Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Electrosprayed core–shell solid dispersions of acyclovir fabricated using an epoxy- coated concentric spray head
Authors Liu Z, Cui L, Yu D ![]() , Zhao Z, Chen L
, Zhao Z, Chen L
Received 20 December 2013
Accepted for publication 17 February 2014
Published 16 April 2014 Volume 2014:9(1) Pages 1967—1977
DOI https://doi.org/10.2147/IJN.S59516
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Zhe-Peng Liu,1 Lei Cui,2 Deng-Guang Yu,3 Zhuan-Xia Zhao,1 Lan Chen1
1School of Medical Instrument and Food Engineering, 2Tin Ka Ping College of Science, 3School of Materials Science and Engineering, University of Shanghai for Science and Technology, Shanghai, People's Republic of China
Abstract: A novel structural solid dispersion (SD) taking the form of core–shell microparticles for poorly water-soluble drugs is reported for the first time. Using polyvinylpyrrolidone (PVP) as a hydrophilic polymer matrix, the SDs were fabricated using coaxial electrospraying (characterized by an epoxy-coated concentric spray head), although the core fluids were unprocessable using one-fluid electrospraying. Through manipulating the flow rates of the core drug-loaded solutions, two types of core–shell microparticles with tunable drug contents were prepared. They had average diameters of 1.36±0.67 and 1.74±0.58 µm, and were essentially a combination of nanocomposites with the active ingredient acyclovir (ACY) distributed in the inner core, and the sweeter sucralose and transmembrane enhancer sodium dodecyl sulfate localized in the outer shell. Differential scanning calorimetry and X-ray diffraction results demonstrated that ACY, sodium dodecyl sulfate, and sucralose were well distributed in the PVP matrix in an amorphous state because of favorable second-order interactions. In vitro dissolution and permeation studies showed that the core–shell microparticle SDs rapidly freed ACY within 1 minute and promoted nearly eightfold increases in permeation rate across the sublingual mucosa compared with raw ACY powders.
Keywords: core–shell microparticle, solid dispersion, coaxial electrospraying, poorly water-soluble drug, epoxy-coated spray head
Introduction
Increasing the solubility of poorly water-soluble drugs remains one of the most challenging aspects of formulation development.1,2 Numerous advanced functional materials, as well as new processes and technologies, have been investigated to provide more effective and versatile ways to address formulation issues associated with poorly water-soluble molecules.3 The development of solid dispersions (SDs), which are mainly obtained by melting or solvent evaporation,4 is one of the more promising strategies for improving the oral bioavailability of poorly water-soluble drugs.
In the traditional solvent-evaporation method, the drug and carrier are first dissolved in an organic solvent or solvent mixture, and the solvent is subsequently removed. Solvent-evaporation methods differ in the ways by which the solvents are removed to solidify the products, and different strategies have been developed for the rapid and effective removal of the solvents. These strategies include spraying (heat- or freeze-spraying) and drying (fluidized bed, freeze, microwave, or vacuum drying). Solvent-evaporation methods take advantage of phase changes and exploit thermal energy, wave energy, or mechanical energy to remove organic solvents.5–8 One concern associated with these methods, however, involves the rapid removal of sufficient amounts of solvent from the coprecipitates. Solvent removal from coprecipitates is challenging, because the latter become increasingly viscous during the “drying” process, further hindering evaporation of the residual solvent. Also, crystal lattices are easily formed during drying, leading to crystalline-particle growth in the later stages of the drying process. Lattice growth arises from the mobility of the drug molecules, and may compromise the properties of the resulting SDs.4,9
Electrohydrodynamic atomization (EHDA) techniques, including electrospinning, electrospraying, and e-jet printing, differ from all other top-down physical methods for nanotechnology, and produce materials at the nanoscale or microscale level directly through the interaction between electrical energy and processed fluids.10,11 This direct production does not involve any energy-transfer process before the energy, often in the form of mechanical, ultrasonic, laser, or microwave energy,12 is exploited to generate nano-objects.
Electrical energy from electrospinning has recently been exploited to remove organic solvents directly and produce polymer nanofibers.13,14 Liquids can readily interact with electrical energy.15 Electrospinning rapidly dries and solidifies microfluid jets, producing nanosized fibers very quickly (often in the order of 10−2 seconds).16,17 Provided favorable secondary interactions are available, the physical state of components in liquid solutions may be propagated into solid nanofibers. The final products have an advantageous 1-D nanoscale structure, and can contain poorly water-soluble drugs in an amorphous state. The reported nanofibers exhibit excellent performance in enhancing the dissolution rates of poorly-water soluble drugs, which suggests that electrospinning is essentially a useful tool for generating SDs of poorly water-soluble drugs.18
Electrospraying, which is based on the principles of electrospinning, has grown in popularity because of its ability to fabricate particles easily. Electrospraying systems generally consist of four components that can take advantage of electrical energy: a syringe pump, a high-power supply, a spray head, and a collector. A significant feature of electrospraying is its ability to generate particles with a mean diameter that can be varied between hundreds of micrometers to tens of nanometers.19 This variation is achieved by careful control of the process parameters, including the flow rate, needle diameter, and applied voltage, as well as the chemical composition and concentration of the spraying solution. The process is simple and straightforward. Electrospraying has been utilized to produce materials with a wide range of applications in the pharmaceutical, ceramics, cosmetics, and food industries; electrospraying is particularly useful for biomedical applications, such as in drug delivery.20
Compared with a single-fluid process, coaxial electrospraying, in which a concentric spray head accommodates two different liquids, can expand the capability of the technique of generating new micro/nanostructures. Using coaxial electrospraying, Loscertales et al first reported the preparation of core–shell microparticles with a droplet encapsulated in a polymer shell.21 The shells in these so-called core–shell nanostructures offer a number of functionalities, including protection of the core from the outside environment, maintenance of compositional and structural integrity, prevention of the core from aggregating or sintering into larger particles, selective percolation of molecules in and out of the interior of the shell, increases in solubility and/or biocompatibility, and the addition of new physical or chemical properties.22 Bottom-up approaches are often believed to be more suitable than top-down approaches for synthesizing core–shell structures.23 However, as a special top-down technology, coaxial EHDA processes have successfully been proven to be capable of generating macro/nanoscale core–shell nanostructures from the template of a concentric spray head, particularly from a concentric spinneret to produce core–sheath nanofibers using coaxial electrospinning.24
Polymer-based SDs have been demonstrated to be suitable for formulating poorly water-soluble drugs in the amorphous state, leading to enhancement of dissolution rates and bioperformance.25 Polyvinylpyrrolidone (PVP), also commonly called polyvidone or povidone, is a water-soluble polymer made from the monomer N-vinylpyrrolidone.26 It has a wide variety of applications in medicine, pharmacy, cosmetics, and industrial production.9 In the pharmaceutical field, PVP series are widely used as excipients, and are particularly suitable for the preparation of SDs by the solvent method. This is due to their good solubility in water and a lot of organic solvents, rapid uptake of water, and their ability to circumscribe crystallization of dispersed drugs.18 In electrospinning applications, PVP has been broadly explored as a filament-forming matrix to fabricate functional nanofibers for such applications as drug-containing nanofibers, inorganic–organic composites, inorganic nanofibers, precursors for organic peroxide nanofibers, and liposomes.9,18,25 To electrospray poorly water-soluble drug and PVP into drug-loaded microparticles may expand their advantages as drug carriers in SDs. In this paper, we report the preparation of SDs in the form of core–shell microparticles via the coaxial electrospraying process using PVP as a polymer matrix and acyclovir (ACY) as a poorly water-soluble model drug.
Materials and methods
Materials
PVP K30 (molecular weight 58,000) was purchased from BASF (Shanghai, People’s Republic of China). ACY was obtained from Zhejiang Charioteer Pharmaceutical (Hangzhou, People’s Republic of China). Methylene blue, sucralose, and sodium dodecyl sulfate (SDS) were provided by Sinopharm Chemical Reagent (Shanghai, People’s Republic of China). N, N-dimethylacetamide (DMAc) and anhydrous ethanol were provided by Shanghai Shiyi Chemicals Reagent (Shanghai, People’s Republic of China). Epoxy (EP) resin and its hardener were purchased from Jiaojiang Qinfeng Chemical Plant (Taizhou, People’s Republic of China). All chemicals used were of analytical grade. Water was double-distilled just before use.
Fabrication of the epoxy-coated concentric spray head and the coaxial electrospraying process
A codissolving solution of 10% (w/v [weight/volume]) PVP and 4% (w/v) ACY in a mixed solvent of DMAc:ethanol (4:6, v:v [volume/volume]) was used as the core fluid. To optimize experimental conditions, 10 parts per million methylene blue was mixed with the core fluids as a marker. The shell liquid consisted of 10% (w/v) PVP, 0.2% (w/v) SDS, and 0.5% (w/v) sucralose in a mixed solvent of water:ethanol (0.5:9.5, v:v).
A homemade EP-coated concentric spray head was developed for the coaxial electrospraying process. A DSA100 drop-shape analysis system (Krüss, Hamburg, Germany) was used in the experiments to investigate surface interactions between the shell fluids and the spray head.
Two syringe pumps (Cole-Parmer® KDS100 and KDS 200; Thermo Fisher Scientific, Waltham, MA, USA) were employed to drive the sheath and core fluids. A metal clip was used to connect the spinneret to a high-voltage supply (Shanghai Sute Electrical Co. Ltd, Shanghai, People’s Republic of China), and the applied voltage was 20 kV. The microparticles were collected on a metal collector wrapped with aluminum foil, and kept at a fixed distance of 20 cm away from the needle tip of the spinneret. The coaxial processes were recorded using a digital video recorder (PowerShot A640; Canon, Tokyo, Japan) under 11× magnification. All of the electrospraying processes were carried out under ambient conditions (22°C±3°C, relative humidity 52%±6%). Other parameters are listed in Table 1. The microparticles obtained were dried for at least 24 hours at 40°C under vacuum (320 Pa) in a DZF-6050 electric vacuum-drying oven (Shanghai Laboratory Instrument Work, Shanghai, People’s Republic of China) to facilitate the removal of residual organic solvent and moisture, and then kept in a desiccator for characterization.
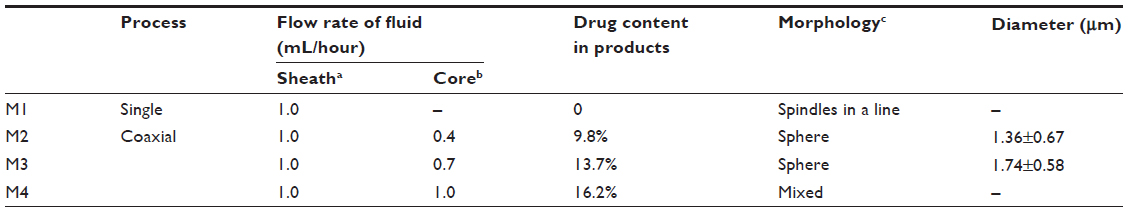 | Table 1 Parameters used for electrospraying, and details of the particle products |
Morphology of the microparticles
The morphology of the core–shell microparticles was assessed using an S-4800 field-emission scanning electron microscope (FESEM; Hitachi, Tokyo, Japan). The average size of the microparticles was determined by measuring the diameters of over 100 microparticles captured by FESEM images using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
The topographies of raw material particles as well as composite microparticles were observed under cross-polarized light using an XP-700 polarized optical microscope (Shanghai Changfang Optical Instrument, Shanghai, People’s Republic of China). Transmission electron microscopy (TEM) images of the samples were taken on a JEM 2100F field-emission transmission electron microscope (JEOL, Tokyo, Japan). TEM samples of core–shell microparticles were prepared by fixing a lacey carbon-coated copper grid to the metal collector. Microparticles were sprayed directly onto the grid.
Physical status of the microparticle components and their compatibility
X-ray diffraction (XRD) analysis was performed on a D/Max-BR diffractometer (Rigaku, Tokyo, Japan) with Cu Kα radiation in the 2θ range of 5°–60° at 40 mV and 300 mA. Differential scanning calorimetry (DSC) was performed using an MDSC 2910 differential scanning calorimeter (TA Instruments, New Castle, DE, USA). Sealed samples were heated at a rate of 10°C · minute−1 from ambient temperature (21°C) to 300°C. The nitrogen gas flow rate was set to 40 mL · minute−1. Attenuated total reflectance–Fourier-transform infrared (ATR–FTIR) analysis was performed on a Nicolet-Nexus 670 FTIR spectrometer (Nicolet Instrument, Madison, WI, USA) over the range of 500–4,000 cm−1 and a resolution of 2 cm−1.
Functional performance of the microparticles
In vitro dissolution tests
In vitro dissolution studies were performed according to the Chinese Pharmacopoeia (2005 edition). Method II, a paddle method using an RCZ-8A dissolution apparatus (Tianjin University Radio Factory, Tianjin, People’s Republic of China), was carried out.
The core–shell microparticles M2 (204 mg) or M3 (146 mg) or 20 mg of crude ACY particles (<100 μm) were placed in 600 mL of phosphate-buffered saline (PBS; pH 6.8, 0.1 M) at 37°C±1°C, 50 rpm, and sink conditions of C<0.2Cs. At predetermined time intervals, samples of 5.0 mL were withdrawn and replaced with fresh medium to maintain a constant volume. After filtration through a 0.22 μm membrane (EMD Millipore, Billerica, MA, USA) and appropriate dilution with PBS, the sample solutions were analyzed at 252 nm using an ultraviolet (UV) spectrophotometer (Unico Instrument, Shanghai, People’s Republic of China). All measurements were carried out six times.
In vitro buccal permeation studies
In vitro permeation studies were performed using an RYJ-6A diffusion test apparatus (Shanghai Huanghai Drug Control Instrument, Shanghai, People’s Republic of China), in which materials were mounted in six Keshary-Chien glass diffusion cells, and a water-bath system maintained a constant temperature of 37°C±0.2°C. Each cell had a diffusion area of 2.60 cm−2, and the receptor compartment had a capacity of 7.2 mL.
Porcine sublingual mucosa samples obtained from a local abattoir within 2 hours of slaughtering were mounted between the donor and receptor compartments of the diffusion cells with the mucosal surface facing up. Each donor compartment was filled with 1.0 mL of PBS, and the hydrodynamics in the receptor compartment were maintained by stirring with a Teflon-coated magnetic bead at 100 rpm. The sublingual membranes were equilibrated for 30 minutes before the permeation tests.
The core–shell microparticles M2 (1.398 mg) or M3 (1.000 mg) or 0.137 mg of crude ACY particles (<100 μm) were placed on the mucosal surface. Samples (1.0 mL) were withdrawn from the receptor compartment at timed intervals and filtered through a 0.22 μm membrane (EMD Millipore). Absorption was measured at 252 nm as previously described. All measurements were carried out six times.
Results and discussion
Epoxy-coated concentric spray head and the coaxial electrospraying process
A schematic of the coaxial electrospraying process is shown in Figure 1A, and a digital picture of the homemade EP-coated concentric spray head is shown in Figure 1B. The EP-coated concentric spray head was prepared by simply inserting a small stainless steel tube (27 G, outer and inner diameters of 0.42 and 0.21 mm, respectively) into a large stainless steel tube (18 G, outer and inner diameters of 1.25 and 0.84 mm, respectively) and then coating the outer steel tube with an EP and hardener mixture. The EP coating facilitates the coaxial process in two ways: 1) as an antistatic polymer, EP can effectively retard the loss of electrical energy from the working fluid to the environment,27 and 2) the small interfacial tension between the EP and the working fluid exerts few negative influences on the formation of the compound Taylor cone and emission of the straight fluid jets. An experiment on the wetting angles of the sheath fluid on an EP-coated plate and a stainless steel plate was conducted, and results are shown in Figure 1C and D. The wetting angles of the sheath fluid on the EP surface and steel plate were 95° and 40°, respectively, which indicates that the surface tension between the EP and shell solution was smaller than that between the steel plate and shell solution.
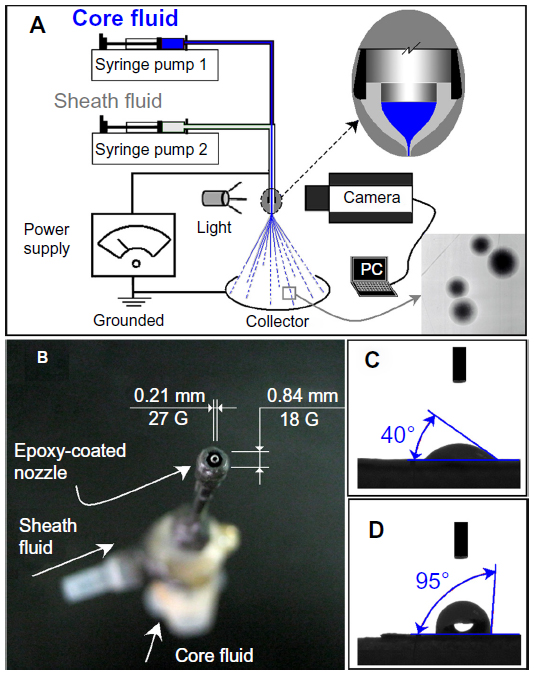 | Figure 1 (A) Diagram of the coaxial electrospraying process. (B) Digital photograph of the epoxy-coated concentric spray head. (C and D) Contact angles of the shell fluid on a stainless steel plate and an epoxy-coated surface, respectively. |
Digital images of the apparatus arrangement and the connection between power supply and spray head are shown in Figure 2A and B. A typical coaxial electrospraying process under an applied voltage of 20 kV with shell and core flow rates of 0.7 and 1.0 mL · hour−1, respectively, is shown in Figure 2C. “A” indicates the explosion processes of the droplets during electrospraying that were faster than the camera-taken photos at a speed of 30 frames per second, where droplets could not be discerned. However, further increased applied voltage resulted in the division of concentric fluids (Figure 2D), which was unfavorable for the formation of core–shell structures and should be avoided. With methylene blue as a color marker, the compound Taylor cone for preparing both M2 and M3 microparticles can be obviously discerned, as shown in Figure 2E and F, respectively.
 | Figure 2 (A–F) Photographs of the coaxial electrospraying process setup and the optimization of parameters. (A) Arrangement of apparatuses used in this work; (B) connection of the spray head with the power supply; (C) typical coaxial electrospraying process under an applied voltage of 20 kV with sheath and core flow rates of 0.7 and 1.0 mL · hour–1, respectively (“A” indicates the faster explosion processes of the droplets than photos taken by a camera); (D) divided electrospraying processes obtained under an excessive voltage of 25 kV; and (E and F) typical compound Taylor cone at a sheath flow rate of 1.0 mL · hour–1 and core flow rates of 0.4 and 0.7 mL · hour–1, respectively. The poorly water-soluble drug acyclovir (ACY) has poor solubility in a series of typical organic solvents, such as methanol, ethanol, chloroform, and acetone. ACY also has a high melting point (257°C). Therefore, reports on the use of this drug in solid dispersions (SDs) using traditional melt and solvent-evaporation technologies are few. Although ACY is soluble in dimethylacetamide (DMAc), polyvinylpyrrolidone (PVP) cannot be electrosprayed into microparticles under these conditions, because of its high boiling point (166°C) and poor volatility. Therefore, preparing microparticulate SDs of ACY by single-fluid electrospraying is impossible, because of the lack of appropriate solvent in which all components can dissolve together. Meanwhile, the solutions can be processed using electrospraying. For coaxial electrospraying, similar to coaxial electrospinning, the core solution does not need to be processable by electrospraying, and the shell solution acts as a guide and surrounds the core liquid. The shell solution is critical, and the selected shell systems should be processable by themselves to facilitate formation of a core–shell structure in the microparticles. Therefore, although the core solution consisted of 10% (w/v) PVP and 4% (w/v) ACY in a mixed solvent of DMAc:ethanol (4:6, v:v), the system was not processable by electrospraying. Nevertheless, the shell fluid consisting of 10% (w/v) PVP, 0.2% (w/v) sodium dodecyl sulfate, and 0.5% (w/v) sucralose in a mixed solvent of water:ethanol (0.5:9.5, v:v) was able to ensure a smooth coaxial electrospraying process and the formation of solid core–shell microparticles. |
Morphologies and core–shell structures of the microparticles
Single-fluid electrospraying of the core fluid did not generate solid products, whereas single-fluid electrospraying of the shell solution resulted in typical spindles on a line morphologies (Figure 3A). When the core fluids were introduced into the shell fluids at an excessive rate of 1.0 · mL hour−1, the core fluids were able to penetrate the shell solutions to generate products with mixed morphologies of clumps, fibers, particles, and beads/spindles on a line (Figure 3B). Therefore, a suitable core-to-shell fluid rate ratio was important for conducting coaxial electrospraying. At rates of 0.4 and 0.7 · mL hour−1, the resultant microparticles had average sizes of 1.36±0.67 and 1.74±0.58 μm (Figure 3C and D). Both M2 and M3 microparticles had many satellites visible on their surface. TEM images clearly demonstrated that M2 and M3 microparticles had core–shell structures (Figure 4). As expected, M2 microparticles (Figure 4A) had a thicker shell than M3 microparticles (Figure 4B) although they had a smaller average size.
 | Figure 3 (A–D) Field-emission scanning electron microscopy images of the microparticles and their diameter distributions. (A) M1, (B) M4, (C) M2, and (D) M3. |
 | Figure 4 Transmission electron microscopy images of (A) M2 and (B) M3. |
Physical status of the microparticle components
XRD and DSC analyses were performed to determine the physical status of the components of core–shell microparticles. As shown in Figure 5A, the presence of numerous distinct peaks in the XRD patterns indicated that ACY, sucralose, and SDS were present as crystalline materials with characteristic diffraction peaks, as also demonstrated by the colorful images of their crude particles (Figure 5B–D) under polarized light. PVP diffraction exhibited a diffused background pattern, with two diffraction halos indicating that the polymer was amorphous. The patterns of the M2 and M3 microparticles showed no characteristic reflections of the crude components, and instead comprised diffuse halos. Unlike the observations on crystalline materials, PVP and the core–shell microparticles showed no bright colors, suggesting that PVP and the microparticles were amorphous (Figure 5E and F).
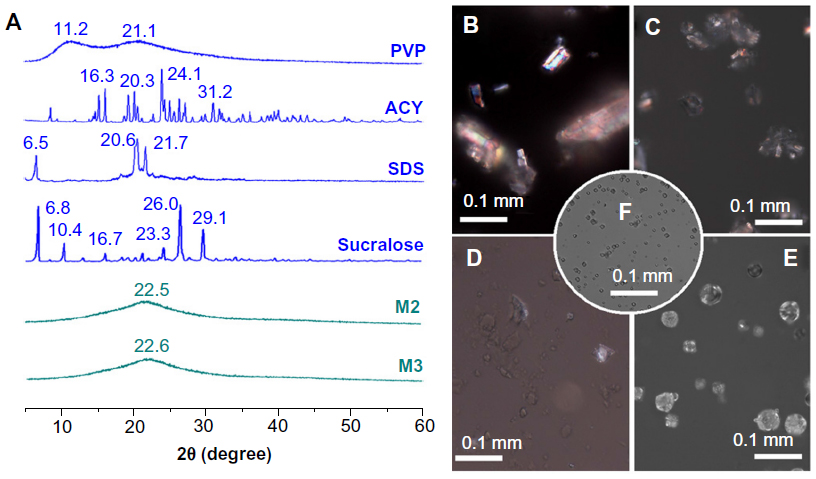 | Figure 5 (A) X-ray diffraction patterns of the raw materials and nanofibers. (B–F) Morphologies of acyclovir (ACY) particles, sucralose particles, sodium dodecyl sulfate (SDS) particles, polyvinylpyrrolidone (PVP) particles, and the prepared core–shell microparticles (M3), respectively, viewed under cross-polarized light. |
The DSC thermograms are shown (Figure 6), and the DSC curve of pure ACY and sucralose exhibited a single endothermic response corresponding to melting points of 257°C and 130°C, respectively. SDS had a melting point of 182°C, followed by a decomposition temperature of 213°C. As an amorphous polymer, PVP K30 did not show any fusion peak or phase transition, apart from a broad endothermic peak caused by dehydration, which lies between 80°C and 120°C with a peak at 85°C. DSC thermograms of the M2 and M3 microparticles did not exhibit any melting peak of small molecules. The microparticles had a broad endothermic peak, ranging within about 60°C–100°C. However, the decomposition bands of SDS in the core–shell microparticles were narrower and higher than those of pure SDS. The peak temperatures of decomposition shifted from 204°C. These results suggested that ACY, SDS, and sucralose were no longer present as a crystalline material, but had been converted into an amorphous state in their respective parts in the core–shell microparticles.
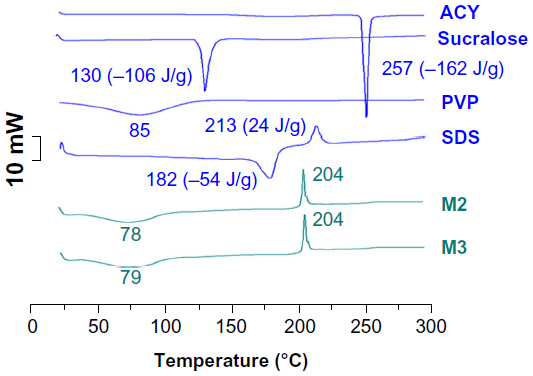 | Figure 6 Differential scanning calorimetry (DSC) thermograms of the raw materials and M2 and M3 microparticles. DSC and X-ray diffraction results demonstrated that the functional molecules (acyclovir [ACY]) and also the excipients (sucralose and sodium dodecyl sulfate [SDS]) were highly distributed throughout the polyvinylpyrrolidone (PVP) matrix, and were present in a complex manner where the original structure of pure materials was lost. Electrospraying, similar to electrospinning, is an inherently appropriate method for preparing solid dispersions. The fast-drying electrospraying process can randomly “freeze” the drug molecules in the solid-polymer matrix into a state comparable to a liquid form. This can prevent phase separation, eg, recrystallization of either drug or matrix, during solvent removal. |
Secondary interactions among the components
Compatibility among components is essential for producing high-quality and stable composite microparticles. Second-order interactions, such as hydrogen bonding, electrostatic interactions, and hydrophobic interactions, can often improve compatibility.9 The molecular structures of the four components are shown in Figure 7. ACY and sucralose molecules possess free hydroxyl groups that could act as potential proton donors for hydrogen bonding. SDS has S=O groups, and PVP has carbonyl groups that could act as proton receptors. Therefore, hydrogen bonding may occur within the core–sheath nanofibers through interactions determined by the specific presence of these groups.
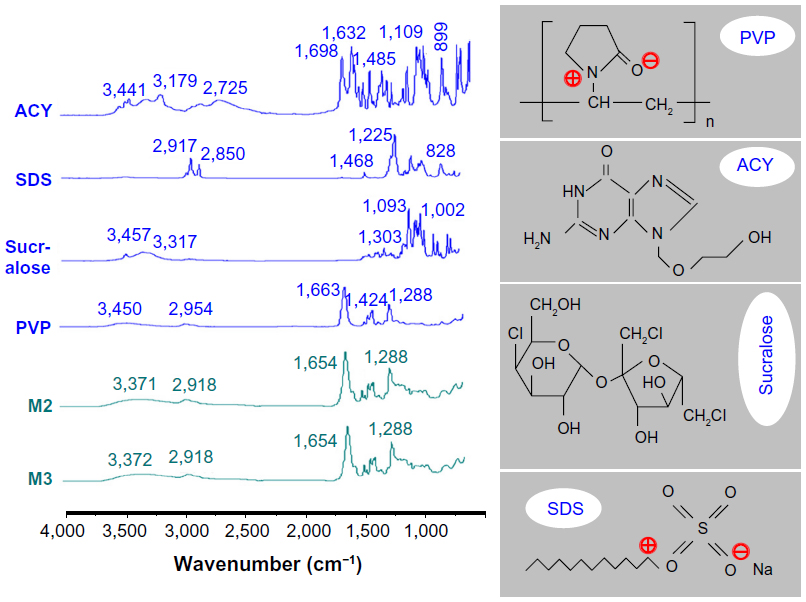 | Figure 7 Attenuated total reflectance–Fourier-transform infrared spectra of the crude materials and microparticles, and molecular structures of polyvinylpyrrolidone (PVP), acyclovir (ACY), sucralose, and sodium dodecyl sulfate (SDS). |
ACY molecules have both −OH and −C=O groups; therefore, hydrogen bonding can form between drug molecules. This phenomenon can be verified from the ATR-FTIR spectra showing sharp peaks for pure crystalline ACY at 1,698 and 1,632 cm−1 (Figure 7), and these peaks represent the stretching vibration of −C=O groups in a different state. However, all peaks of ACY were absent in the core–shell microparticles when samples were evaluated using ATR-FTIR spectra. Only one large peak at 1,654 cm−1 could be identified for the core–shell microparticles. This result indicated that hydrogen bonding between the PVP carbonyl and oxydryl groups of the ACY molecule occurred. Meanwhile, interaction of ACY with the polymer renders the structure less likely to form the dimers that are necessary for constructing a crystal lattice.
The occurrence of hydrogen bonding was supported by the following findings: 1) disappearance of the absorbance of −C=O groups in ACY, 2) shift to lower wavenumbers of peaks assigned to C=O stretching vibrations in PVP (from 1,663 cm−1 to 1,654 cm−1 for the core–shell microparticles), 3) disappearance of multiple peaks within 3,000–4,000 cm−1 (related to O−H stretching vibrations in sucralose and ACY), and 4) presence of some peaks in the finger region of the ACY spectrum in decreased intensities, or even total disappearance in the spectra of microparticles M2 and M3.
The absorption of SDS (C−H stretching) at 2,917 and 2,850 cm−1 and PVP (C−H stretching) at 2,954 cm−1 were not observed in the core–sheath nanofibers compared with single SDS and PVP. However, the broader and weaker band at 2,918 cm−1 indicated hydrophobic interactions between PVP and SDS molecules. Although electrostatic interactions cannot be determined from the ATR-FTIR spectra, they existed between the negatively charged SDS head group and the nitrogen atom on the pyrrolidone ring of PVP,28 as well as between the negatively charged PVP oxygen (N+=C−O−) and the electron-poor C-1´ of SDS,29 as shown in Figure 7.
These results indicated that a combination of hydrogen bonding and some hydrophobic and electrostatic forces can provide an environment that stabilizes the structure to give a high degree of compatibility among the components that make up the core–shell microparticles. In turn, a homogeneous structure that influences both the stability of SDs and the even dispersion of the drug is created, thereby facilitating the dissolution of ACY from the microparticles.
Functional performances of the microparticles
In vitro dissolution tests
ACY has a UV absorbance peak at 252 nm, whereas the other components PVP, SDS, and sucralose have no absorbance at this wavelength. Using this parameter, the ACY content was determined by UV spectroscopy, and samples were calculated from the calibration curve A=0.05908C + 0.00515 (R=0.9999), where C is the concentration of ACY (μg · mL−1), and A is absorbance at 252 nm (linear range 2–20 μg · mL−1).
A comparison of the in vitro dissolution profiles of ACY from core–shell microparticles with those of crude ACY particles (≤100 μm) is shown in Figure 8. Using a slide glass to collect 10 minutes of microparticles M2 and M3 and then placing a drop of water on them revealed that all microparticles were simultaneously dissolved. After natural drying, observations under cross-polarized light of the transparent membranes revealed larger and brighter dots in M3 than in M2, suggesting the recrystallization of ACY and a high ACY content in M3 (Figure 8B and C). The microparticles released all encapsulated ACY within 1 minute, whereas only 37.7% of drug was released from the ACY particles after dissolution in media for 1 hour.
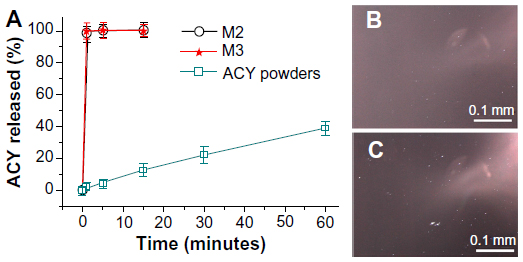 | Figure 8 (A) In vitro drug-release profiles (n=6). (B and C) Recrystallization of acyclovir (ACY) after natural drying and fast dissolution of the spread M2 and M3 microparticles, respectively, when a drop of water was placed on them. |
The core–shell microparticles had the following essential properties for improving the dissolution rates of poorly water-soluble drugs. First, PVP was hygroscopic and hydrophilic, and polymer–solvent interactions were stronger than polymer–polymer attraction forces. Therefore, the polymer chain rapidly absorbed solvent molecules to increase the volume of the polymer matrix and allow the polymer chains to loosen out from their coiled shape. Furthermore, the microparticles had a small diameter and a high surface-to-volume ratio, which indicated a large surface area for the drug to be in contact with the dissolution media. Second, the surfactant SDS facilitated the electrospraying process by reducing the surface tension of the shell fluids and enhanced the hydrophilicity and wettability of the core–shell microparticles, thereby promoting their fast-disintegration processes to release ACY contained in the core parts. Third, the fast dissolution of the core–shell microparticles augmented the local concentrations of supersaturated drug solution, especially in the presence of PVP and SDS. Correspondingly, drug gradients increased for the rapid diffusion of molecules to the dissolution media. Fourth, the drug and matrix polymer formed composites at the molecular level to avoid the crystal lattice energy that must be overcome to dissolve the raw ACY powders.
The synergistic actions of the aforementioned factors can almost simultaneously dissolve ACY molecules with PVP and SDS molecules. In other words, the capability of these microparticles to significantly improve the dissolution rate of poorly water-soluble drugs can be attributed to the reasonable selections of drug carriers, unique properties of microparticles, and amorphous drug status in the filament-forming matrix.
In vitro permeation properties
A comparison of the in vitro permeation profiles of ACY from core–shell microparticles with those of raw ACY particles is shown in Figure 9. The cumulative permeation percentages after half an hour for the core–shell M2 and M3 microparticles and ACY powders were 46.7%, 47.4%, and 6.6%, respectively. Regression values from the linear release time equations were estimated for the core–shell M2 and M3 microparticles and raw ACY powders, ie, QM2 =2.1524t + 2.5953 (R=0.9866, t≤30 minutes), QM3 =2.1619t + 2.4762 (R=0.9877, t≤30 minutes) and QACY =0.2734t + 2.0292 (R=0.9941, t≤30 minutes), respectively (Q is the total permeation of ACY in micrograms, and t is the time in minutes). Therefore, the core–shell M2 and M3 microparticles and raw ACY powders had permeation rates of 0.823, 0.831, and 0.105 μg · minute−1 · cm−2, respectively.
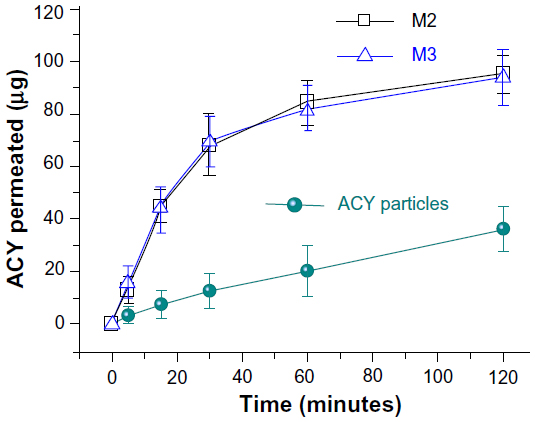 | Figure 9 In vitro permeation profiles of acyclovir (ACY) powders and core–shell microparticles solid dispersions (n=6). |
The core–shell microparticles yielded a permeation rate of ACY that was almost eightfold faster than that of crude ACY powders. The faster permeation rates from the core–shell microparticles can be explained as follows. Permeability across the sublingual mucosa is a passive diffusion process, and thus the faster dissolution of ACY leads to an increased drug-concentration gradient on the mucosal surface. This phenomenon facilitates rapid partitioning of drug into the sublingual mucosae and subsequent permeation. Meanwhile, the SDS released from the shell of the microparticles also improved permeation by extracting intercellular lipids acting as a rate-limiting barrier to the transport of ACY molecules.30 The ACY-loaded core–sheath nanofiber SDs with an average diameter of 410±94 nm have been reported to rapidly free drug within 1 minute and provide more than sixfold-increased permeation rates across sublingual mucosa compared to crude ACY particles.18 Although the present core–shell microparticles were more than threefold larger than the nanofibers, they exhibited a better effect in enhancing the dissolution and permeation of ACY. This should be attributed to the fact that PVP K30 (as a matrix of microparticles) has a smaller molecular weight than PVP K60 (as a matrix of nanofibers), and thus can dissolve faster correspondingly.
In addition to the advantages of improving the solubility behavior and permeation properties of ACY, the core–shell SDs were easier to postprocess, because they have good drug stability, attributed to the steric constraint provided by the small diameter of microparticles and the “nanocoating” effect of the shell on the core drug-loaded region. Therefore, the recrystallization and possible growth of a crystal lattice can be effectively avoided. Within this context, the artificial sweetener sucralose has recently gained popularity because it is 600 times more effective by weight than sucrose, and does not have the bitter aftertaste attributed to many nonnutritive sweeteners. Sucralose is US Food and Drug Administration-approved, and is now used as a general-purpose sweetener in foods, beverages, dietary supplements, and medical foods. The positioning of sucralose in the shell part effectively provides an acceptable taste for patients if SDs are further developed as oral drug-delivery systems.31
Conclusion
A new coaxial electrospraying process was successfully developed, which was characterized by the usage of a new type of concentric spray head with an EP coating. Novel structural SDs of ACY in the form of core–shell microparticles were generated using the developed coaxial processes, despite the core mixed solutions being unprocessable using one-fluid electrospraying. The drug contents of the microparticles are tunable simply through the flow rates of the core drug-loaded solutions within a suitable range. The two types of microparticles prepared in this study had average diameters of 1.36±0.67 and 1.74±0.58 μm, respectively. They were essentially a combination of nanocomposites with the active ingredient ACY distributed in the inner core, and the sweeter and transmembrane enhancer localized in the outer shell. The components of both the core and the shell presented amorphous states, because of the favorable interactions between them and the polymer matrix. The microparticles provided faster dissolution and permeation rates of ACY, because of the core–shell structure, the tailored positional distributions of components, the amorphous state of ACY, and the physicochemical properties of the selected polymer matrix and the other excipients. This study provided an example of the systematic design, preparation, characterization, and application of a novel type of SD consisting of multiple components and with unique structural characteristics. This microparticle structure can enhance the dissolution and permeation performance of a series of poorly water-soluble drugs, and should thus be further developed for novel oral or sublingual drug-delivery systems.
Acknowledgments
This work was supported by the National Science Foundation of China (51373101), the Natural Science Foundation of Shanghai (13ZR1428900), and the Key Project of the Shanghai Municipal Education Commission (13ZZ113).
Disclosure
The authors report no conflicts of interest in this work.
References
Bikiaris DN. Solid dispersions, part II: new strategies in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin Drug Deliv. 2011;8(12):1663–1680. | |
Tran PH, Tran TT, Park JB, Lee BJ. Controlled release systems containing solid dispersions: strategies and mechanisms. Pharm Res. 2011;28(10):2353–2378. | |
Brettmann B, Bell E, Myerson A, Trout B. Solid-state NMR characterization of high-loading solid solutions of API and excipients formed by electrospinning. J Pharm Sci. 2012;101(4):1538–1545. | |
Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water-soluble drugs. Drug Discov Today. 2007;12(23–24):1068–1075. | |
Pasquali I, Bettini R, Giordano F. Supercritical fluid technologies: an innovative approach for manipulating the solid-state of pharmaceuticals. Adv Drug Deliv Rev. 2008;60(3):399–410. | |
Qian F, Huang J, Hussain MA. Drug-polymer solubility and miscibility: stability consideration and practical challenges in amorphous solid dispersion development. J Pharm Sci. 2010;99(7):2941–2947. | |
Myerson AS, Trout BL. Electrospun formulations containing crystalline active pharmaceutical ingredients. Pharm Res. 2013;30(1):238–246. | |
Nagy ZK, Balogh A, Drávavölgyi G, et al. Solvent-free melt electrospinning for preparation of fast dissolving drug delivery system and comparison with solvent-based electrospun and melt extruded systems. J Pharm Sci. 2013;102(2):508–517. | |
Yu DG, Branford-White C, White K, Li XL, Zhu LM. Dissolution improvement of electrospun nanofiber-based solid dispersions for acetaminophen. AAPS PharmSciTech. 2010;11(2):809–817. | |
Chakraborty S, Liao IC, Adler A, Leong KW. Electrohydrodynamics: a facile technique to fabricate drug delivery systems. Adv Drug Del Rev. 2009;61(12):1043–1054. | |
Ding L, Lee T, Wang CH. Fabrication of monodispersed taxol-loaded particles using electrohydrodynamic atomization. J Control Release. 2005;102(2):395–413. | |
Yu DG, Williams GR, Wang X, Liu XK, Li HL, Bligh SW. Dual drug release nanocomposites prepared using a combination of electrospraying and electrospinning. RSC Adv. 2013;3(14):4652–4658. | |
Zamani M, Prabhakaran MP, Ramakrishna S. Advances in drug delivery via electrospun and electrosprayed nanomaterials. Int J Nanomedicine. 2013;8:2997–3017. | |
Duan H, Feng B, Guo X, et al. Engineering of epidermis skin grafts using electrospun nanofibrous gelatin/polycaprolactone membranes. Int J Nanomedicine. 2013;8:2077–2084. | |
Salata OV. Tools of nanotechnology: electrospraying. Curr Nanosci. 2005;1(1):25–33. | |
Li D, Xia Y. Electrospinning of nanofibers: reinventing the wheel? Adv Mater. 2004;16(14):1151–1170. | |
Seil JT, Webster TJ. Spray deposition of live cells throughout the electrospinning process produces nanofibrous three-dimensional tissue scaffolds. Int J Nanomedicine. 2011;6:1095–1099. | |
Yu DG, Zhu LM, Branford-White C, et al. Solid dispersions in the form of electrospun core-sheath nanofibers. Int J Nanomedicine. 2011;6:3271–3280. | |
Nath SD, Son S, Sadiasa A, Min YK, Lee BT. Preparation and characterization of PLGA microspheres by the electrospraying method for delivering simvastatin for bone regeneration. Int J Pharm. 2013;443(1–2):87–94. | |
Enayati M, Ahmad Z, Stride E, Edirisinghe M. One step electrohydrodynamic production of drug-loaded micro- and nanoparticles. J R Soc Interface. 2010;7(45):667–675. | |
Loscertales IG1, Barrero A, Guerrero I, Cortijo R, Marquez M, Gañán-Calvo AM. Micro/nano encapsulation via electrified coaxial liquid jets. Science. 2002;295(5560):1695–1698. | |
Liang X, Li J, Joo JB, et al. Diffusion through the shells of yolk-shell and core-shell nanostructures in the liquid phase. Angew Chem Int Ed. 2012;51(32):8034–8036. | |
Chaudhuri RG, Paria S. Core/shell nanoparticles: classes, properties, synthesis mechanisms, characterization, and applications. Chem Rev. 2012;112(4):2373–2433. | |
Yu DG, Liu F, Cui L, Liu ZP, Wang X, Bligh SW. Coaxial electrospinning using a concentric Teflon spinneret to prepare biphasic-release nanofibers of helicid. RSC Adv. 2013;3(39):17775–17783. | |
Bikiaris DN. Solid dispersions, part I: recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin Drug Deliv. 2011;8(11):1501–1519. | |
Bühler V. Kollidon: Polyvinylpyrollidone for the Pharmaceutical Industry. 2nd ed. Ludwigshafen: BASF; 1998. | |
Li W, Yu DG, Chen K, Wang G, Williams GR. Smooth preparation of ibuprofen/zein microcomposites using an epoxy-coated electrospraying head. Mater Lett. 2013;93(2):125–128. | |
Pongpeerapat A, Higashi K, Tozuka Y, Moribe K, Yamamoto K. Molecular interaction among probucol/PVP/SDS multicomponent system investigated by solid-state NMR. Pharm Res. 2006;23(11):2566–2574. | |
Roscigno P, Asaro F, Pellizer G, Ortana O, Paduano L. Complex formation between PVP and sodium decyl sulfate. Langmuir. 2003;19(23):9638–9644. | |
Nicolazzo JA, Reed BL, Finnin BC. Buccal penetration enhancers – how do they really work? J Control Release. 2005;105(1–2):1–15. | |
Yu DG, Yang JM, Branford-White C, Lu P, Zhang L, Zhu LM. Third generation solid dispersions of ferulic acid in electrospun composite nanofibers. Int J Pharm. 2010;400(1–2):158–164. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.