Back to Journals » Cancer Management and Research » Volume 9
Emerging combination therapies for metastatic colorectal cancer – impact of trifluridine/tipiracil
Authors Puthiamadathil JM, Weinberg BA ![]()
Received 22 July 2017
Accepted for publication 5 September 2017
Published 3 October 2017 Volume 2017:9 Pages 461—469
DOI https://doi.org/10.2147/CMAR.S113320
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Video abstract presented by Benjamin A Weinberg
Views: 482
Jeevan M Puthiamadathil,1 Benjamin A Weinberg1,2
1Department of Medicine, 2Ruesch Center for the Cure of Gastrointestinal Cancers, Lombardi Comprehensive Cancer Center, Georgetown University, Washington, DC, USA
Abstract: Patients with metastatic colorectal cancer (mCRC) are surviving longer now than ever before, but mortality rates are still high and more effective therapies are clearly needed. For patients with disease that is refractory to fluoropyrimidines, oxaliplatin, irinotecan, and biologic agents targeting the vascular endothelial growth factor and epidermal growth factor receptor pathways, novel treatment options trifluridine/tipiracil (TAS-102) and regorafenib can be effective disease stabilizers. However, objective clinical responses are rare and toxicities are manageable but common. In order to tackle poor clinical responses to TAS-102, there is an ongoing effort to effectively combine this drug with other agents, particularly those targeting angiogenesis. Certain subpopulations appear to benefit more than others from TAS-102; those that benefit often have underlying genetic defects in DNA repair pathways and/or develop neutropenia. In this review, we focus on the role of TAS-102 in the treatment of mCRC, including its use in combination with other agents, potential predictive biomarkers of response to TAS-102, and possible future directions.
Keywords: metastatic colorectal cancer, trifluridine, tipiracil, TAS-102, regorafenib
Introduction
In 2017, it is estimated that there will be 135,430 new cases of colorectal cancer (CRC) and 50,260 deaths resulting from this disease, making it the second leading cause of cancer mortality in the USA.1 At diagnosis, about 21% of patients have metastatic disease, and these individuals have a 5-year survival rate of only 13.9%.2 Due to advances in chemotherapy for patients with metastatic colorectal cancer (mCRC), median overall survival (mOS) has increased to ≥30 months, compared with <12 months in the era of treatment with single-agent fluoropyrimidines.3–5
First-line chemotherapeutic options for mCRC involve fluoropyrimidines, such as 5-fluorouracil (5-FU) or capecitabine, combined with oxaliplatin and/or irinotecan. Standard treatment regimens include FOLFOX (leucovorin/5-FU/oxaliplatin), CAPOX (capecitabine/oxaliplatin), FOLFIRI (leucovorin/5-FU/irinotecan), and FOLFOXIRI (leucovorin/5-FU/oxaliplatin/irinotecan).3,6–9 The advent of monoclonal antibodies (mAbs) bevacizumab (Avastin®; Genentech, South San Francisco, CA, USA),9 an anti-vascular endothelial growth factor A (VEGF-A) antibody, and cetuximab (Erbitux; Eli Lilly and Company, Indianapolis, IN, USA),10 an anti-epidermal growth factor receptor (EGFR) antibody, given in combination with fluoropyrimidine/oxaliplatin/irinotecan regimens, gave rise to the most promising treatment options available in the first line. EGFR targeting agents are restricted to patients with RAS wild-type tumors.11,12 When first-line therapies fail, patients are often treated with yet unused cytotoxic therapies combined with continued bevacizumab therapy,13,14 the anti-EGFR antibody, panitumumab (Vectibix®; Amgen, Thousand Oaks, CA, USA),15 or other antiangiogenic agents including ziv-aflibercept (Zaltrap®; Sanofi/Regeneron, Tarrytown, NY, USA) and ramucirumab (Cyramza®; Eli Lilly and Company).16,17 Pembrolizumab, an anti-programmed death 1 (PD1) mAb immune checkpoint inhibitor, is now Food and Drug Administration (FDA) approved for microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) mCRC following treatment with fluoropyrimidine, oxaliplatin, and irinotecan regimens.18–20 Previously, patients who progressed on standard chemotherapy and biologics were often left to rely on best supportive care (BSC) or participate in a clinical trial. However, now there are new and approved treatment options available with proven efficacy and tolerability in patients with refractory mCRC.
One such treatment is trifluridine/tipiracil (TAS-102, Lonsurf®; Taiho Oncology, Princeton, NJ, USA), which contains the oral cytotoxic agent trifluridine that was developed more than half a century ago. In this review, we discuss the pharmacology and clinical data of TAS-102; compare TAS-102 with regorafenib (Stivarga®, Bayer AG, Leverkusen, Germany), an alternative treatment option for patients with refractory mCRC; and discuss other therapies used in combination with TAS-102. Additionally, we consider potential biomarkers that could predict response to therapy with TAS-102 and examine clinical trials of TAS-102 in patients with refractory mCRC.
Pharmacology
TAS-102, which consists of trifluridine and tipiracil in a 1:0.5 molar ratio, was approved by the FDA for the treatment of refractory mCRC in September 2015.21 Trifluridine (5-trifluoro-2′-deoxythymidine) was developed by Heidelberger et al in 1964 as an alternative to 5-FU.22 It is a thymidine analog that inhibits cell growth by affecting DNA synthesis (Figure 1). Thymidine kinase (TK) phosphorylates trifluridine to its monophosphate form, trifluridine monophosphate (TF-TMP), which then inhibits thymidylate synthase (TS) and prevents methylation of 2′-deoxyuridine-5′-monophosphate (dUMP) to 2′-deoxythymidine-5′-monophosphate.23–26 dUMP is then alternatively phosphorylated to its triphosphate form, 2′-deoxyuridine-5′-triphosphate, which causes the incorporation of uracil into DNA, inhibiting DNA synthesis and leading to cell death.27,28 Additionally, in a separate pathway, TF-TMP is further phosphorylated to trifluridine triphosphate (TF-TTP), which causes cell death via direct incorporation into DNA.29,30 Despite the evident clinical activity of trifluridine in patients with CRC and breast cancer, its short half-life (12–18 minutes) and significant myelosuppression were found to limit its clinical utility.31
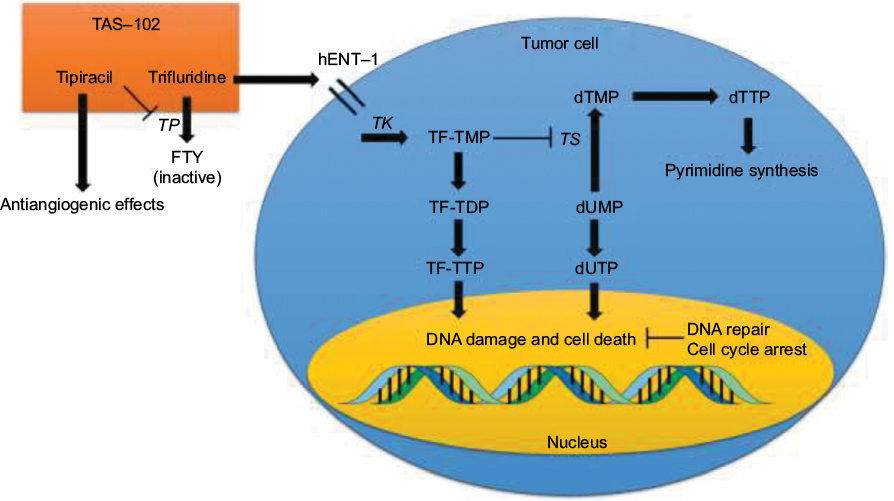 | Figure 1 Mechanism of action of TAS-102. Note: Enzymes are italicized. Abbreviations: TP, thymidine phosphorylase; FTY, 5-trifluoromethyl-2,4(1H,3H)-pyrimidinedione; hENT, human equilibrative nucleoside transporter; TK, thymidine kinase; TF-TMP, trifluorothymidine monophosphate; TF-TDP, trifluorothymidine diphosphate; TF-TTP, trifluorothymidine triphosphate; TS, thymidylate synthase; dTMP, 2′-deoxythymidine-5′-monophosphate; dTTP, 2′-deoxythymidine-5′-triphosphate; dUMP, 2′-deoxyuridine-5′-monophosphate; dUTP, 2′-deoxyuridine-5′-triphosphate. |
In the 1990s, it was discovered that the combination of tipiracil (5-chloro-6-[(2-iminopyrrolidin-1-yl)methyl]-1H-pyrimidine-2,4-dione) with trifluridine increased the bioavailability of trifluridine by inhibiting thymidine phosphorylase (TP), the enzyme that converts trifluridine into inactive 5-trifluoromethyl-2,4(1H,3H)-pyrimidinedione.32 Inhibition of TP, a known platelet-derived endothelial cell growth factor, also has indirect antiangiogenic benefits.33–35 TAS-102 was effective in 5-FU-resistant CRC cell lines, due to direct TF-TTP incorporation into DNA and the resistance of trifluridine to degradation by DNA glycosylase, suggesting efficacy in 5-FU-refractory mCRC.32,36–38
Clinical trials of TAS-102
Hong et al published the initial Phase I trial of TAS-102 in 14 patients with refractory mCRC (Table 1).39 The maximally tolerated dose (MTD) was 50 mg/m2/day administered once daily on days 1–14 of a 21-day cycle (grade 3–4 granulocytopenia was the dose limiting toxicity [DLT] in 3 of 6 patients treated at the 60 mg/m2/day dose level). Of the 12 patients evaluable for response, the best response was in 4 patients with stable disease. Another Phase I study reported by Overman et al examined 2 TAS-102 dosing schedules.40 In the 63 patients studied (52 with refractory mCRC), the recommended Phase II doses (RP2Ds) were 100 mg/m2/day administered once daily on days 1–5 and 8–12 of a 28-day cycle or 160 mg/m2/day once daily on days 1–5 of a 21-day cycle. Granulocytopenia was again the DLT in most cases. Thirty percent of patients had stable disease, but no objective clinical responses were seen.
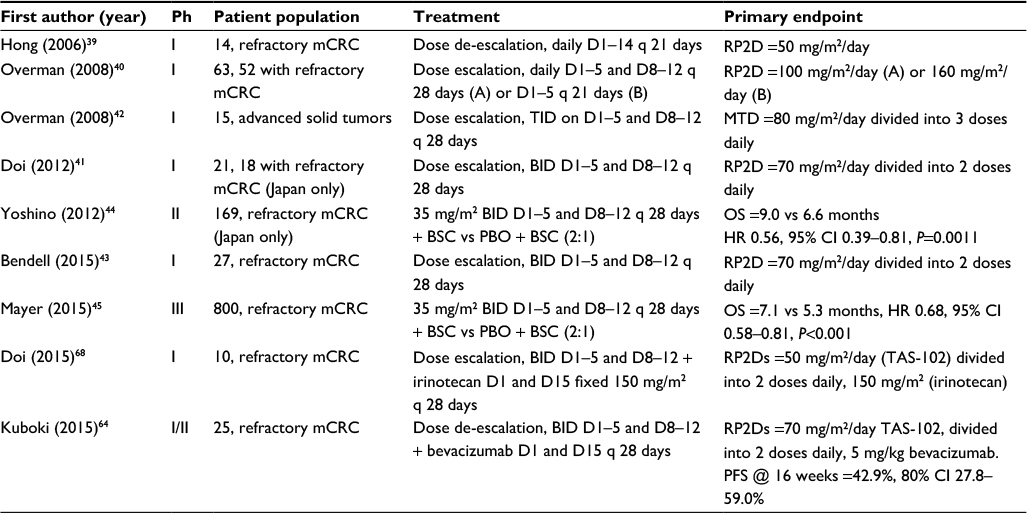 | Table 1 Completed TAS-102 trials in mCRC Abbreviations: Ph, phase; mCRC, metastatic colorectal cancer; RP2D, recommended Phase II dose; D, day; TID, three times daily; MTD, maximum tolerated dose; BID, twice daily; BSC, best supportive care; PBO, placebo; OS, overall survival; HR, hazard ratio; CI, confidence interval; PFS, progression-free survival. |
Doi et al conducted another Phase I study using twice daily dosing in 21 Japanese patients (18 with refractory mCRC).41 In this trial, the RP2D was 70 mg/m2/day split into 2 daily doses on days 1–5 and 8–12 of a 28-day cycle (the DLT again was grade 4 neutropenia). No objective responses were observed in patients with mCRC, but 50% had stable disease. Median progression-free survival (mPFS) was 2.4 months and mOS was 9.8 months. Three times daily dosing was also studied in a separate Phase I trial of 15 patients with advanced solid tumors. On a schedule of TAS-102 split into 3 daily doses on days 1–5 and 8–12 of a 28-day cycle, the MTD was 80 mg/m2/day and the DLT was granulocytopenia. Nine patients had prolonged stable disease.42 Finally, Bendell et al conducted a Phase I study involving TAS-102 treatment of 27 US patients with refractory mCRC. The RP2D in this study was 70 mg/m2/day, split into 2 daily doses, administered on days 1–5 and 8–12 of a 28-day cycle, at which dose 70% of evaluable patients had stable disease lasting at least 6 weeks, and mPFS and mOS of the entire population were 5.3 and 7.5 months, respectively.43
In the J003 Phase II trial published by Yoshino et al, 169 Japanese patients with refractory mCRC were randomized 2:1 to 35 mg/m2 TAS-102 or placebo, both administered twice daily on days 1–5 and 8–12 of a 28-day cycle.44 The primary endpoint of the study was mOS, which was significantly longer in the TAS-102 group compared to placebo: 9.0 versus 6.6 months (hazard ratio [HR] for death 0.56, 80% confidence interval [CI] 0.44–0.71; P=0.0011). mPFS was also significantly longer in the TAS-102 group: 2.0 versus 1.0 month (HR 0.41, 95% CI 0.28–0.59; P<0.0001). In the subgroup analysis, patients with KRAS exon 2 mutated tumors were found to have a greater overall survival (OS) benefit (13.0 vs 6.9 months with placebo, P=0.0056) than patients with KRAS exon 2 wild-type tumors (7.2 vs 7.0 months, P=0.191). Regarding adverse events, patients who received TAS-102 had significantly more hematological adverse events (including 50% with grade 3–4 neutropenia), as well as diarrhea, nausea, and anorexia than those receiving placebo.
These early phase trials led to the randomized, double-blind, Phase III study of TAS-102 plus BSC versus placebo plus BSC in patients with mCRC refractory to standard chemotherapies (RECOURSE). The RECOURSE trial randomized 800 patients 2:1 to receive 35 mg/m2 TAS-102 or placebo twice daily on days 1–5 and 8–12 every 28 days, stratified by KRAS exon 2 mutation status, time between diagnosis of metastatic disease and randomization (<18 months or not), and region (Japan or US, Europe, and Australia).45 mOS, the primary endpoint, and mPFS were significantly longer following TAS-102 treatment compared with placebo (mOS, 7.1 vs 5.3 months; HR 0.68, 95% CI 0.58–0.81; P<0.001; mPFS, 2.0 vs 1.7 months; HR 0.48, 95% CI 0.41–0.57; P<0.001). Response rates were low, with an objective response rate (ORR) following TAS-102 treatment of 1.6% compared with 0.4% following placebo (P=0.29). Contrary to results from the Phase II study by Yoshino et al,44 there was no significant difference in OS benefit between patients with KRAS exon 2 wild-type and patients with KRAS exon 2 mutant tumors. In the study population as a whole, grade 3–4 neutropenia was more common with TAS-102 than placebo (38% vs 0%), and febrile neutropenia was rare (4% vs 0%). Time to an Eastern Cooperative Oncology Group performance status of ≥2 was also longer following TAS-102 therapy: 5.7 versus 4.0 months (HR 0.66, 95% CI 0.56–0.78; P<0.001). This trial led to the FDA approval of TAS-102 for patients with mCRC refractory to standard therapies.21
Comparison with regorafenib
TAS-102 is often compared with regorafenib, another oral agent approved for refractory mCRC. Regorafenib is a polytyrosine kinase inhibitor that blocks many targets promoting angiogenesis (VEGF receptors 1–3, TIE2, platelet-derived growth factor receptor [PDGFR], and fibroblast growth factor receptor [FGFR]) along with other oncogenic kinases (including KIT, BRAF, and RET).46 The landmark regorafenib monotherapy for previously treated mCRC (CORRECT) trial randomized 760 patients with refractory mCRC 2:1 to regorafenib 160 mg daily or placebo administered on days 1–21 of a 28-day cycle.47 The primary endpoint of mOS was statistically significantly longer following regorafenib therapy: 6.4 versus 5.0 months (HR 0.77, 95% CI 0.64–0.94; P=0.0052). mPFS was also significantly extended by regorafenib (1.9 vs 1.7 months; HR 0.49, 95% CI 0.42–0.58; P<0.0001), but the observed increase in ORR was not significant (1.0% of patients treated with regorafenib had an objective response compared with 0.4% of those receiving placebo; P=0.19). Fifty-four percent of patients who received regorafenib had grade 3–4 adverse events compared with only 14% of those receiving placebo; adverse events included hand–foot skin reaction (17% vs <1%), fatigue (10% vs 5%), diarrhea (7% vs 1%), hypertension (7% vs 1%), and rash (6% vs 4%).
Although both oral agents are approved by the FDA for the treatment of refractory mCRC, TAS-102 and regorafenib have important differences in their side effect profiles; thus, 2 retrospective analyses from Japan demonstrated that TAS-102 and regorafenib have similar efficacy but that regorafenib leads to more hand–foot skin reaction and a greater elevation of liver enzymes and bilirubin, whereas TAS-102 causes more neutropenia, febrile neutropenia, leukopenia, and nausea.48,49 The decision of how to best sequence the order of the 2 drugs is typically based on their side effect profiles. Although efficacy has not been prospectively compared head-to-head, both agents are associated with similar survival outcomes and very low ORRs. Thus, these drugs can be effective at controlling disease but are not expected to elicit tumor shrinkage as single agents.
Predictive biomarkers
A biomarker that is able to predict which patients might benefit most from TAS-102 would be highly useful, and research on potential candidates is ongoing. Early neutropenia following initiation of TAS-102 may be predictive of improved OS. A retrospective study of 95 Japanese patients treated with TAS-102 demonstrated that patients who had grade 2 or greater chemotherapy-induced neutropenia (absolute neutrophil count <1,500/mm3) during the first cycle had an improved disease control rate (52.6% vs 29.2%; odds ratio 2.67, 95% CI 1.01–7.24; P=0.045) and mPFS (2.7 vs 2.0 months; HR 0.59, 95% CI 0.38–0.91; P=0.017), although improvements in mOS were not statistically significant (6.7 vs 5.0 months; HR 0.68, 95% CI 0.41–1.10; P=0.11).50 One prospective cohort study of 149 US patients with refractory mCRC who were chosen for TAS-102 treatment compared subjects who had grade 2 or greater chemotherapy-induced neutropenia at 1 month with those who did not.51 mOS (14.0 vs 5.6 months; P<0.0001) and mPFS (3.0 vs 2.4 months; P=0.0096) were both longer in the neutropenic group. While these results are intriguing, the etiology of this association between neutropenia and improved survival with TAS-102 remains unclear. The authors of the US study postulate that the patients in the non-neutropenic group are rapid TAS-102 metabolizers and therefore are not receiving the same therapeutic benefit. In a post hoc analysis of the Phase II J003 study, neutropenia during the first 2 cycles was also associated with an OS benefit.52 Data from the RECOURSE trial confirm the association between neutropenia and survival: patients who received TAS-102 and had grade 3 or greater neutropenia during any cycle had an OS benefit compared with patients who did not develop neutropenia; additionally, delays between TAS-102 cycles due to neutropenia were also associated with an OS benefit.53 Therefore, intensifying dose and schedule for patients not experiencing grade 2–4 neutropenia may need to be considered, although this strategy should be first evaluated in a prospective trial.
Another interesting post hoc analysis of the RECOURSE trial sorted patients according to number of lines of prior treatment due to progression of disease between lines of therapy. Patients who had >3 prior lines of treatment had significantly improved OS compared with placebo (HR 0.59, 95% CI 0.44–0.78) as opposed to patients with <3 prior lines (HR 0.89, 95% CI 0.65–1.22).54 This study suggests that a greater number of prior lines of therapy may predict improved survival of patients later treated with TAS-102. Also, a prespecified analysis of the RECOURSE trial looked at TAS-102 efficacy in patients aged ≥65 years compared with younger patients (<65 years of age). There were no significant differences in adverse event profiles, and the OS benefit of TAS-102 was similar regardless of age group (≥65 years: n=352, 7.0 months [TAS-102] vs 4.6 months [placebo], HR 0.62, 95% CI 0.48–0.80; P=0.0002; <65 years: n=448, 7.1 vs 5.7 months, HR 0.74, 95% CI 0.59–0.94; P=0.0130).55 Thus, patient age does not appear to be a predictive biomarker of survival with TAS-102.
Defects in the DNA repair pathway within CRC cells may predict susceptibility to TAS-102. When TF-TTP is incorporated into DNA, it induces single-strand breaks that ultimately cause double-strand breaks.37 In response to DNA damage, the homologous recombination repair (HRR) pathway mediates DNA repair mechanisms and interacts with cell cycle checkpoints to halt DNA synthesis.56 Suenaga et al analyzed genomic DNA from 181 mCRC patients treated with TAS-102 and 52 patients treated with regorafenib, specifically looking at single nucleotide polymorphisms (SNPs) in HRR pathway genes (ATM, BRCA1, BRCA2, XRCC3, FANCD2, H2AX, and RAD51) and cell cycle checkpoint genes (ATR, CHEK1, CHEK2, CDKN1A, TP53, CHE1, PIN1, and PCNA).57 Genetic variants in the HRR pathway genes—ATM rs609429 and XRCC3 rs861539—were found to be predictive biomarkers of OS upon TAS-102 treatment. Interestingly, there was also an association between neutropenia and ATM rs609429. This field requires further investigation, but combination therapies with TAS-102 and HRR pathway targeting agents have a rational scientific basis.
As it is known that TK phosphorylates trifluridine to TF-TMP, leading to both inhibition of TS and, in a separate pathway, the formation of TF-TTP, which is directly incorporated into DNA, the finding that TK and TS are frequently overexpressed in CRC cells is promising for using TAS-102 in patients with CRC.58 In studies using CRC cell lines, TAS-102 resistance mechanisms included decreased expression of TK and human equilibrative nucleoside transporter (hENT), the transmembrane transport protein responsible for allowing cellular influx of trifluridine.59 Thus, tumors with high TK expression should be more susceptible to TAS-102 due to increased formation of TF-TMP and TF-TTP. In a pooled retrospective analysis of patients with refractory mCRC, patients with high tumor TK1 expression (cytoplasmic TK) had significantly improved mOS upon treatment with TAS-102 compared with placebo. Interestingly, it was observed that patients with high TK1 who received placebo had an overall worse prognosis than those with low TK1.60 Therefore, it was concluded that high TK1 expression is a negative prognostic factor in general but a positive predictor of TAS-102 efficacy. Similarly, the hENT-1 germline SNPs rs760370 and rs9394992 have also been shown to be candidate prognostic and predictive biomarkers in retrospective studies.61 TK1 and hENT-1 expression should be validated as predictive and prognostic biomarkers in future prospective studies of TAS-102.
Combination therapies with TAS-102 and ongoing clinical trials
Given the low response rate associated with TAS-102, there is significant interest in combining it with other effective therapies. Preclinical CRC xenograft models have demonstrated synergy when combining TAS-102 with bevacizumab, panitumumab, or cetuximab.62,63 High quality data from the ML18147 and BRiTE studies support the use of bevacizumab beyond disease progression and into later lines of CRC therapy.16,17 In a Phase I/II trial of TAS-102 combined with bevacizumab, Kuboki et al confirmed that standard dosing of TAS-102 (35 mg/m2 twice daily on days 1–5 and 8–12 of a 28-day cycle) and bevacizumab (5 mg/kg every 14 days) were the RP2Ds, and the study met its primary endpoint; thus, the PFS rate at 16 weeks was 42.9% (95% CI 27.8%–59.0%) in 21 evaluable patients with refractory mCRC.64 mPFS and mOS were relatively impressive at 5.6 and 11.2 months, respectively, although these findings should be validated in a randomized trial. One ongoing randomized Phase II trial is comparing TAS-102 plus bevacizumab therapy with capecitabine plus bevacizumab therapy in patients with untreated mCRC deemed unsuitable for intensive therapy (defined as combination 5-FU with oxaliplatin or irinotecan, TASCO1, NCT02743221, Table 2). While both capecitabine and TAS-102 inhibit TS, TAS-102 could be more effective in untreated mCRC patients because of direct TF-TTP incorporation into DNA and resistance to degradation by DNA glycosylase as described in the “Pharmacology” section above. Our institution has an ongoing Phase II trial of TAS-102 plus bevacizumab as maintenance therapy for patients with mCRC and stable disease or better after first-line chemotherapy (ALEXANDRIA, NCT02654639).65
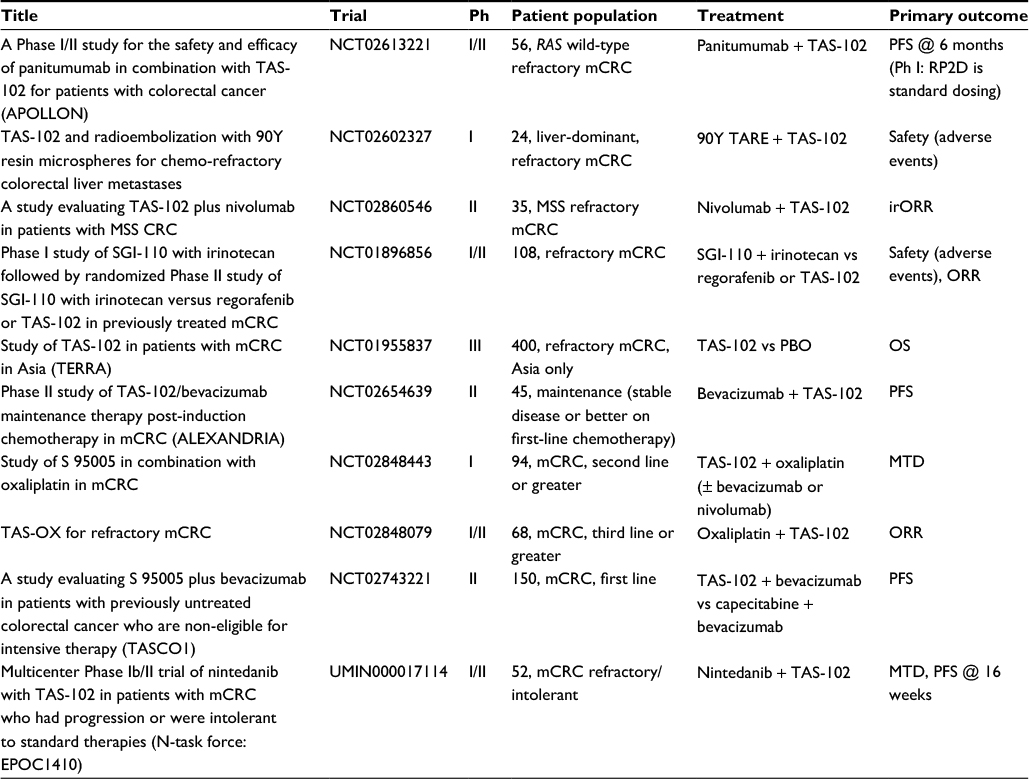 | Table 2 Ongoing TAS-102 trials in mCRC Abbreviations: mCRC, metastatic colorectal cancer; PFS, progression-free survival; RP2D, recommended Phase II dose; TARE, transarterial radioembolization; MSS, microsatellite stable; irORR, immune-related objective response rate; CRC, colorectal cancer; ORR, objective response rate; PBO, placebo; OS, overall survival; Ph, phase; MTD, maximum tolerated dose. |
Another potentially exciting clinical strategy is to combine TAS-102 with the anti-EGFR mAbs cetuximab and panitumumab. Kato et al are conducting the APOLLON trial, a Japanese Phase I/II study of panitumumab combined with TAS-102 in patients with RAS wild-type, refractory mCRC (NCT02613221). Phase I results confirmed that the RP2Ds are standard panitumumab (6 mg/kg every 14 days) and TAS-102 dosing (35 mg/m2 twice daily on days 1–5 and 8–12 of a 28-day cycle), and Phase II is ongoing.66
Aside from the use of combination biological agents in the form of mAbs, there are also ongoing studies using chemotherapy in combination with TAS-102. The combination of TAS-102 and oxaliplatin is supported in mouse xenograft studies.67 TAS-102 and oxaliplatin are being evaluated in an ongoing Phase I/II study of patients with refractory mCRC (NCT02848079). In a Phase I study of combination irinotecan and TAS-102, the RP2D of irinotecan was 150 mg/m2 on days 1 and 15, and the RP2D of TAS-102 was 50 mg/m2/day split into 2 daily doses on days 1–5 and 8–12, both on a 28-day cycle.68 There is also an ongoing Phase I study evaluating TAS-102 in combination with irinotecan and bevacizumab (NCT01916447).
Nintedanib is an oral antiangiogenic agent that inhibits VEGF receptors 1–3, PDGFRs-α/β, FGFR1–3, RET, FLT3, Lck, and Lyn.69,70 An ongoing Phase III trial is comparing the efficacy of nintedanib (200 mg twice daily) plus BSC compared with that of placebo plus BSC, both in 21-day courses (LUME-Colon 1, NCT02149108).71 Additionally, nintedanib is being studied in combination with TAS-102 in a Phase I/II Japanese study. In the Phase I portion of the study, the RP2D of TAS-102 was established at 50 mg/m2/day on days 1–5 and 8–12, and the RP2D of nintedanib was established at 200 mg twice daily, both on a 28-day cycle; the Phase II part of the study is ongoing (UMIN000017114).72
Finally, there is considerable effort underway to combine immune checkpoint inhibitors with other agents to treat microsatellite stable (MSS) mCRC. While pembrolizumab is effective in dMMR or MSI-H mCRC, no objective responses have been seen in MSS mCRC.18,19 Ongoing studies include a Phase II study of nivolumab, an anti-PD1 mAb, combined with TAS-102 in refractory MSS mCRC (NCT02860546) and a Phase I study of TAS-102 and oxaliplatin with or without bevacizumab or nivolumab (NCT02848443). Results from these studies will help inform future combination strategies with TAS-102.
Conclusion
TAS-102 is an excellent addition to the armamentarium of treatments for mCRC. However, valid biomarkers of potential efficacy need to be established to improve patient selection for TAS-102 therapy. Given the better outcomes of patients who develop neutropenia, future studies might evaluate dose and schedule intensification in patients without neutropenia. In addition, there are limited prospective data regarding combination strategies. The increased efficacy of TAS-102 in combination with bevacizumab is supported by Phase II data and is frequently utilized in our clinical practice, although the benefit of bevacizumab in this setting has not been confirmed in a randomized trial. Ongoing studies are evaluating a range of TAS-102 combinations and the use of TAS-102 in earlier lines of therapy. Important strides are being made in the treatment of mCRC, but more effective therapies are still needed.
Acknowledgment
We would like to thank Marion L Hartley for her edits to this review.
Disclosure
The authors report no conflicts of interest in this work.
References
American Cancer Society. Cancer facts & figures 2017. 2017. Available from: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf. Accessed June 30, 2017. | ||
SEER. Cancer of the colon and rectum – cancer stat facts. 2017. Available from: https://seer.cancer.gov/statfacts/html/colorect.html. Accessed June 30, 2017. | ||
de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18(16):2938–2947. | ||
Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015;16(13):1306–1315. | ||
Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1065–1075. | ||
Cheeseman SL, Joel SP, Chester JD, et al. A ‘modified de Gramont’ regimen of fluorouracil, alone and with oxaliplatin, for advanced colorectal cancer. Br J Cancer. 2002;87(4):393–399. | ||
Tournigand C, Andre T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22(2):229–237. | ||
Loupakis F, Cremolini C, Masi G, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371(17):1609–1618. | ||
Hochster HS, Hart LL, Ramanathan RK, et al. Safety and efficacy of oxaliplatin and fluoropyrimidine regimens with or without bevacizumab as first-line treatment of metastatic colorectal cancer: results of the TREE Study. J Clin Oncol. 2008;26(21):3523–3529. | ||
Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–1417. | ||
Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–1034. | ||
Van Cutsem E, Lenz HJ, Kohne CH, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol. 2015;33(7):692–700. | ||
Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30(28):3499–3506. | ||
Tabernero J, Yoshino T, Cohn AL, et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015;16(5):499–508. | ||
Douillard JY, Siena S, Cassidy J, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25(7):1346–1355. | ||
Bennouna J, Sastre J, Arnold D, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 2013;14(1):29–37. | ||
Grothey A, Sugrue MM, Purdie DM, et al. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE). J Clin Oncol. 2008;26(33):5326–5334. | ||
Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. | ||
Le DT, Durham JN, Smith KN, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017; 357(6349):409–413. | ||
FDA. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2017. Available from: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm560040.htm. Accessed July 12, 2017. | ||
Marcus L, Lemery SJ, Khasar S, et al. FDA approval summary: TAS-102. Clin Cancer Res. 2017;23(12):2924–2927. | ||
Heidelberger C, Parsons DG, Remy DC. Syntheses of 5-trifluoromethyluracil and 5-trifluoromethyl-2′-deoxyuridine. J Med Chem. 1964;7:1–5. | ||
Heidelberger C, Boohar J, Kampschroer B. Fluorinated pyrimidines. XXIV. In vivo metabolism of 5-trifluoromethyluracil-2-C-14 and 5-trifluoromethyl-2′-deoxyuridine-2-C-14. Cancer Res. 1965;25:377–381. | ||
Reyes P, Heidelberger C. Fluorinated pyrimidines. XXVI. Mammalian thymidylate synthetase: its mechanism of action and inhibition by fluorinated nucleotides. Mol Pharmacol. 1965;1(1):14–30. | ||
Santi DV, Sakai TT. Thymidylate synthetase. Model studies of inhibition by 5-trifluoromethyl-2′-deoxyuridylic acid. Biochemistry. 1971;10(19):3598–3607. | ||
Eckstein JW, Foster PG, Finer-Moore J, Wataya Y, Santi DV. Mechanism-based inhibition of thymidylate synthase by 5-(trifluoromethyl)-2′-deoxyuridine 5′-monophosphate. Biochemistry. 1994;33(50):15086–15094. | ||
Aherne GW, Hardcastle A, Raynaud F, Jackman AL. Immunoreactive dUMP and TTP pools as an index of thymidylate synthase inhibition; effect of tomudex (ZD1694) and a nonpolyglutamated quinazoline antifolate (CB30900) in L1210 mouse leukaemia cells. Biochem Pharmacol. 1996;51(10):1293–1301. | ||
Webley SD, Welsh SJ, Jackman AL, Aherne GW. The ability to accumulate deoxyuridine triphosphate and cellular response to thymidylate synthase (TS) inhibition. Br J Cancer. 2001;85(3):446–452. | ||
Emura T, Nakagawa F, Fujioka A, Ohshimo H, Kitazato K. Thymidine kinase and thymidine phosphorylase level as the main predictive parameter for sensitivity to TAS-102 in a mouse model. Oncol Rep. 2004;11(2):381–387. | ||
Markley JC, Chirakul P, Sologub D, Sigurdsson ST. Incorporation of 2′-deoxy-5-(trifluoromethyl)uridine and 5-cyano-2′-deoxyuridine into DNA. Bioorg Med Chem Lett. 2001;11(18):2453–2455. | ||
Heidelberger C, Dexter DL, Wolberg WH. Clinical pharmacology of 5-trifluoromethyl-2′-deoxyuridine (F3TDR). Proc Am Assoc Cancer Res. 1970;11:35. | ||
Fukushima M, Suzuki N, Emura T, et al. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′-deoxyribonucleosides. Biochem Pharmacol. 2000;59(10):1227–1236. | ||
Moghaddam A, Bicknell R. Expression of platelet-derived endothelial cell growth factor in Escherichia coli and confirmation of its thymidine phosphorylase activity. Biochemistry. 1992;31(48):12141–12146. | ||
Brown NS, Bicknell R. Thymidine phosphorylase, 2-deoxy-D-ribose and angiogenesis. Biochem J. 1998;334(Pt 1):1–8. | ||
van Triest B, Pinedo HM, Blaauwgeers JL, et al. Prognostic role of thymidylate synthase, thymidine phosphorylase/platelet-derived endothelial cell growth factor, and proliferation markers in colorectal cancer. Clin Cancer Res. 2000;6(3):1063–1072. | ||
Temmink OH, Emura T, de Bruin M, Fukushima M, Peters GJ. Therapeutic potential of the dual-targeted TAS-102 formulation in the treatment of gastrointestinal malignancies. Cancer Sci. 2007;98(6):779–789. | ||
Suzuki N, Nakagawa F, Nukatsuka M, Fukushima M. Trifluorothymidine exhibits potent antitumor activity via the induction of DNA double-strand breaks. Exp Ther Med. 2011;2(3):393–397. | ||
Suzuki N, Emura T, Fukushima M. Mode of action of trifluorothymidine (TFT) against DNA replication and repair enzymes. Int J Oncol. 2011;39(1):263–270. | ||
Hong DS, Abbruzzese JL, Bogaard K, et al. Phase I study to determine the safety and pharmacokinetics of oral administration of TAS-102 in patients with solid tumors. Cancer. 2006;107(6):1383–1390. | ||
Overman MJ, Varadhachary G, Kopetz S, et al. Phase 1 study of TAS-102 administered once daily on a 5-day-per-week schedule in patients with solid tumors. Invest New Drugs. 2008;26(5):445–454. | ||
Doi T, Ohtsu A, Yoshino T, et al. Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumours. Br J Cancer. 2012;107(3):429–434. | ||
Overman MJ, Kopetz S, Varadhachary G, et al. Phase I clinical study of three times a day oral administration of TAS-102 in patients with solid tumors. Cancer Invest. 2008;26(8):794–799. | ||
Bendell JC, Rosen LS, Mayer RJ, et al. Phase 1 study of oral TAS-102 in patients with refractory metastatic colorectal cancer. Cancer Chemother Pharmacol. 2015;76(5):925–932. | ||
Yoshino T, Mizunuma N, Yamazaki K, et al. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2012;13(10):993–1001. | ||
Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372(20):1909–1919. | ||
Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245–255. | ||
Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–312. | ||
Masuishi T, Taniguchi H, Hamauchi S, et al. Regorafenib versus trifluridine/tipiracil for refractory metastatic colorectal cancer: a retrospective comparison. Clin Colorectal Cancer. 2017;16(2):e15–e22. | ||
Sueda T, Sakai D, Kudo T, et al. Efficacy and safety of regorafenib or TAS-102 in patients with metastatic colorectal cancer refractory to standard therapies. Anticancer Res. 2016;36(8):4299–4306. | ||
Hamauchi S, Yamazaki K, Masuishi T, et al. Neutropenia as a predictive factor in metastatic colorectal cancer treated with TAS-102. Clin Colorectal Cancer. 2017;16(1):51–57. | ||
Kasi PM, Kotani D, Cecchini M, et al. Chemotherapy induced neutropenia at 1-month mark is a predictor of overall survival in patients receiving TAS-102 for refractory metastatic colorectal cancer: a cohort study. BMC Cancer. 2016;16:467. | ||
Nishina T, Yoshino T, Shinozaki E, et al. Onset of neutropenia as an indicator of treatment response in the randomized phase II study of TAS-102 vs placebo in Japanese patients with metastatic colorectal cancer (study J003-10040030). J Clin Oncol. 2016;34(Suppl):Abstr 3557. | ||
Ohtsu A, Yoshino T, Falcone A, et al. Onset of neutropenia as an indicator of treatment response in the phase 3 RECOURSE trial of trifluridine/tipiracil (TAS-102) versus placebo in patients with metastatic colorectal cancer. J Clin Oncol. 2017;35(Suppl 4S):Abstr 775. | ||
Van Cutsem E, Ohtsu A, Leger C, et al. Effect of number of prior lines versus prior regimens in pts receiving trifluridine/tipiracil (TAS-102). J Clin Oncol. 2017;35(Suppl 4S):Abstr 720. | ||
Van Cutsem E, Benedetti FM, Mizuguchi H, et al. TAS-102 versus placebo (PBO) in patients (pts) ≥ 65 years (y) with metastatic colorectal cancer (mCRC): an age-based analysis of the recourse trial. J Clin Oncol. 2016;34(Suppl 4S):Abstr 638. | ||
Hustedt N, Durocher D. The control of DNA repair by the cell cycle. Nat Cell Biol. 2016;19(1):1–9. | ||
Suenaga M, Schirripa M, Cao S, et al. Genetic variants of DNA repair-related genes predict efficacy of TAS-102 in patients with refractory metastatic colorectal cancer. Ann Oncol. 2017;28(5):1015–1022. | ||
Shintani M, Urano M, Takakuwa Y, Kuroda M, Kamoshida S. Immunohistochemical characterization of pyrimidine synthetic enzymes, thymidine kinase-1 and thymidylate synthase, in various types of cancer. Oncol Rep. 2010;23(5):1345–1350. | ||
Temmink OH, Bijnsdorp IV, Prins HJ, et al. Trifluorothymidine resistance is associated with decreased thymidine kinase and equilibrative nucleoside transporter expression or increased secretory phospholipase A2. Mol Cancer Ther. 2010;9(4):1047–1057. | ||
Yoshino T, Shinozaki E, Yamazaki K, et al. Effect of thymidine kinase 1 expression on prognosis and treatment outcomes in refractory metastatic colorectal cancer: results from two randomized studies of TAS-102 versus a placebo. J Clin Oncol. 2017;35(Suppl 4S):Abstr 529. | ||
Zhang W, Suenaga M, Schirripa M, et al. Genetic variants of hENT-1 to predict efficacy of TAS-102 in patients with refractory metastatic colorectal cancer. J Clin Oncol. 2016;34(Suppl):Abstr 3580. | ||
Tsukihara H, Nakagawa F, Sakamoto K, et al. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, together with bevacizumab, cetuximab, or panitumumab on human colorectal cancer xenografts. Oncol Rep. 2015;33(5):2135–2142. | ||
Baba Y, Tamura T, Satoh Y, et al. Panitumumab interaction with TAS-102 leads to combinational anticancer effects via blocking of EGFR-mediated tumor response to trifluridine. Mol Oncol. 2017;11(8):1065–1077. | ||
Kuboki Y, Nishina T, Shinozaki E, et al. An investigator initiated multicenter phase I/II study of TAS-102 with bevacizumab for metastatic colorectal cancer refractory to standard therapies (C-TASK FORCE). J Clin Oncol. 2015;33(Suppl):Abstr 3544. | ||
Salem ME, Wang H, Fakih M, et al. An open-label, multi-center, phase 2 study of switch maintenance with TAS-102 plus bevacizumab following oxaliplatin or irinotecan-based fluoropyrimidine-containing induction chemotherapy in patients with metastatic colorectal cancer: ALEXANDRIA study. J Clin Oncol. 2016;34(Suppl):Abstr TPS3624. | ||
Kato T, Kagawa Y, Komatsu Y, et al. A phase I/II study for panitumumab combined with TAS-102 in patients with RAS wild-type metastatic colorectal cancer (APOLLON study): phase I results. J Clin Oncol. 2017;35(Suppl 4S):Abstr 770. | ||
Nukatsuka M, Nakagawa F, Takechi T. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, with oxaliplatin on human colorectal and gastric cancer xenografts. Anticancer Res. 2015;35(9):4605–4615. | ||
Doi T, Yoshino T, Fuse N, et al. Phase I study of TAS-102 and irinotecan combination therapy in Japanese patients with advanced colorectal cancer. Invest New Drugs. 2015;33(5):1068–1077. | ||
Mross K, Buchert M, Frost A, et al. Vascular effects, efficacy and safety of nintedanib in patients with advanced, refractory colorectal cancer: a prospective phase I subanalysis. BMC Cancer. 2014;14:510. | ||
Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774–4782. | ||
Van Cutsem E, Yoshino T, Hocke J, Oum’Hamed Z, Studeny M, Tabernero J. Rationale and design for the LUME-colon 1 study: a randomized, double-blind, placebo-controlled phase III trial of nintedanib plus best supportive care versus placebo plus best supportive care in patients with advanced colorectal cancer refractory to standard treatment. Clin Colorectal Cancer. 2016;15(1):91–94. | ||
Nishina T, Kuboki Y, Shinozaki S, et al. A multicentre phase I/II study of TAS-102 with nintedanib in patients with metastatic colorectal cancer refractory to standard therapies (N-task force: EPOC1410); phase I results. Ann Oncol. 2016;27(Suppl 6):Abstr 472P. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.