Back to Journals » International Journal of Nanomedicine » Volume 12
Electrostatic interactions between polyglutamic acid and polylysine yields stable polyion complex micelles for deoxypodophyllotoxin delivery
Authors Wang Y, Huang L, Shen Y, Tang L, Sun R, Shi D ![]() , Webster TJ
, Webster TJ ![]() , Tu J, Sun C
, Tu J, Sun C
Received 27 April 2017
Accepted for publication 21 June 2017
Published 30 October 2017 Volume 2017:12 Pages 7963—7977
DOI https://doi.org/10.2147/IJN.S140573
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Israel Rubinstein
Yutong Wang,1–3,* Liping Huang,1,2,* Yan Shen,1,2,* Lidan Tang,1,2,4 Runing Sun,1,5 Di Shi,6 Thomas J Webster,6 Jiasheng Tu,1,2 Chunmeng Sun1,2
1Center for Research Development and Evaluation of Pharmaceutical Excipients and Generic Drugs, China Pharmaceutical University, 2State Key Laboratory of Natural Medicines, Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, 3Department of Pharmacy, Nanjing University of Chinese Medicine, Nanjing, 4Changzhou Second People’s Hospital, Changzhou, 5School of Engineering, China Pharmaceutical University, Nanjing, People’s Republic of China; 6Department of Chemical Engineering, Northeastern University, Boston, MA, USA
*These authors contributed equally to this work
Abstract: To achieve enhanced physical stability of poly(ethylene glycol)-poly(D,L-lactide) polymeric micelles (PEG-PDLLA PMs), a mixture of methoxy PEG-PDLLA-polyglutamate (mPEG-PDLLA-PLG) and mPEG-PDLLA-poly(L-lysine) (mPEG-PDLLA-PLL) copolymers was applied to self-assembled stable micelles with polyion-stabilized cores. Prior to micelle preparation, the synthetic copolymers were characterized by 1H-nuclear magnetic resonance (NMR) and infrared spectroscopy (IR), and their molecular weights were calculated by 1H-NMR and gel permeation chromatography (GPC). Dialysis was used to prepare PMs with deoxypodophyllotoxin (DPT). Transmission electron microscopy (TEM) images showed that DPT polyion complex micelles (DPT-PCMs) were spherical, with uniform distribution and particle sizes of 36.3±0.8 nm. In addition, compared with nonpeptide-modified DPT-PMs, the stability of DPT-PCMs was significantly improved under various temperatures. In the meantime, the pH sensitivity induced by charged peptides allowed them to have a stronger antitumor effect and a pH-triggered release profile. As a result, the dynamic characteristic of DPT-PCM was retained, and high biocompatibility of DPT-PCM was observed in an in vivo study. These results indicated that the interaction of anionic and cationic charged polyionic segments could be an effective strategy to control drug release and to improve the stability of polymer-based nanocarriers.
Keywords: polyion complex micelles, electrostatic interaction, oligopeptide, stability, pharmacokinetics
Introduction
Polymeric micelles (PMs) have been widely applied as nanocarriers to deliver insoluble drugs due to their amphiphilic properties.1,2 PMs are usually formed by the self-assembling of amphiphilic block copolymers, among which poly(D,L-lactide) (PDLLA) as the hydrophobic segment and poly(ethylene glycol) (PEG) as the hydrophilic segment are the most popular candidates based on their biodegradability and biocompatibility.3,4 With the addition of PEGylation, which is a typical approach to reduce nonspecific cellular uptake and therefore offers a stealth immune effect,5,6 methoxy PEG (mPEG)-PDLLA micelles have been evaluated as biodegradable and biocompatible nanocarriers in numerous in vitro and in vivo studies.7–10 In addition, due to their ease of modification, PEG-PDLLA-based nanoparticles can achieve both passive and active targeting effects via different pathways, among which the enhanced permeability and retention (EPR) effect and receptor-mediated cellular uptake are frequently discussed.11–14
Although it is well known that in vitro and in vivo stability is crucial for a drug delivery system to withstand dissociation and premature release of cargo after entry into the bloodstream, the inherent instability of PMs still remains a significant challenge, inhibiting their clinical use. Improving their stability under physiological conditions can lead to dramatic improvements in pharmacokinetics and thereby break the bottleneck to expand clinical applications of PMs.15 For this, core-crosslinked strategies have been proposed and polymerizable acrylate,16 methacrylate,17 or cinnamyl groups18 have been introduced into the terminal or side chains of core-forming hydrophobic blocks. As a result, micelles exhibited high stability against sodium dodecyl sulfate (SDS) surfactant as well as organic solvents, and drug loading efficiency and thermodynamic stability were significantly enhanced against dilution. In addition to chemical cross-linking, the stability of micelles can also be improved by introducing physical interactions such as isomer interactions,19 π-π interactions,20 and electrostatic reactions21 inside the micelle core. Among all the interactions, the electrostatic reaction is an ideal option as it can deliver small and large charged molecules by reacting with block ionomers to form a stable core.22,23
Usually, the PMs synthesized by electrostatic reactions consist of 2 oppositely charged polyelectrolytes. This electrostatic complex has a core–shell structure, which is termed as polyion complex micelle (PCM).24,25 Unlike common micelles that are formed only with amphiphilic block copolymers, electronic segments are conjugated onto the lipophilic terminus of the block copolymers and the PCM is formed with a complex of cationic and anionic segments through electrostatic interactions. Through this unique method, the synthesized PCM will not only be more stable than the normal PMs but also exhibit a better ability to respond to external stimuli such as pH,26 ionic strength, and temperature.27,28
In the current study, in order to deliver hydrophobic deoxypodophyllotoxin (DPT), which is a promising microtubule-targeted antitumor agent and a cell cycle-regulatory protein modulator,29,30 2 block copolymers with oppositely charged tails, namely, mPEG-block-PDLLA-block-poly(glutamic acid) (mPEG-PDLLA-PLG) and mPEG-block-PDLLA-block-poly(L-lysine) (mPEG-PDLLA-PLL) were engineered. These copolymers can be self-assembled into micelles through electrostatic interactions between PLG and PLL. In the meantime, DPT can be entrapped inside the micelles by a lipophilic PDLLA chain. Here, for the first time, the physical stability and drug release of DPT-PCM were investigated and the biocompatability properties of DPT-PCM and DPT-PM compared.
Materials and methods
Materials and animals
DPT was received from the Institute of Chemical Industry, China Pharmaceutical University. D,L-lactic acid was purchased from Ling Feng Chemical Reagent Co (Shanghai, China). mPEG-OH, with a molecular weight of 2,000 g/mol, was purchased from Sigma-Aldrich (Sigma-Aldrich, St Louis, MO, USA). Dicyclohexylcarbodiimide (DCC), dimethylaminopyridine (DMAP) and N-(tert-butoxycarbonyl)-L-phenylalanine (Phe-NBOC) were purchased from J&K Chemical Ltd (Beijing, China). Cyclic carboxylic acid anhydrides (N-carboxyanhydrides or NCAs) of glutamic acid and lysine were obtained from Nanjing Chemlin Chemical Industry Co, Ltd (China). New Zealand rabbits and male Sprague Dawley rats were purchased from Qinglongshan Animal Center (Nanjing, China). All animal experiments were conducted in accordance with the terms regulated by the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Ethics Committee of China Pharmaceutical University.
Synthesis of mPEG-PDLLA-PLG and mPEG-PDLLA-PLL
Synthesis of mPEG-PDLLA
mPEG-PDLLA copolymers were synthesized by ring-opening polymerization (Figure 1B).31 Briefly, as shown in Figure 1A, mPEG (50.0 g) and D,L-lactide (50.0 g, 1:1 w/w%) were added into a 3-necked flask, and the system was vacuumed and protected with nitrogen flow. The mixture was completely melted at 130°C and then cooled to 110°C. Next, 1 mL of stannous octoate in methylbenzene (20%, v/v%) was added under a N2 stream. Subsequently, the sealed flask was vacuum-dried to remove the water and methylbenzene and subsequently kept in a silicone oil bath at 130°C with continuous stirring for 6 h. The synthesized polymer was dissolved in methylene chloride, followed by precipitation in ice-cold diethyl ether 3 times. The resultant precipitates were collected and dried in vacuum at room temperature for 24 h.
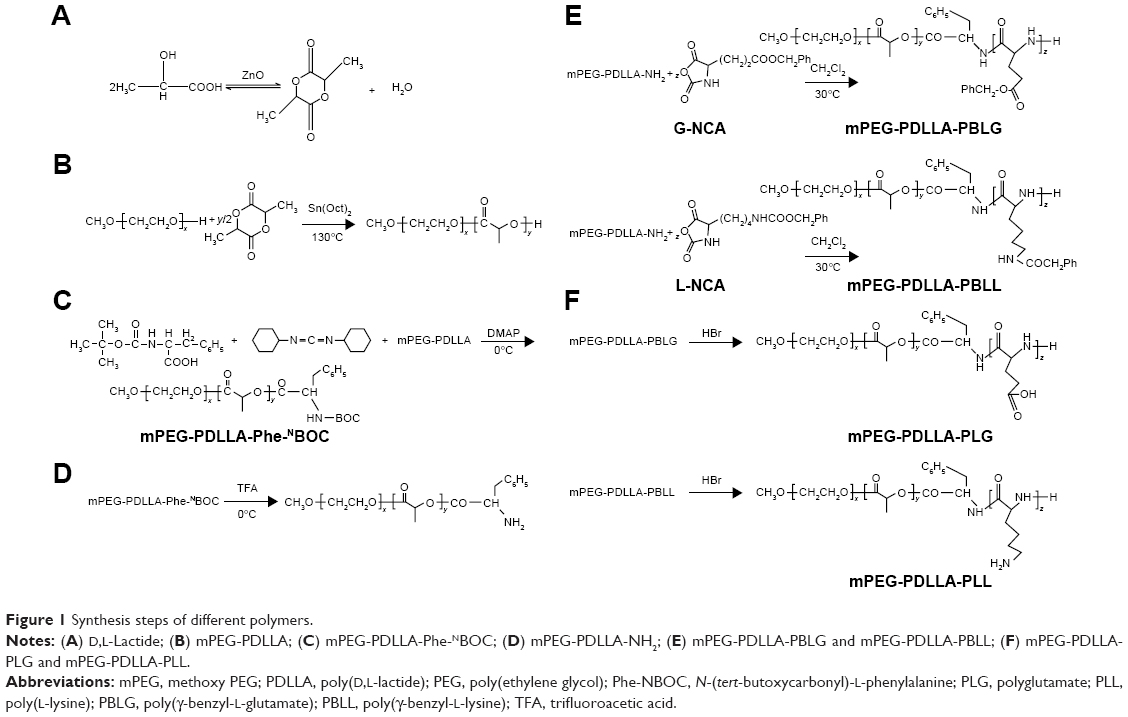 | Figure 1 Synthesis steps of different polymers. |
Synthesis of mPEG-PDLLA-Phe-NBOC
For the synthesis of mPEG-PDLLA-Phe-NBOC32,33 (Figure 1C), mPEG-PDLLA (10.0 g) and Phe-NBOC (4.0 g) were dissolved in 80 mL of cold CH2Cl2 in a predried flask. Subsequently, DCC (3.0 g) and DMAP (0.1 g) were added into the flask for esterification at 0°C under protection of a N2 stream. After a 48 h reaction, excessive DCC was removed by filtration. The filtrate was washed with 80 mL of saturated aqueous NaHCO3 twice, followed by 80 mL of H2O twice. Vacuum-rotary evaporation was carried out to remove the residual DCC. Cold ethyl ether was applied to precipitate the resultant product in CH2Cl2, and the precipitated polymers were collected by filtration and then vacuum-dried at room temperature for 24 h.
Synthesis of mPEG-PDLLA-NH2
In this procedure (Figure 1D), mPEG-PDLLA-Phe-NBOC (6.0 g) was dissolved in 30 mL of CH2Cl2. The solution was cooled to 0°C in an ice bath and made to react with trifluoroacetic acid (TFA) (15 mL) for 2 h under a N2 flow. The resultant solution was then washed with 50 mL of saturated aqueous NaHCO3 twice, followed with 50 mL H2O twice. Next, the CH2Cl2 layer was collected and evaporated to a volume of 20 mL and was mixed with cold ethyl ether for precipitation; the precipitate was dried in a vacuum for 24 h at room temperature.32,33
Synthesis of triblock copolymer mPEG-PDLLA-poly(γ-benzyl-L-glutamate) (PBLG)/poly(γ-benzyl-L-lysine) (PBLL)
In this procedure (Figure 1E), mPEG-PDLLA-NH2 (4.0 g) and either γ-benzyl-L-glutamate-N-carboxyanhydride (G-NCA) (1.3 g) or γ-benzyl-L-lysine-N-carboxyanhydride (L-NCA) (1.5 g) were dissolved in chloroform (75 mL), and the solution was stirred for 72 h at 30°C under a N2 flow. Next, the CH2Cl2 layer was collected and evaporated to a volume of 20 mL and was mixed with cold ethyl ether for precipitation; the precipitate was then vacuum-dried for 24 h at room temperature.
Synthesis of mPEG-PDLLA-PLG and mPEG-PDLLA-PLL
Deprotection of mPEG-PDLLA-PBLG/PBLL was carried out as shown in Figure 1F. For this, mPEG-PDLLA-PBLG/PBLL (1.0 g) and 33 wt% HBr (33 wt%) were dissolved in 10 mL of acetic acid in a 3-necked flask. After stirring for 1 h under N2 flow, the deprotected copolymers were precipitated by treating with an excess of cold methanol, followed by vacuum-drying at room temperature for 24 h.32
Characteristics of mPEG-PDLLA-PLG and mPEG-PDLLA-PLL
Molecular weight calculation
As shown in Figure 2, the integrated areas of methylene protons of “g” and “p” at 3.5–3.7 ppm and 4.9–5.1 ppm, as well as the methyne protons of “i” at 5.1–5.3 ppm, were recorded. The composition of the copolymers was calculated using the following equations.
 | Figure 2 Structural representations of mPEG-PDLLA-PBLG and mPEG-PDLLA-PBLL. |
Areai:Areag:Areap = y:4x:2z x = 2,000/44 = 45.5 y = x × 4 × Areai/Areag z = x × 2 × Areap/Areag Molecular weight of mPEG − PDLLA − PBLG = 44x + 72y + 219z Molecular weight of mPEG − PDLLA − PBLL = 44x + 72y + 246z |
where x, y, and z are the numbers of the different blocks in the polymer, as shown in Figure 2.
1H-nuclear magnetic resonance (NMR) and Fourier transform near-infrared (FT-NIR) spectra measurements
1H-NMR measurements were performed with dimethyl sulfoxide (DMSO)-d6 and CDCl3 using a Varian 300 MHz instrument (Bruker, Billerica, MA, USA). Sample temperature at 25°C was used for all measurements. FT-NIR spectra were recorded on a TENSOR 27 spectrometer (Bruker) by the KBr method. The average molecular weight of mPEG-PDLLA-PBLG/PBLL and mPEG-PDLLA-PLG/PLL was determined by 1H-NMR and gel permeation chromatography (GPC) using a Shimadzu (Kyoto, Japan) high-performance liquid chromatography (HPLC) system at 35°C. Tetrahydrofuran was used as the mobile phase. Seven polystyrene standards (Polymer Laboratories Ltd, Church Stretton, Shropshire, UK), with peak molecular weights of 1,930, 3,070, 5,120, 6,930, 10,680, 12,830, and 19,760 Da were used for molecular weight calibration. Accordingly, 20 μL of the sample was injected, and the mobile phase was pumped at a flow rate of 1 mL/min (LC-20AT). Detection was performed using a RID-10A refractive index detector. The obtained chromatogram was interpreted using GPC software.34
Preparation of DPT-PCM and DPT-PM
The PCM was prepared by a well-established dialysis method.35,36 Briefly, mPEG-PDLLA-PLG (50 mg) and mPEG-PDLLA-PLL (50 mg) at a 1:1 (w/w%) ratio were separately dissolved in 2 mL of DMSO. Then, 10 mL of DPT was added to each solution. Subsequently, 8 mL of phosphate-buffered saline solution (PBS; 10 mmol/L, pH 7.4) was dropped slowly into each solution. Finally, the PCM was prepared by mixing the 2 solutions dropwise in a new bottle. The PCM solution was stirred for 0.5 h, then dialyzed in a dialysis bag (with a molecular weight cutoff of 3.5 kDa) against PBS for 1 h, followed by dialysis against water for 4 h. Subsequently, the PCM solution was centrifuged at 10,000 rpm for 10 min. After filtration of the supernatant through a 0.22 μm pore size membrane, DPT-loaded PCM (DPT-PCM) was collected by lyophilization. DPT-loaded mPEG-PDLLA micelles (DPT-PMs) were prepared in the same manner and served as the control.
Loading (LE%) and entrapment (EE%) efficiency of DPT
For LE% and EE% assays, an appropriate amount of DPT-PCM was dissolved in water (1.0 mg/mL), followed by the addition of methanol to make a 20-times dilution. Subsequently, 20 μL of the solution was separated by a reversed-phase C18 column (Hedera ODS-2, 250×4.6 mm, 5 μm) at 30°C using HPLC (Ultimate 3000; Dionex Corporation, Sunnyvale, CA, USA).37 The mobile phase was a mixture of methanol–water (75:25, v/v) at a flow rate of 1.0 mL/min. Detection was performed at 294 nm (ultraviolet detector). LE% and EE% of DPT in PCM and PM were calculated using the following equations.38,39
|

Morphology and differential scanning calorimetry (DSC) analysis
Particle size was measured using a particle size and zeta potential analyzer (90 Plus Zeta; Brookhaven Instruments Corporation, Holtsville, NY, USA). The morphology of DPT-PCM and DPT-PM was observed using transmission electron microscopy (TEM) (H-7650; Hitachi, Tokyo, Japan). Both lyophilized powders were dissolved and diluted to 1.0 mg/mL with pure water in an individual tube before TEM measurements. A drop of each sample was applied to a copper grid coated with a carbon film and air-dried; a 2% (w/v) solution of phosphotungstic acid (PTA) was then dropped onto the grids. After negative staining and spontaneous drying at room temperature, the samples were examined under a microscope.
Thermal analysis of DPT, the polyion complexes (PICs), the physical mixture of DPT and PIC (1:5, w/w%), and DPT-PCM (weight ratio of DPT/PIC =1:5, w/w%) was carried out by DSC (DSC 204 F1 Phoenix; Netzsch, Selb, Germany) at a heating rate of 10°C/min.
Stability assay
DPT-PCM and DPT-PM were dissolved in 5% glucose injections (1.0 mg/mL). Drug content was determined by the aforementioned HPLC method after incubation at 4°C, 25°C, and 37°C for different times. The drug content was calculated using the following equation:
|
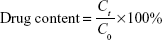
where C0 represents the original concentration of DPT in PCM or PM, and Ct represents the concentration of DPT in PCM or PM at different times.
In vitro cytotoxicity
An MTT assay was performed to study the cytotoxicity of the 2 kinds of micelles and DPT. A549 cells were purchased from Cell Bank of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, and were seeded in 96-well plates at a density of 2,000–5,000 cells per well and cultured in an incubator at 37°C with 5% CO2 for 24 h. Cell culture medium (100 μL) containing DPT, DPT-PM, or DPT-PCM at various concentrations was added to the 96-well plates, followed by incubation for either 24 h or 48 h. Then, 20 μL of MTT in PBS (5 mg/mL) was added to each well, and the cells were incubated for another 4 h. The blue–purple formazan crystals in each well were dissolved with 150 μL of DMSO, and cell viability was monitored by recording the absorbance at 490 nm. Blank PM and blank PCM were used as controls. Results were expressed as percentage cell viability.
In vitro drug release
An appropriate amount of drug-loaded micelles was dissolved in 5% glucose injection and diluted to a final concentration of DPT at 1 mg/mL. Then, 1 mL of the solution was sealed in dialysis bags (cellulose membrane, 3.5 kDa cutoff) and incubated in 100 mL PBS with 1% Polysorbate-80 at different pH values (5.0, 6.8, and 7.4) at 37°C in a water bath shaking at 75 rpm. At each predetermined time interval, 1 mL of sample from each tube was collected and the same volume of corresponding medium was replenished immediately. DPT concentration was then determined using HPLC as previously described. A control experiment was also conducted to identify the release profile of DPT-PM under the same condition. All tests were conducted in triplicate.
Hemolytic assay
To compare the hemocompatibility of DPT-PCM and DPT-PM, a hemolytic study was conducted using rabbit blood. DPT-PCM and DPT-PM were dissolved and diluted with a glucose solution (5%) to a concentration of 1.5 mg/mL. An 82% erythrocyte suspension and normal saline were then added. After incubation for 3 h at 37°C, the samples were centrifuged at 1,500 rpm for 15 min, and absorbance was measured at 540 nm. The hemolysis rate was calculated using the following equation:40
|
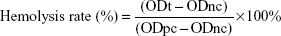
where ODt is the absorbance of the sample, ODnc is the absorbance of the negative control, and ODpc is the absorbance of the positive control.
Injection irritation
Ten New Zealand rabbits (1.8–2.0 kg) were equally randomized into 2 groups (n=5). DPT-PCM and DPT-PM were dissolved and diluted with a 5% glucose solution to a final concentration of 1.5 mg/mL. The 2 groups of rabbits were then treated with DPT-PCM and DPT-PM solutions at a dose of 5 mg/kg via a left marginal ear vein at a rate of 1 mL/min for 3 d. In the meantime, the same volume of glucose solution (5%) was given via the right marginal ear vein as the control. Any paradoxical reactions were recorded by an experienced unbiased observer. Injection sites and proximal regions were fixed with a 10% formaldehyde solution. The fixed tissues were embedded in paraffin and 4 μm-thick sections were obtained using a microtome. The sectioned tissues were mounted on glass slides, followed by staining with hematoxylin and eosin (H–E) for evaluating the cellular reaction at the injection site.40–42
In vivo pharmacokinetics
Twelve rats were equally randomized into 2 groups, which were the DPT-PCM group (Group A) and the DPT-PM group (Group B). DPT-PCM and DPT-PM were diluted with glucose solution (5%) to a concentration of 6 mg/mL and were injected at a dose of 15 mg/kg. Blood samples from each group were collected at predetermined time intervals and stored in heparinized tubes. All samples were centrifuged at 8,000 rpm for 5 min, and the supernatant was obtained and frozen at −20°C for HPLC analysis. Plasma data were subjected to a 2-compartment model for pharmacokinetic analysis using PKSlover version 3.0.43 The maximum plasma concentration (Cmax) was obtained directly from the concentration–time plot. The mean residence time (MRT) was calculated as AUMC0–inf/AUC0–inf, where AUMC0–inf represents the area under the first moment plasma concentration–time curve and AUC0–inf represents the total area under the curve.
Statistical analysis
All data are presented as mean value ± SD. Each experiment was conducted at least in triplicate and differences between mean values were determined using analysis of variance (ANOVA) and Students t-tests.
Results and discussion
Characterization of mPEG-PDLLA-PLG and mPEG-PDLLA-PLL
The 1H-NMR spectra in Figure S1A showed that mPEG-PDLLA-PLG and mPEG-PDLLA-PLL copolymers were successfully synthesized. Small peaks, which were assigned to phenyl and methyl protons from Phe-NBOC units, at 7.25 and 1.30 ppm (Figure S1B) confirmed the successful conjugation of Phe-NBOC to mPEG-PDLLA. The undetectable peaks at 1.30 ppm (Figure S1C) confirmed the removal of the tertiary butyl from mPEG-PDLLA-Phe-NBOC. The peaks at 4.20 ppm (g, Figure S1D), 2.04 ppm (h, Figure S1D), 2.22 ppm (i, Figure S1D), 5.11 ppm (j, Figure S1D), 7.33 ppm (k, Figure S1D), 4.18 ppm (l, Figure S1E), 1.65 ppm (m, Figure S1E), 2.98 ppm (n, Figure S1E), 5.00 ppm (o, Figure S1E), and 7.34 ppm (p, Figure S1E) confirmed the successful conjugation of PBLG and PBLL. Peaks at 5.11 ppm (j, Figure S1F), 7.33 ppm (k, Figure S1F), 5.00 ppm (o, Figure S1G), and 7.34 ppm (p, Figure S1G) were not detected because of the removal of the carbobenzoxy group from PBLG and PBLL.
The IR spectra in Figure S2A also confirmed that the PEG-PDLLA-PLG and mPEG-PDLLA-PLL copolymers were successfully synthesized. Results showed that mPEG-PDLLA-PBLG and mPEG-PDLLA-PBLL (Figure S2B and C) had characteristic peaks at 3,323, 1,690, 1,637 and 1,548 cm−1, which were attributed to symmetrical and asymmetrical stretching vibrations (υN–H), 2 kinds of (υC=O) and (υCO-NH) of PBLG and PBLL, respectively. Characteristic peaks at 751 and 699 cm−1 were attributed to stretching vibrations in phenyl groups. In Figure S2D and E, the peaks at 751 and 699 cm−1 were not detected, which confirmed the successful detachment of phenyl in PBLG/PBLL and the successful synthesis of PEG-PDLLA-PLG and mPEG-PDLLA-PLL copolymers.
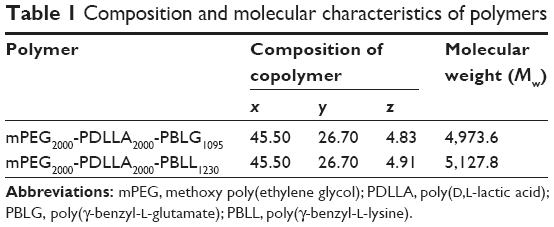
The parameters x, y, and z were calculated from 1H-NMR. The z-values of the 2 copolymers were 4.83 and 4.91, which were close to the theoretical value of 5.0 (Table 1). The molecular weights of the copolymers were calculated by 1H-NMR and GPC. Molecular weights calculated from 1H-NMR were consistent with the theoretical molecular weights (Table 2). Results also showed that the molecular weights of the conjugated polymers measured by GPC displayed a narrow polydispersity, implying that the product has high purity.
 | Table 1 Composition and molecular characteristics of polymers |
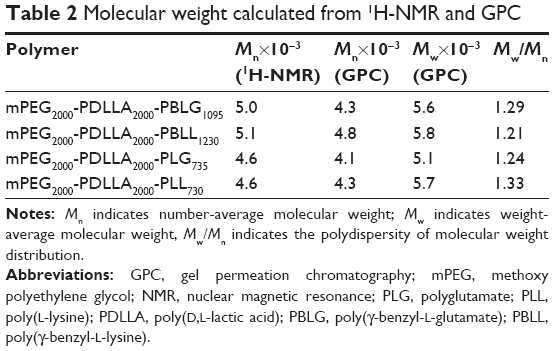 | Table 2 Molecular weight calculated from 1H-NMR and GPC |
Herein, 2 pairs of oppositely charged copolymers were synthesized. It should be noted that the lengths of PLG and PLL were almost equal in each pair of copolymers (Table 1). This is important for DPT-PCM preparation since it has been proven that DPT-PCM is formed by matched pairs with the same block lengths of polyanions and polycations under stoichiometric conditions.35
Characterization of DPT-PCM and DPT-PM
As shown in Table 3, similar drug loading and encapsulation efficiencies for DPT were found between PCM and PM, suggesting that the introduction of polyelectrolytes in PCM had no negative effects on these properties.
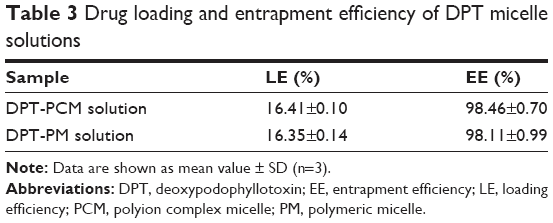 | Table 3 Drug loading and entrapment efficiency of DPT micelle solutions |
Particle size, zeta potential, and morphology results are shown in Table 4 and Figure 3A. The sizes of DPT-PCM and DPT-PM obtained from the zeta potential analyzer were 36.3±0.8 nm and 27.3±0.4 nm, respectively. Compared to DPT-PM, the increased particle size of DPT-PCM may be due to the existence of a polyionic core that consisted of prolonged segments of PLG and PLL; however, this needs further investigation. TEM results showed that the prepared micelles exhibited a uniform spherical shape with a narrow polydispersity. In addition, zeta potential results showed no difference between the 2 micelle types.
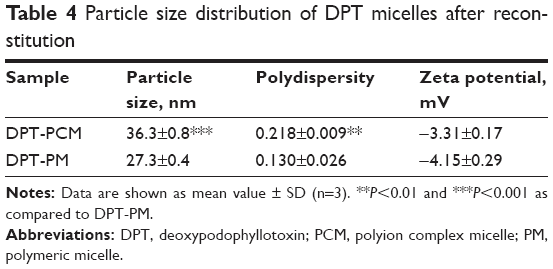 | Table 4 Particle size distribution of DPT micelles after reconstitution |
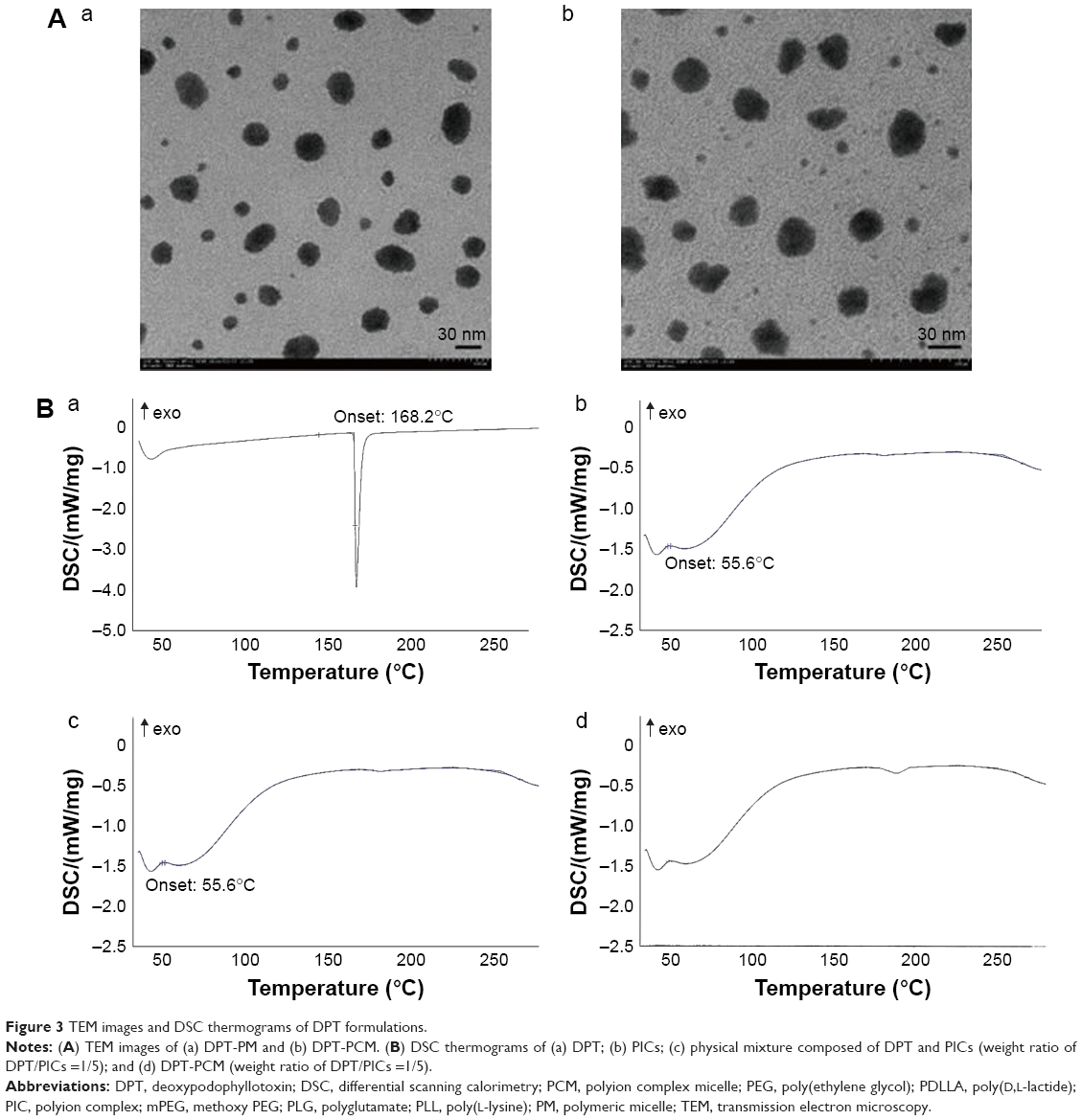 | Figure 3 TEM images and DSC thermograms of DPT formulations. |
From the DSC thermograms (Figure 3B), the endothermic peaks around 55.6°C and 171.8°C suggested dehydration of PICs and DPT. The shapes of the observed peaks were identical with that of the single substance in Figure 3Ba and Figure 3Bb/c. However, compared to the physical mixture, DPT-PCM revealed a strikingly contrasting DSC thermogram in Figure 3Bd, whereby no obvious melting peak of DPT appeared and the PIC peak was not as sharp as the counterpart in Figure 3Bb. This indicated that DPT was loaded in the micelles and therefore appeared in an amorphous state.44
Stability of DPT-PCM and DPT-PM
As shown in Figure 4A, the particle size of DPT-PCM was sustained at 40.8 nm at 4°C in the first 7 d. The particle size increased gradually and reached 81.2 nm on the 15th day. However, the size of DPT-PM increased from the 2nd day and reached 789.2 nm on the 15th day. When the storage temperature increased to 25°C (Figure 4B), DPT-PCM remained stable at 35.6 nm for 3 d, while DPT-PM only displayed a relatively stable particle size within the first 6 h after preparation and then increased to 725.7 nm after 12 h. When the temperature was raised to 37°C (Figure 4C), DPT-PCM showed a 10 times higher stability than DPT-PM (ie, 20 h for DPT-PCM and 2 h for DPT-PM). Moreover, at 4°C (Figure 4D), the drug content in DPT-PCM decreased slowly from the 7th day to the 14th day and remained at 93.9% of the original content. However, for DPT-PM, the drug content decreased to 82% by the 14th day. To assess whether high temperature aggravated the instability of the micelles, the final drug content was measure at 25°C (Figure 4E) and 37°C (Figure 4F). Results showed that after only 20 h of incubation at 25°C, only 40.2% of the original content from DPT-PM was detected, while the drug content from DPT-PCM remained constant for up to 3 d. At 37°C (Figure 4F), the drug content from DPT-PM decreased rapidly in the first 2 h, and only 22.1% of the original content was detected by the 12th hour, while no drug leaked from DPT-PCM in the first 24 h.
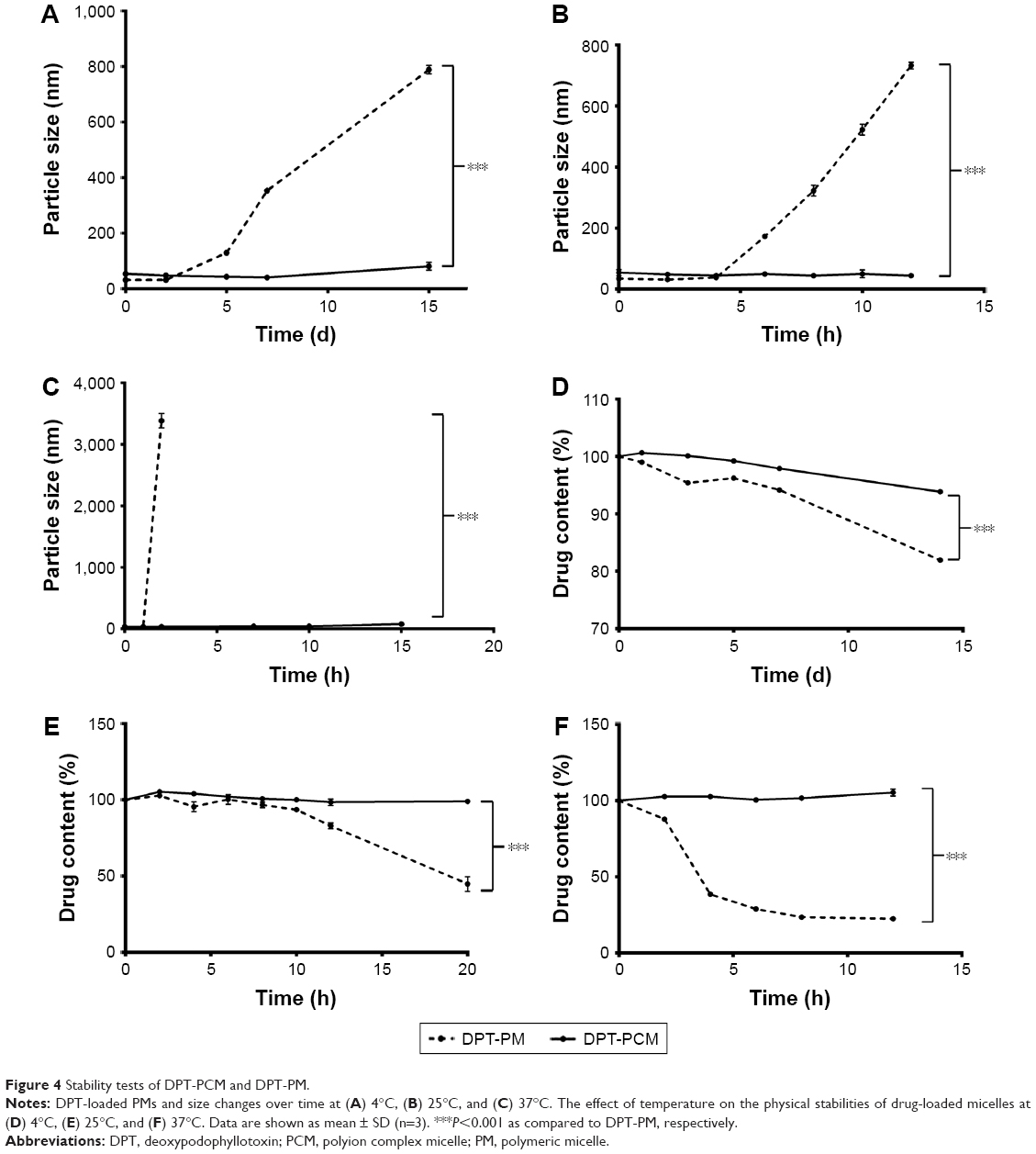 | Figure 4 Stability tests of DPT-PCM and DPT-PM. |
Due to the electrostatic interactions in the core, DPT-PCM showed much less temperature-dependent variation in particle sizes and drug contents throughout the incubation period when compared to DPT-PM, hence having enhanced stability. In addition, the presence of PLL in the alpha helix conformation and the existence of hydrogen bonds that created hydrophobic domains might have also contributed to the increase of stability of the complex structure.45
Cell viability assay
The antitumor activities of DPT, DPT-PM, DPT-PCM and blank micelles against A549 cells were tested. As shown in Figure 5A, both blank PM and blank PCM showed negligible cytotoxicity, demonstrating satisfactory safety of the polymers to these cells. As is well known, DPT is a promising microtubule-targeting antitumor agent and a cell cycle-regulatory protein modulator, thereby exhibiting a dose- and time-dependent cytotoxic effect toward A549 cells. In addition, DPT-PCM and DPT-PM showed greater cytotoxicity than DPT (P<0.001) at both 24 h and 48 h. It should be noted that compared with DPT-PM, DPT-PCM exhibited a stronger antitumor effect at 48 h than at 24 h (P<0.01) (Figure 5B), indicating that DPT-PCM may have prolonged action in cancer therapy. These data indicated that PCM may serve as a potential carrier for antitumor drug delivery and, thus, deserves further study.
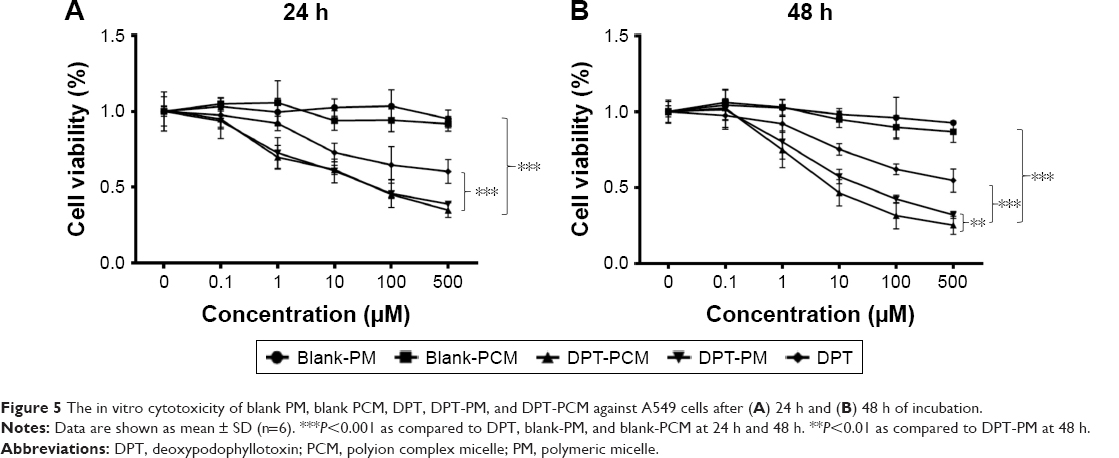 | Figure 5 The in vitro cytotoxicity of blank PM, blank PCM, DPT, DPT-PM, and DPT-PCM against A549 cells after (A) 24 h and (B) 48 h of incubation. |
In vitro drug release
In vitro drug release of DPT-PCM and DPT-PM was studied under simulated physiological conditions (PBS, pH 7.4, 1% Polysorbate-80), an acidic tumor extracellular environment (PBS, pH 6.8, 1% Polysorbate-80), and a lysosomal microenvironment (PBS, pH 5.0, 1% Polysorbate-80) (Figure 6Aa). It was obvious that the environmental pH had a strong effect on DPT release from the micelles, which was caused by the hydrolysis of the ester linkage in the PDLLA structure under acidic conditions. At pH 7.4, DPT-PCM and DPT-PM showed sustained DPT release, with <50% of the initial DPT amount after 48 h. However, at slightly acidic pH (eg, pH 6.8, resembling that of a tumor cell microenvironment), DPT-PCM released 94.07%±0.54% of the initial DPT amount within 48 h, while DPT-PM only released 80.49%±0.39%.
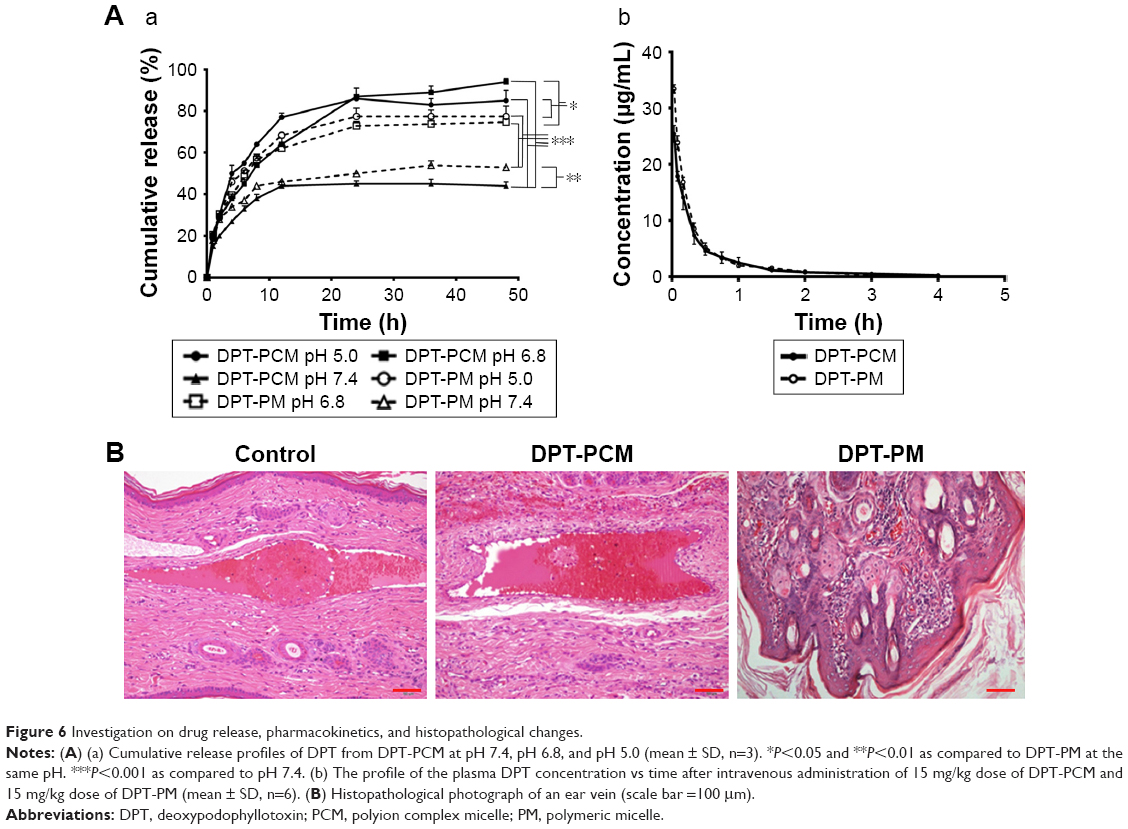 | Figure 6 Investigation on drug release, pharmacokinetics, and histopathological changes. |
Since the stability of PIC micelles is highly dependent on the charge balance between the counterions, the abrupt ionic change can induce the destabilization of the PIC micelles and cause them to release their internal cargo.46 Under physiological conditions (pH 7.4), the protonation process of lysine and the deprotonation process of the glutamic acid stabilized the micelles and thereby prevented drug release. However, in the acid environment, the peptides in mPEG-PDLLA-PLG were protonated, which led to instability of the electrostatic interactions, followed by accelerated dissociation of DPT-PCM. In the late endosomal or lysosomal microenvironments (pH ~5.0), the protonation process of lysine became faster; therefore, the release rate in the first 12 h was higher than that at pH 6.8. In conclusion, the protonation and deprotonation processes of peptides was the main reason for the pH-sensitive release of DPT from DPT-PCM, compared to DPT-PM, at pH 6.8 and pH 5.0. Such pH-triggered release will decrease drug loss during systemic blood circulation and improve intracellular drug release at/in tumor cells, which will contribute to the overall therapeutic efficacy.47
Hemocompatibility
Blood compatibility is considered one of the most important issues for in vivo applications of polymer–based formulations. Here, hemolysis ratios were quantified based on spectrophotometric measurements of hemoglobin released from red blood cells (RBCs). After RBC suspensions were treated with DPT-PM and DPT-PCM, results showed that all hemolysis ratios were <5% throughout the concentration range tested (0–0.3 mg/mL) (data not shown), which indicated excellent blood compatibility and safety for intravenous administration. In addition, low hemolysis results indicated that the micelles did not significantly disassemble into the unimers, which is responsible for hemolysis.48,49
Rabbit ear vein irritation test
Rabbit ears were sectioned and stained with H–E (Figure 6B). Compared with the DPT-PCM group, a mild inflammatory response was observed in the DPT-PM group after injection, as was indicated by a focal minimal foreign body reaction with a minimal number of neutrophils in subcutaneous tissue. In contrast, negligible venous irritation was noted in the DPT-PCM group and no obvious visible damage was found. As a result, it could be concluded that DPT-PCM produced minimal injection irritation as compared to DPT-PM.
Pharmacokinetic study
The pharmacokinetic profile of the plasma DPT concentration versus time obtained from the in vivo study is shown in Figure 6Ab and Table 5. After administration with a dose of 15 mg/kg DPT-PCM or DPT-PM, the C0 obtained was 25.43±1.48 μg/mL and 33.83±1.00 μg/mL, respectively. The area under the plasma concentration–time curve (AUC) of DPT-PCM and DPT-PM was 588.62±119.59 μg/mL·min versus 660.66±33.47 μg/mL·min. A significant difference in AUCs, half-times (t1/2), clearance rates (CL), and mean residence times (MRT) between the 2 micelles was not found (P>0.05). It is hypothesized that pharmacokinetic results only showed the dynamic characteristics of DPT-PCM, which were not affected by the addition of the amino acid chain segments. However, due to the similar surface and size of the 2 micelles, the CL of PCM did not change (P>0.05). The enhanced stability of PCM can stabilize the micelles during the preparation process, which is helpful for reducing drug leakage before freeze-drying. In addition, as the pharmacokinetic assay was conducted in healthy rats rather than in tumor-baring rats, PCM may release similar amounts of DPT compared to PM. Finally, the morphology and structure of DPT-PCM, compared to those of DPT-PM, are supposed to be more intact during circulation, which are not reflected in the pharmacokinetic results. Collectively, these results showed that the dynamic characteristics of DPT-PCM were not affected by the addition of amino acid chain segments.
 | Table 5 Pharmacokinetic parameters of DPT-PCM and DPT-PM |
Conclusion
In this study, oppositely charged oligopeptides, PLG and PLL, were conjugated to the carboxyl terminus of mPEG-PDLLA. These 3-block copolymers, mPEG-PDLLA-PLG and mPEG-PDLLA-PLL, were self-assembled into PCMs, which showed high stability in vitro. In addition, the drug release profile indicated that DPT-PCM was pH sensitive and can dissociate more quickly at pH 6.8 and 5.0 than DPT-PM, implying a great potential for cancer therapy. Moreover, DPT-PCM and DPT-PM exhibited comparable biocompatibility. These results demonstrated that the newly engineered PCMs can serve as a promising nanoplatform with superior stability for delivering hydrophobic antitumor therapeutic agents.
Acknowledgments
This work was funded by the Ministry of Science and Technology of the People’s Republic of China (2017ZX09101001-005, 2017ZX09101001-006), the Chinese Pharmacopeia Commission (ZG2017-5-01), the National Natural Science Foundation of China (81501579, 81673364), the Natural Science Foundation of Jiangsu Province (BK20150702), the Six Talents Summit Program of Jiangsu Province (YY-043), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Disclosure
The authors report no conflicts of interest in this work.
References
Giacomelli C, Schmidt V, Borsali R. Specific interactions improve the loading capacity of block copolymer micelles in aqueous media. Langmuir. 2007;23(13):6947–6955. | ||
Gaucher G, Dufresne M-H, Sant VP, Kang N, Maysinger D, Leroux J-C. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109(1):169–188. | ||
Andrianov AK, Payne LG. Polymeric carriers for oral uptake of microparticulates. Adv Drug Deliv Rev. 1998;34(2):155–170. | ||
Tan C, Wang Y, Fan W. Exploring polymeric micelles for improved delivery of anticancer agents: recent developments in preclinical studies. Pharmaceutics. 2013;5(1):201–219. | ||
Wörz A, Berchtold B, Moosmann K, Prucker O, Rühe J. Protein-resistant polymer surfaces. J Mater Chem. 2012;22(37):19547–19561. | ||
Schöttler S, Becker G, Winzen S, et al. Protein adsorption is required for stealth effect of poly (ethylene glycol)-and poly (phosphoester)-coated nanocarriers. Nat Nanotechnol. 2016;11(4):372–377. | ||
Burt HM, Zhang X, Toleikis P, Embree L, Hunter WL. Development of copolymers of poly (D, L-lactide) and methoxypolyethylene glycol as micellar carriers of paclitaxel. Colloids Surf B Biointerfaces. 1999;16(1):161–171. | ||
Shi J, Zhang J, Shen Y, et al. Arginine-stabilized mPEG-PDLLA (50/50) polymeric micelles of docetaxel by electrostatic mechanism for tumor-targeted delivery. Drug Deliv. 2015;22(2):168–181. | ||
Ouahab A, Shao C, Shen Y, Tu J. Development and characterization of stabilized double loaded mPEG-PDLLA micelles for simultaneous delivery of paclitaxel and docetaxel. Drug Dev Ind Pharm. 2014;40(7):860–868. | ||
Ling X, Huang Z, Wang J, et al. Development of an itraconazole encapsulated polymeric nanoparticle platform for effective antifungal therapy. J Mater Chem B. 2016;4(10):1787–1796. | ||
Maeda H, Bharate G, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. | ||
Sun G, Hagooly A, Xu J, et al. Facile, efficient approach to accomplish tunable chemistries and variable biodistributions for shell cross-linked nanoparticles. Biomacromolecules. 2008;9(7):1997–2006. | ||
Gao H, Xiong J, Cheng T, et al. In vivo biodistribution of mixed shell micelles with tunable hydrophilic/hydrophobic surface. Biomacromolecules. 2013;14(2):460–467. | ||
Ouahab A, Cheraga N, Onoja V, Shen Y, Tu J. Novel pH-sensitive charge-reversal cell penetrating peptide conjugated PEG-PLA micelles for docetaxel delivery: in vitro study. Int J Pharm. 2014;466(1–2):233–245. | ||
Owen SC, Chan DP, Shoichet MS. Polymeric micelle stability. Nano Today. 2012;7(1):53–65. | ||
Iijima M, Nagasaki Y, Okada T, Kato M, Kataoka K. Core-polymerized reactive micelles from heterotelechelic amphiphilic block copolymers. Macromolecules. 1999;32(4):1140–1146. | ||
Rijcken CJ, Snel CJ, Schiffelers RM, van Nostrum CF, Hennink WE. Hydrolysable core-crosslinked thermosensitive polymeric micelles: synthesis, characterisation and in vivo studies. Biomaterials. 2007;28(36):5581–5593. | ||
Yan LS, Yang LX, He HY, et al. Photo-cross-linked mPEG-poly(gamma-cinnamyl-L-glutamate) micelles as stable drug carriers. Polym Chem. 2012;3(5):1300–1307. | ||
Kang N, Perron ME, Prud’homme RE, Zhang YB, Gaucher G, Leroux JC. Stereocomplex block copolymer micelles: core-shell nanostructures with enhanced stability. Nano Lett. 2005;5(2):315–319. | ||
Carstens MG, Bevernage JJL, van Nostrum CF, et al. Small oligomeric micelles based on end group modified mPEG-oligocaprolactone with monodisperse hydrophobic blocks. Macromolecules. 2007;40(1):116–122. | ||
Yuan X, Harada A, Yamasaki Y, Kataoka K. Stabilization of lysozyme-incorporated polyion complex micelles by the ω-end derivatization of poly (ethylene glycol)-poly (α, β-aspartic acid) block copolymers with hydrophobic groups. Langmuir. 2005;21(7):2668–2674. | ||
Kim HJ, Zheng M, Miyata K, Kataoka K. Preparation of polyion complex micelles using block copolymers for siRNA delivery. Methods Mol Biol. 2016;1364:89–103. | ||
Huo H, Gao Y, Wang Y, et al. Polyion complex micelles composed of pegylated polyasparthydrazide derivatives for siRNA delivery to the brain. J Colloid Interface Sci. 2015;447:8–15. | ||
Harada A, Kataoka K. Formation of polyion complex micelles in an aqueous milieu from a pair of oppositely-charged block copolymers with poly (ethylene glycol) segments. Macromolecules. 1995;28(15):5294–5299. | ||
Bayó-Puxan N, Dufresne M-H, Felber AE, Castagner B, Leroux J-C. Preparation of polyion complex micelles from poly (ethylene glycol)-block-polyions. J Control Release. 2011;156(2):118–127. | ||
Chen J, Ding J, Zhang Y, Xiao C, Zhuang X, Chen X. Polyion complex micelles with gradient pH-sensitivity for adjustable intracellular drug delivery. Polym Chem. 2015;6(3):397–405. | ||
Luo Y-L, Yuan J-F, Shi J-H, Gao Q-Y. Synthesis and characterization of polyion complex micelles and their controlled release of folic acid. J Colloid Interface Sci. 2010;350(1):140–147. | ||
Luo Y, Wang A, Yuan J, Gao Q. Preparation, characterization and drug release behavior of polyion complex micelles. Int J Pharm. 2009;374(1):139–144. | ||
Tian H, Ip L, Luo H, Chang DC, Luo KQ. A high throughput drug screen based on fluorescence resonance energy transfer (FRET) for anticancer activity of compounds from herbal medicine. Br J Pharmacol. 2007;150(3):321–334. | ||
Imbert T. Discovery of podophyllotoxins. Biochimie. 1998;80(3):207–222. | ||
Ouahab A, Shen Y, Tu J-S. Novel oral delivery system of indomethacin by solidified mPEG-PDLLA micelles: in vivo study. Drug Deliv. 2012;19(4):232–237. | ||
Deng C, Rong G, Tian H, Tang Z, Chen X, Jing X. Synthesis and characterization of poly (ethylene glycol)-b-poly (L-lactide)-b-poly (L-glutamic acid) triblock copolymer. Polymer. 2005;46(3):653–659. | ||
Liu P, Yu H, Sun Y, Zhu M, Duan Y. A mPEG-PLGA-b-PLL copolymer carrier for adriamycin and siRNA delivery. Biomaterials. 2012;33(17):4403–4412. | ||
Wang Y, Sun T, Zhang Y, et al. Exenatide loaded PLGA microspheres for long-acting antidiabetic therapy: preparation, characterization, pharmacokinetics and pharmacodynamics. RSC Adv. 2016;6(44):37452–37462. | ||
He P, Zhao C-W, Xiao C-S, Tang Z-H. Thermosensitive polyion complex micelles prepared by self-assembly of two oppositely charged diblock copolymers. Chin J Polym Sci. 2013;31(2):318–324. | ||
Yokoyama M, Fukushima S, Uehara R, et al. Characterization of physical entrapment and chemical conjugation of adriamycin in polymeric micelles and their design for in vivo delivery to a solid tumor. J Control Release. 1998;50(1):79–92. | ||
Zhao J, Shi J-F, Xiong Y-R, TU J-S. Pharmacokinetics of deoxypodophyllotoxin polymeric micelles in rats. Chin J New Drugs. 2014;22:021. | ||
Qi F, Wu J, Fan Q, et al. Preparation of uniform-sized exenatide-loaded PLGA microspheres as long-effective release system with high encapsulation efficiency and bio-stability. Colloids Surf B Biointerfaces. 2013;112:492–498. | ||
Samadi N, Abbadessa A, Di Stefano A, et al. The effect of lauryl capping group on protein release and degradation of poly (d, l-lactic-co-glycolic acid) particles. J Control Release. 2013;172(2):436–443. | ||
Ling X, Zhao C, Huang L, et al. Synthesis and characterization of hyaluronic acid–platinum (iv) nanoconjugate with enhanced antitumor response and reduced adverse effects. RSC Adv. 2015;5(99):81668–81681. | ||
Li C, Li SS, Tu TJ, et al. Paclitaxel-loaded cholesterol-conjugated polyoxyethylene sorbitol oleate polymeric micelles for glioblastoma therapy across the blood-brain barrier. Polym Chem. 2015;6(14):2740–2751. | ||
Xiong Y, Jiang W, Shen Y, et al. A poly(gamma, L-glutamic acid)-citric acid based nanoconjugate for cisplatin delivery. Biomaterials. 2012;33(29):7182–7193. | ||
Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–314. | ||
Chen W, Gu B, Wang H, Pan J, Lu W, Hou H. Development and evaluation of novel itraconazole-loaded intravenous nanoparticles. Int J Pharm. 2008;362(1):133–140. | ||
Boudier A, Aubert-Pouëssel A, Louis-Plence P, et al. The control of dendritic cell maturation by pH-sensitive polyion complex micelles. Biomaterials. 2009;30(2):233–241. | ||
Lee Y, Kataoka K. Biosignal-sensitive polyion complex micelles for the delivery of biopharmaceuticals. Soft Matter. 2009;5(20):3810–3817. | ||
Lv S, Song W, Tang Z, et al. Charge-conversional PEG-polypeptide polyionic complex nanoparticles from simple blending of a pair of oppositely charged block copolymers as an intelligent vehicle for efficient antitumor drug delivery. Mol Pharm. 2014;11(5):1562–1574. | ||
Shi C, Zhang Z, Shi J, Wang F, Luan Y. Co-delivery of docetaxel and chloroquine via PEO–PPO–PCL/TPGS micelles for overcoming multidrug resistance. Int J Pharm. 2015;495(2):932–939. | ||
Manganiello MJ, Cheng C, Convertine AJ, Bryers JD, Stayton PS. Diblock copolymers with tunable pH transitions for gene delivery. Biomaterials. 2012;33(7):2301–2309. |
Supplementary materials
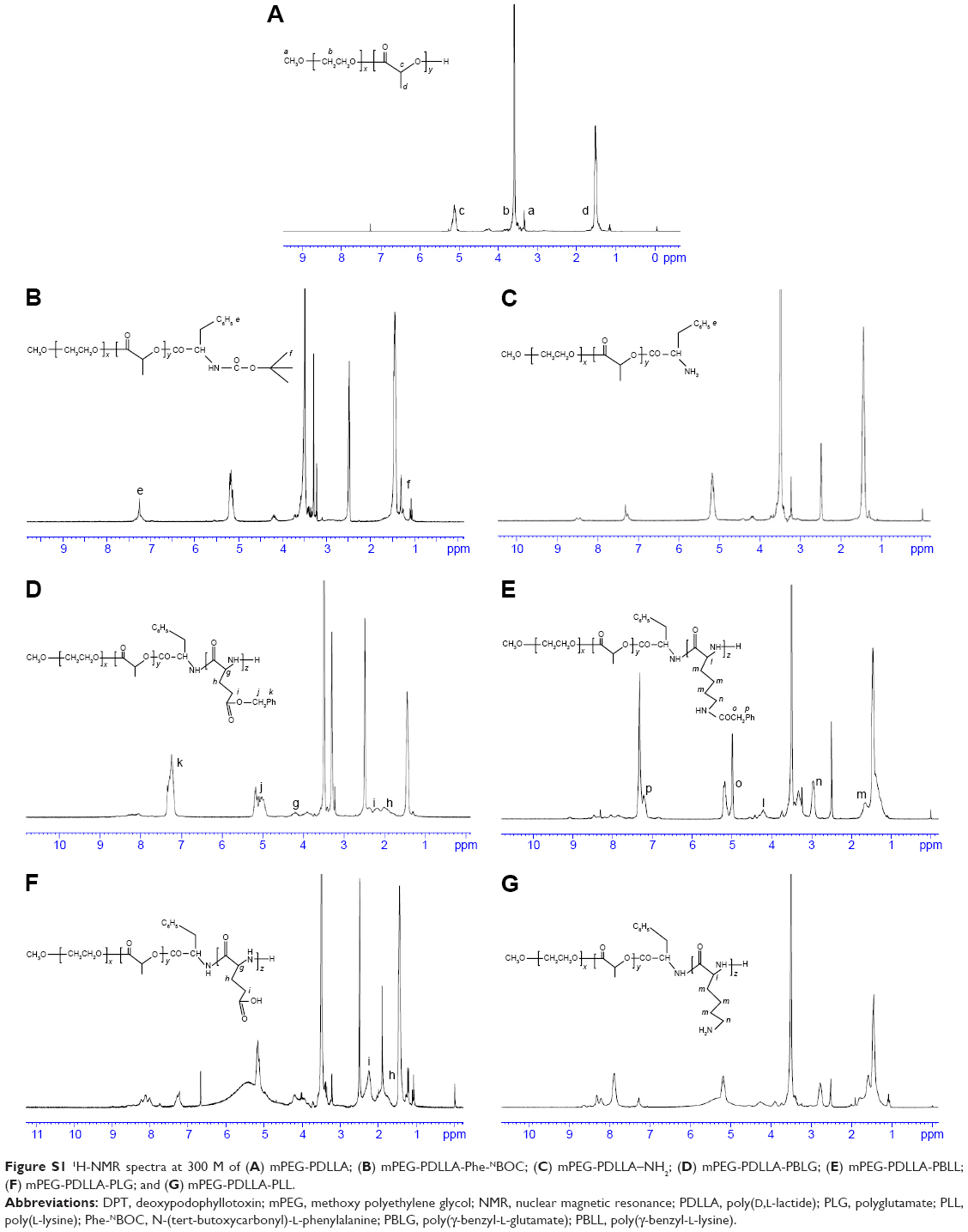 | Figure S1 1H-NMR spectra at 300 M of (A) mPEG-PDLLA; (B) mPEG-PDLLA-Phe-NBOC; (C) mPEG-PDLLA–NH2; (D) mPEG-PDLLA-PBLG; (E) mPEG-PDLLA-PBLL; (F) mPEG-PDLLA-PLG; and (G) mPEG-PDLLA-PLL. |
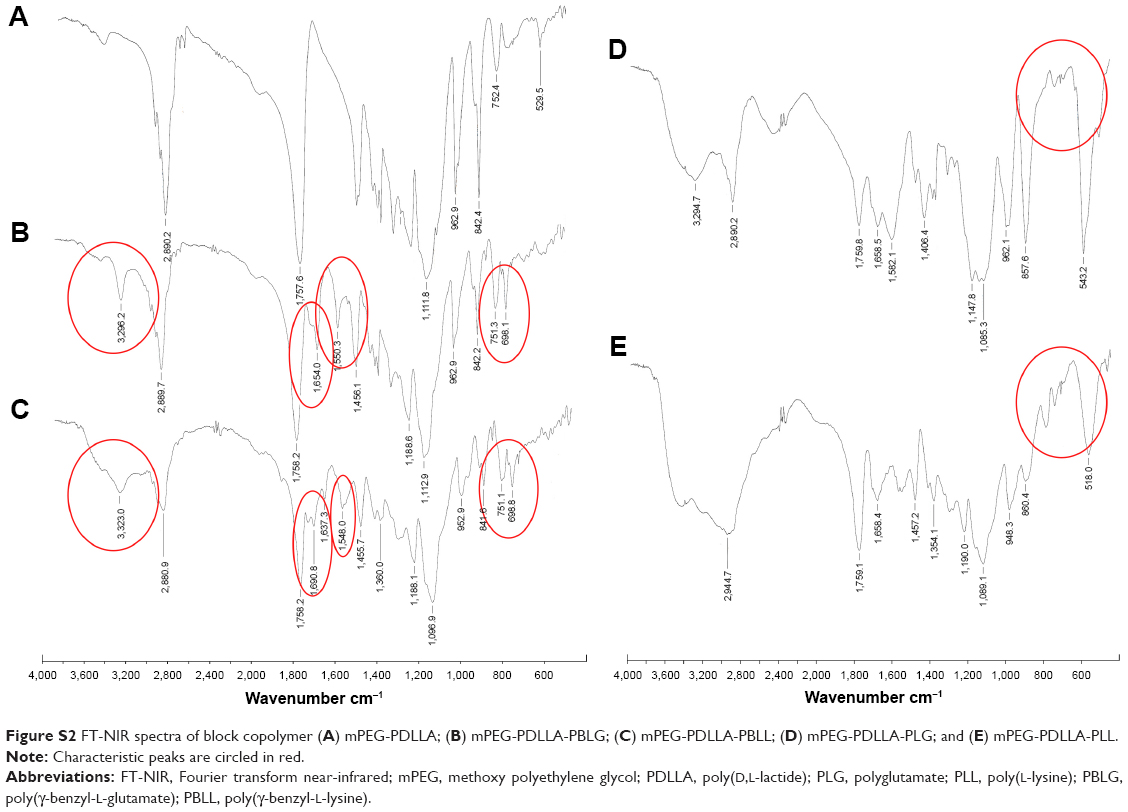 | Figure S2 FT-NIR spectra of block copolymer (A) mPEG-PDLLA; (B) mPEG-PDLLA-PBLG; (C) mPEG-PDLLA-PBLL; (D) mPEG-PDLLA-PLG; and (E) mPEG-PDLLA-PLL. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.