubinska-Magiera M, Rzepecki R. Laminopathies: the molecular background of the disease and the prospects for its treatment. Cell Mol Biol Lett. 2011;16:114–148. | |
Cenni V, Capanni C, Columbaro M, et al. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur J Histochem. 2011;55:e36. | |
Ullrich NJ, Kieran MW, Miller DT, et al. Neurologic features of Hutchinson-Gilford progeria syndrome after lonafarnib treatment. Neurology. 2013;81:427–430. | |
Wang Y, Ostlund C, Worman HJ. Blocking protein farnesylation improves nuclear shape abnormalities in keratinocytes of mice expressing the prelamin A variant in Hutchinson-Gilford progeria syndrome. Nucleus. 2010;1:432–439. | |
Varela I, Pereira S, Ugalde AP, et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14:767–772. | |
Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford Progeria Syndrome. Proc Natl Acad Sci U S A. 2012;109:16666–16671. | |
Gordon LB, Massaro J, D’Agostino RB, et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130:27–34. | |
Ibrahim MX, Sayin VI, Akula MK, et al. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science. 2013;340:1330–1333. | |
Choi JC, Muchir A, Wu W, et al. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. 2012;4:144ra102. | |
Choi JC, Worman HJ. Reactivation of autophagy ameliorates LMNA cardiomyopathy. Autophagy. 2013;9:110–111. | |
Cenni V, Capanni C, Mattioli E, et al. Rapamycin treatment of mandibuloacral dysplasia cells rescues localization of chromatin-associated proteins and cell cycle dynamics. Aging (Albany NY). 2014;6:755–770. | |
Huang S, Chen L, Libina N, et al. Correction of cellular phenotypes of Hutchinson-Gilford progeria cells by RNA interference. Hum Genet. 2005;118:444–450. | |
Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–445. | |
Fong LG, Vickers TA, Farber EA, et al. Activating the synthesis of progerin, the mutant prelamin A in Hutchinson-Gilford progeria syndrome, with antisense oligonucleotides. Hum Mol Genet. 2009;18:2462–2471. | |
McClintock D, Gordon LB, Djabali K. Hutchinson-Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-lamin A G608G antibody. Proc Natl Acad Sci U S A. 2006; 103:2154–2159. | |
Osorio FG, Navarro CL, Cadiñanos J, et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3:106ra107. | |
Luo YB, Mitrpant C, Adams AM, et al. Antisense oligonucleotide induction of progerin in human myogenic cells. PLoS One. 2014;9:e98306. | |
Anjos-Afonso F, Siapati EK, Bonnet D. In vivo contribution of murine mesenchymal stem cells into multiple cell-types under minimal damage conditions. J Cell Sci. 2004;117:5655–5664. | |
Griffin M, Greiser U, Barry F, O’Brien T, Ritter T. Genetically modified mesenchymal stem cells and their clinical potential in acute cardiovascular disease. Discov Med. 2010;9:219–223. | |
Epting CL, López JE, Shen X, Liu L, Bristow J, Bernstein HS. Stem cell antigen-1 is necessary for cell-cycle withdrawal and myoblast differentiation in C2C12 cells. J Cell Sci. 2004;117:6185–6195. | |
Rivier F, Alkan O, Flint AF, et al. Role of bone marrow cell trafficking in replenishing skeletal muscle SP and MP cell populations. J Cell Sci. 2004;117:1979–1988. | |
Scharner J, Figeac N, Ellis JA, Zammit PS. Ameliorating pathogenesis by removing an exon containing a missense mutation: a potential exon-skipping therapy for laminopathies. Gene Ther. 2015;22:503–515. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.