Back to Journals » Research Reports in Clinical Cardiology » Volume 8
Eisenmenger syndrome: current perspectives
Authors Nashat H, Kempny A, McCabe C, Price LC, Harries C, Alonso-Gonzalez R, Gatzoulis MA, Wort SJ, Dimopoulos K ![]()
Received 21 July 2016
Accepted for publication 25 October 2016
Published 2 February 2017 Volume 2017:8 Pages 1—12
DOI https://doi.org/10.2147/RRCC.S117838
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Kones
Heba Nashat,1–3 Aleksander Kempny,1–3 Colm McCabe,1 Laura C Price,1–3 Carl Harries,1 Rafael Alonso-Gonzalez,1–3 Michael A Gatzoulis,1–3 Stephen J Wort,1–3 Konstantinos Dimopoulos1–3
1Adult Congenital Heart Centre and National Centre for Pulmonary Hypertension, Royal Brompton Hospital, 2National Heart and Lung Institute (NHLI), Imperial College, 3National Institute for Health Research (NIHR) Cardiovascular Biomedical Research Unit, Royal Brompton Hospital and National Heart and Lung Institute, Imperial College, London, UK
Abstract: Eisenmenger syndrome (ES) is the most severe form of pulmonary arterial hypertension (PAH) related to congenital heart disease (CHD). It results from a cardiac defect allowing significant systemic-to-pulmonary (left-to-right) shunting, which triggers the development of pulmonary vascular disease (PVD) if the defect is not repaired in a timely fashion. Once severe PVD has developed, the defect cannot be repaired. With advances in pediatric cardiology and surgery, the prevalence of ES is steadily falling in developed countries; nonetheless, there will always be patients who are unsuitable for repair at the time of diagnosis, or emigrating from countries with less advanced healthcare, who will develop ES. ES is a multisystem disorder causing chronic hypoxemia and reduced cardiac output resulting in significant morbidity and mortality. While lung (plus defect repair) or combined heart and lung transplantation is thought be the definitive treatment for ES, transplant organs are a limited resource and long-term results are still suboptimal. PAH pharmacotherapy was, until quite recently, largely directed at symptomatic relief and had no impact on morbidity and mortality. Targeted PAH therapies have recently been proven to be beneficial in various forms of PAH in terms of functional status, progression of disease, and prognosis. Data on the effect of PAH therapies in the ES cohort remain limited, but available studies demonstrate evidence of improvement in symptoms, exercise capacity, and some evidence of survival benefit. ES patients should be followed in specialized centers, by means of an interdisciplinary approach by clinicians experienced in PAH and CHD. However, local physicians working in cardiology, respiratory medicine, primary care, and emergency services are likely to encounter ES patients and need to be aware of the main issues and pitfalls in their care. The authors present an overview of the management of ES, focusing on the most common issues and complications.
Keywords: Eisenmenger syndrome, pulmonary arterial hypertension, congenital heart disease, cyanosis, pulmonary arterial hypertension therapies, hyperviscosity, thrombosis, bleeding, pregnancy, Down syndrome
Introduction
Congenital heart disease (CHD) is the most common inborn defect, occurring in ~0.8% of neonates.1 With perinatal care and screening programs, including advances in fetal echocardiography, affected children are increasingly born into a “prepared” environment and undergo surgery in a timely fashion, with increasing numbers surviving to adulthood.2 It is estimated that up to 10% of adults with CHD develop pulmonary arterial hypertension (PAH) of any severity, a disease process that is the result of numerous pathological pathways affecting the pulmonary vascular bed, resulting in a rise in pulmonary vascular resistance (PVR), right heart failure, and premature death.3
Eisenmenger syndrome (ES) is the most advanced form of PAH associated with CHD (PAH-CHD). In 1897, Dr Victor Eisenmenger first reported the case of a man with cyanotic heart disease and severe pulmonary hypertension (PH). During the years that followed, the anatomy, pathophysiology, and diagnostic features of the abnormalities described in his report became better understood and increasingly relevant to clinical practice. In 1958, Dr Paul Wood created a structured classification of the clinical characteristics of this condition based on his observations of 127 patients, and demonstrated that ES can develop as a result of large shunts at various locations: atrial or ventricular, patent ductus, or aortopulmonary window.4 Long-term exposure of the pulmonary bed to increased pulmonary flow and pressure from a post-tricuspid (eg, ventricular septal defect [VSD]) left–right shunt results in vascular remodeling and dysfunction. This, in turn, leads to a rise in PVR, which, if severe enough, results in reversal of the shunt and the clinical cyanosis characteristic of ES. Once ES is established, the defect is no longer surgically correctable, as it is felt to act as a relief valve for the right ventricle (RV).5
The exact prevalence of ES is not known. According to historical data and before the advent of timely intervention, ~8% of patients with CHD and 11% of those with left-to-right shunts developed ES.6,7 Enhanced understanding and timely surgical or interventional repair of defects has resulted in a substantial reduction in the numbers of patients developing ES. Nevertheless, this condition will continue to occur in patients unsuitable for early repair and individuals from countries where availability of tertiary pediatric cardiology and cardiothoracic surgery is limited.
Anatomical variations in patients with ES and outcome
The classification of PH was first developed in 1998 and the condition was categorized into groups that shared similar pathological, hemodynamic characteristics, and therapeutic approaches. The current classification in use was finalized at the fifth World Health Organization (WHO) meeting in 2013 by Simonneau et al.8 The classification comprises of five groups. Group 1 is PAH, to which PAH-CHD belongs as a type of “associated PAH”. PAH-CHD is further divided into four main anatomic subgroups, of which ES is one (Table 1).8 The frequency of PH in these types of CHD and the subsequent development of right-to-left shunting vary depending on the size and location of the defect and whether previous intervention has been performed.9 ES is typically associated with large, nonrestrictive intra- or extra-cardiac communications. These include VSDs, atrial septal defects (ASDs), an aortopulmonary window, or a patent ductus arteriosus.
 | Table 1 Clinical classification of pulmonary arterial hypertension associated with congenital heart disease Abbreviations: ASD, atrial septal defect; PAH, pulmonary arterial hypertension; PVR, pulmonary vascular resistance; VSD, ventricular septal defect. |
Observational studies have shown that there may be differences in the natural course and ventricular adaptation to long-standing pulmonary vascular disease, depending on the location of the defect in ES patients and the presence of a uni- or biventricular heart. In an echocardiographic study of 191 patients with ES and noncomplex CHD, the location of the defect was associated with different physiological adaptation of the RV to the PAH and, possibly, a different prognosis: pre-tricuspid shunts were older, had larger RVs, and a trend toward worse prognosis when compared to post-tricuspid lesions.10,11 Indeed, the mechanisms behind shunt reversal in ES ASD patients differs significantly to post-tricuspid shunts, depending significantly on the relative compliance of the left and right ventricles and, perhaps, less so on the ratio between systemic and pulmonary resistances.
General management of Eisenmenger syndrome
Although patients with ES can survive into the fourth and fifth decades of life, their condition is associated with high morbidity, reduced functional status, and frequent hospitalizations. In ES, the chronic hypoxemia, together with a low cardiac output and PAH, has significant multisystem effects, resulting in a vast spectrum of complications (Table 2).5,12–15 Cardiac arrhythmias, hemoptysis, infections, and right heart failure are important late complications and are a frequent cause of death. Other complications such as pulmonary artery (PA) dilatation and in situ thrombosis are frequently encountered in this cohort (Figure 1).
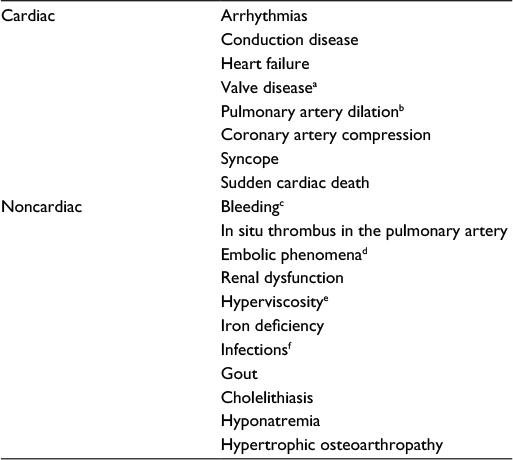 | Table 2 Complications of Eisenmenger syndrome Notes: aTricuspid or pulmonary valve regurgitation. bMay cause extrinsic compression of coronary arteries or PA dissection. cMay be pulmonary, gastrointestinal, or cerebral. dMay be pulmonary or cerebral/systemic. eSymptoms may include headache, dizziness, visual disturbance, altered mental state, tinnitus, and fatigue. fImportant to consider endocarditis or cerebral abscess. Abbreviation: PA, pulmonary artery. |
 | Figure 1 Computed tomography of a patient with a patent ductus arteriosus demonstrating dilated PA compared to the aorta (A), typically seen in Eisenmenger syndrome. In situ thrombosis (B, arrows) is also a common complication in patients with Eisenmenger syndrome. Abbreviation: PA, pulmonary artery. |
The anticipation and timely management of potentially specific complications arising from cyanosis due to the right-to-left shunt and associated morbidities, such as iron deficiency, thrombosis, gout, renal dysfunction, cholelithiasis, and cerebral infections (Figure 2), are just as important to consider as the specific treatment of the PAH with therapies.
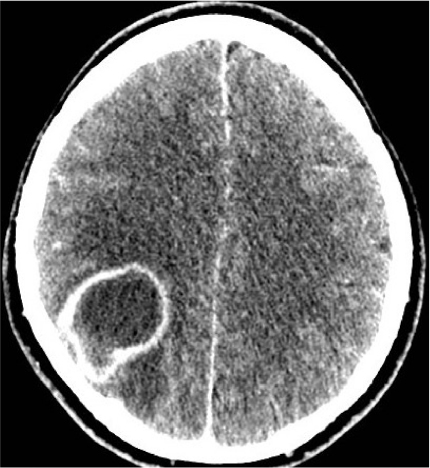 | Figure 2 Cerebral abscess with surrounding edema and midline shift in a patient with VSD and ES presenting with syncope and a mild fever. Abbreviation: ES, Eisenmenger syndrome; VSD, ventricular septal defects. |
Current guidelines in the management of patients with ES recommend regular consultations with experienced physicians in CHD and PH.15 Areas that require routine consideration are detailed in the following sections, but also include adequate contraception, endocarditis prophylaxis, up-to-date immunization against influenza, and pneumococcal infections.16
Before the advent of PAH-specific therapies, medical treatment was restricted to diuretics, anticoagulation, digoxin, β-blockers, and antiarrhythmics, mainly for symptomatic relief with limited evidence. β-blockers and calcium channel blockers are generally contraindicated in PAH due to the negative inotropic effect on the RV.17 Unlike left-sided heart failure, β-blockers have not yet been shown to improve RV function but may be used in specialist PH centers for other indications such as prevention of recurrent arrhythmias in small doses. More recently, animal studies have shown a potential role for β-blocker therapy to protect RV function and their use in idiopathic PAH (iPAH) is currently being assessed in clinical trials.18,19 Moreover, patients with ES often have diastolic impairment, if there is a need for heart rate control, then β-blockers are at times favored over digoxin as they improve diastolic filling time and potentially optimize symptoms, albeit with no robust evidence. Interestingly, in a study of 153 patients with ES, Diller et al noted a trend toward better survival in patients on β-blockers in their analysis.20
While the natriuretic peptide, sympatho-adrenergic, and renin–angiotensin–aldosterone systems are activated as a result of abnormal distension of cardiac chambers, these are not currently targets for therapy in ES, with the exception of aldosterone antagonists.21 Aldosterone antagonists (eg, spironolactone) and loop diuretics are often used in these patients. However, the use of diuretics should be carefully monitored as dehydration and significant fluid shifts may exacerbate a right-to-left shunt, thus worsening the cyanosis, potentiating renal dysfunction, and increasing the risk of thromboembolism. Hyperaldosteronism is common in RV dysfunction in PH and is often used in conjunction with other diuretics, albeit with very limited evidence. Traditionally, angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) have been avoided in patients with ES because of the potential worsening of cyanosis from an increase in a right-to-left shunt as a consequence of the reduction of the systemic vascular resistance (SVR).22 However, evidence in this is limited and ambiguous. A study of 60 patients with cyanotic heart disease examined the effect of ACE inhibition and did not report a significant reduction in oxygen saturation, but a potential improvement in functional class and quality of life.23 Moreover, in a German registry of 153 patients with ES of various cardiac anatomies, ~10% were on ACE-i/ARBs with no significant association between ACE-i/ARBs and their outcome.24
Hyperuricemia and gout are also common in ES and administration of uricosuric or uricostatic agents is indicated in patients with recurrent gouty arthritis. In the acute phase, symptoms can be relieved by oral colchicine or intraarticular corticosteroids, as indicated. Asymptomatic hyperuricemia is not an indication for routine drug therapy.25
Secondary erythrocytosis, iron deficiency, and venesection
The effect of chronic hypoxemia causes a compensatory secondary erythrocytosis aimed at increasing the blood’s oxygen-binding capacity. This process requires substantial iron stores for the production of hemoglobin (Hb) and is likely the main cause of the iron deficiency commonly found in cyanotic patients.26 Prophylactic or routine venesection to maintain a hematocrit level within an arbitrary predetermined level (hematocrit <65%) is not indicated.27,28 This is one of the major misconceptions in the management of patients with cyanotic CHD and routine venesection can be harmful to these patients, as they can result in iron deficiency, reduced exercise tolerance, impaired oxygen transport capacity due to “relative anemia”, and may increase the risk of cerebrovascular events.29,30 Venesection should only be offered to patients with moderate-to-severe hyperviscosity symptoms (Table 3) due to significant secondary erythrocytosis (Hb level well above 24 g/dL) in the absence of volume depletion/dehydration.31 Also, venesection can be considered preoperatively for autologous blood donation if the hematocrit level is >65% and for boosting platelet production.32
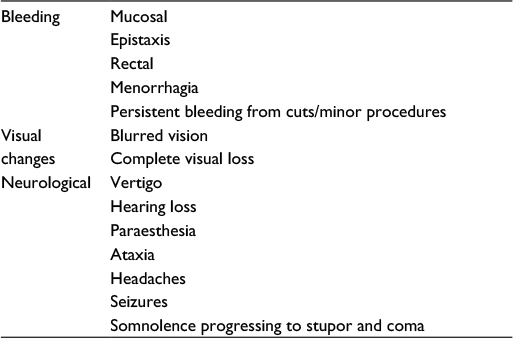 | Table 3 Hyperviscosity symptoms Note: Hyperviscosity symptoms consist principally of the triad of mucosal bleeding, visual changes, and neurologic symptoms. |
Iron deficiency is a common complication in patients with ES.33 “Relative anemia” is often not recognized in cyanotic patients: a Hb concentration, which drops to levels that are normal for the general population, is often suboptimal for cyanotic individuals.27
Beyond high iron requirements and inappropriate venesections, other contributors to iron deficiency and relative anemia include bleeding and the use of oral anticoagulants (OACs). Hemoptysis, epistaxis, menorrhagia, and gastrointestinal bleeding are not uncommon in ES and may be difficult to manage. Iron supplementation is recommended, especially when Hb concentration is significantly below the level expected for the patient’s oxygen saturation, but excessive erythrocytosis causing hyperviscosity symptoms (Table 3) should be monitored. A study of 68 patients by Van De Bruaene et al found that patients with resting oxygen saturations of <83% often reached Hb concentrations in the range that may be considered “at risk” for hyperviscosity symptoms when treated for iron deficiency and suggested closer monitoring for hyperviscosity symptoms in this group.34 As iron deficiency is strongly related to adverse outcomes, particularly in those on anticoagulation or those having inappropriate venesection, it is important to regularly screen patients for this and treat when appropriate.
Oxygen therapy
The use of oxygen supplementation in patients with ES is controversial. It has been shown to have no impact on exercise capacity and survival in adult patients with this condition.35 However, some may benefit from nocturnal supplementation for symptomatic relief, and oxygen may be useful in patients with advanced disease and in those awaiting heart–lung transplants. Air travel appears to be safe, as long as airplanes are adequately pressurized.29,36 Supplemental oxygen during commercial air travel is not routinely required, especially for shorter flights up to 4 hours. It can, however, be administered for symptomatic benefit on for longer flights.32
Anticoagulation
Patients with ES are at an increased risk of PA thrombosis. A retrospective analysis by Silversides et al found that, of 34 patients with ES, 21% had evidence of proximal PA thrombus. These patients were more likely to be women and have lower oxygen saturations and advanced disease with worse ventricular dysfunction, exercise intolerance, and natriuretic peptide levels.37 On the other hand, ES patients are also at an increased risk of bleeding, including potentially life-threatening pulmonary hemorrhage. It is not routine to anticoagulate patients with ES, unless there are coexisting conditions such as atrial fibrillation, pulmonary thromboembolic, congestive heart failure, or embolic events in the absence of significant hemoptysis or other bleeding risks. OACs are associated with an increased risk of iron deficiency anemia and patients should be routinely investigated for this.34 Also, endothelin receptor antagonists (ERAs) can reduce the efficacy of anticoagulants such as warfarin; therefore, necessitate close monitoring of the international normalized ratio is needed to maintain an adequate therapeutic index. Novel OACs have not been tested in this group and there are concerns regarding the limited availability of direct reversing agents. With the recent Food and Drug Administration (FDA) approval of iarucizumab for reversal of dabigatran, this may become an option in the future.38
Exercise training
Exercise training (ET) should not be discouraged in patients with ES.39 In patients with severe heart failure, regular training improves endothelial function in systemic vessels and long-term outcome.40,41 Impaired skeletal muscle function has been described in CHD and is a potential target for rehabilitation/ET.42 Mild-to-moderate aerobic activity and low-level resistance exercises can be safely performed in most clinically stable patients. The recommendation for patients with ES is low dynamic sport leisure activities with a low isometric (static) component, while competitive sports are contraindicated. Highly isometric exercise (eg, weight lifting) should be avoided. Patients with a persistent shunt should avoid scuba diving.43
Noncardiac surgery
PAH, CHD, and ES are known risk factors for perioperative complications.44 Patients with PAH are often counseled against elective surgery because of the risk of early and sudden postoperative deaths. In PAH, the RV cannot accommodate large alterations in preload or afterload induced by fluid shift, anesthetic medication, insufflation of gas in the abdomen during laparoscopic procedures, or autonomic changes precipitated by hypoxia or hypercapnia, which are magnified by stress or pain. RV ischemia, resulting in worsening RV function, can be precipitated by systemic hypotension and arrhythmias during surgery.45 Therefore, the choice of anesthetic is crucial for patients with PAH. The same principles of risk management apply to ES.
The two main principles of perioperative risk management are the prevention of systemic hypotension and avoidance of an increase in PVR (PH crisis). Specific risks during surgery include arrhythmias, thromboembolism, and bleeding and sudden changes in SVR. Close monitoring, optimization of systemic blood pressure, pain control, oxygenation and ventilation, avoidance of exacerbating factors, and use of vasopressors and pulmonary therapies, as necessary, are essential elements of perioperative management. In a study of 33 ES patients undergoing general or regional anesthesia, 26% experienced profound hypotension and 17% a drop in oxygen saturation. Bennett et al found that vasopressor agents given during induction significantly reduced the incidence of hypotension.46
Patients with an underlying cardiac condition, which includes any cyanotic CHD, are at higher risk of infective endocarditis and should receive prophylaxis. This, however, only applies to high-risk procedures, which are mainly invasive dental procedures, and not for respiratory tract procedures, gastrointestinal/urogenital, or skin/soft tissue procedures.16
Transplantation
The only definitive treatment for ES is lung transplantation (LT) with shunt closure or heart and lung transplantation (HLT). In a a worldwide registry of 605 transplanted ES patients over a 10-year period, Waddell et al found that, in this heterogeneous group, post-transplantation survival related to the underlying cause of the ES, and HLT appeared to be better than LT with shunt correction: 30 days and 1-year survival rates were 80.7% and 70.1% compared with 68% and 55.2%, respectively.47 In another study of 51 patients with ES transplanted in the UK, the 1-, 5-, and 10-year survival rates for ES were 72.6%, 51.3%, and 27.6%, respectively, compared to non-ES of 74%, 48.1%, and 26%, respectively with no difference in survival overall.48 Even though it has been suggested that patients with ES may have a better post-transplantation prognosis than patients with iPAH or other types of congenital heart defects, the general lack of organs and suboptimal survival after LT or HLT underline the need for alternative therapeutic options.49
Disease-specific targeted PAH therapy
The mainstay of PAH treatment is, nowadays, specific disease-targeting pulmonary therapies, which have only been in use since the 1990s. Acute vasoreactivity testing with nitric oxide (NO) and subsequent therapy with calcium channel blockers in responders is established in iPAH, but no evidence is available in PAH-CHD. The use of calcium channel blockers is, in fact, discouraged in ES, as they may cause significant peripheral vasodilatation and hypoxia.50
So far, only three pathomechanistic pathways in PAH have been translated into clinical practice: prostacyclin, nitric oxide, and endothelin pathways. Oral and parenteral therapies have been designed to target these pathways, namely prostanoids, phosphodiesterase type-5 inhibitors (PDE-5i), and ERAs. Other substances, such as soluble guanylate cyclase inhibitors, which target the same pathways as PDE-5i are becoming available; however, their role in ES has not been established. There are very few randomized data on specific pulmonary therapies in ES, with further support from a number of prospective, open-label, and retrospective studies.51–54
Despite the associated functional limitations and multitude of comorbidities, survival prospects in adults with ES are generally considered better than in other forms of PAH, but mortality remains high. In a systematic review of literature and reanalysis of data in treatment-naïve patients with ES, Diller et al found that after adjusting for immortal time bias, the 10-year mortality in 1,131 patients from 12 studies was 30%–40% and survival prospects have not considerably improved over the last decade in untreated patients.20 However, registry data have shown better survival prospects in ES patients on pulmonary therapies disease when compared to those who were treatment naïve.24,55
Endothelin-1 receptor antagonists
ET-1 (Endothelin-1) is a powerful vasoconstrictor, produced in endothelial cells, with elevated concentrations seen in patients with PAH. ET-1 plays a key role in the pathogenesis of PAH by inducing proliferation, fibrosis, and inflammation.56 These effects are mediated through the endothelin receptor subtypes A (ETA) and B (ETB) on which orally active ERAs selectively target either a single receptor (ETA) or both receptors (ETA and ETB). Commercially available ERAs are ambrisentan, which targets a single receptor ETA, and bosentan and macitentan that target both receptors. A fourth ERA targeting a single receptor, sitaxentan, was withdrawn in 2010 due to two cases of fatal hepatic failure.
Bosentan was the first drug in this category to be approved by the FDA and the European Medicines Agency in 2002 for the treatment of PAH. Its specific use in patients with ES is supported by the first ever randomized control trial (RCT) of PAH therapies in this cohort. Bosentan Randomized Trial of Endothelin Antagonist Therapy-5 was a multicenter, double-blind, randomized, placebo-controlled study of 54 patients with ES, assessing the safety and efficacy of bosentan as an endpoint in this cohort. Sixteen weeks of therapy resulted in significant improvement in hemodynamics and 6-minute walk distance (6MWD), without adversely affecting systemic arterial oxygenation.52 These positive findings were maintained in the open-label extension up to 40 weeks.57 Bosentan is currently endorsed as the first-line choice of treatment in patients with ES in the WHO functional class III–IV. A caution with this drug is that in 10% of cases there is an increase in hepatic aminotransferases, which is reversible by dose reduction or discontinuation. For this reason, monthly liver function tests should be performed in patients receiving bosentan. Furthermore, interaction with progesterone-based contraception means that double contraception is recommended.
Ambrisentan is a selective mono-antagonist of the ETA receptor with a longer half-life allowing for once-a-day dosing. Unlike bosentan, there are no RCTs or prospective studies of ambrisentan in ES. A retrospective study by Zuckerman et al in patients with CHD showed improvement in 6MWD without any adverse outcome in arterial saturations or serious adverse events.58 Liver function abnormalities are less common with this ERA, ranging from 0.8% to 3%: monthly blood tests are required in the UK, but not in the United States.
Macitentan, similar to bosentan, is a dual ET receptor antagonist and has been evaluated in a large event-driven RCT on patients with PAH (not ES). The Study with Endothelin Receptor Antagonist in Pulmoanry Arterial Hypertension to Improve Clinical Outcome (SERAPHIN) included 565 patients with PAH and ~8% of these had PAH after repair of a congenital shunt. When compared to placebo, macitentan significantly reduced morbidity/mortality and increased exercise capacity. There were no cases of liver toxicity, but a reduction in Hb was commonly seen.59 Concurrently, the clinical study to evaluate the effects of macitentan on exercise capacity in subjects with Eisenmenger syndrome; MAESTRO, an RCT is underway with an aim to evaluate the effect of macitentan on 6 minute walk distances in patients with ES.60
Phosphodiesterasetype-5 inhibitors and guanylate cyclase stimulators
Phosphodiesterases are a group of enzymes that inactivate cyclic adenosine monophosphate (cAMP) and guanosine monophosphate (cGMP), which are expressed abundantly in lung tissue and are upregulated in patients with PAH.61 Inhibition of these enzymes results in vasodilation through NO/cGMP pathways in sites in which they are most dense. PDE-5i clinically acts as potent vasodilators and also has an anti-proliferative effect. Tadalafil and sildenafil selectively inhibit the phosphodiesterase type-5 enzymes. Oral administration is not associated with hepatic toxicity; therefore, patients do not require regular blood testing. As the CYP3A4 pathway metabolizes these drugs, caution should be exercised when prescribing them with CYP3A4 inhibitors and inducers.62
Two small RCTs have been published in ES evaluating the effect of PDE-5i in a total of 48 patients. These studies demonstrated improvements in 6MWD, functional class, PA pressures invasively, and by echocardiographic parameters.53,63 Further prospective studies have supported the enhancing effects of PDE-5i in patients with ES.64,65 However, because the evidence level is lower than that of ERAs, PDE-5i are often used as second-line disease targeting therapy.
Riociguat is a relatively new drug in PAH. Unlike PDE-5i that acts on the NO/cGMP pathway to slow down cGMP degradation, guanylate cycle stimulators (sGCs) enhance the production of cGMP, a potent vasodilator. It has been tested by means of an RCT and has shown favorable results in exercise capacity, hemodynamics, functional class, and time to clinical worsening in patients with PAH.66 It was most beneficial when combined with other disease-targeted therapy, but combination with a PDE-5i is contraindicated due the risk of profound hypotension.67 Rosenkranz et al explored the efficacy and safety of riociguat in the subgroup of 35 patients with repaired CHD included in the PATENT-1 trial and its open-label extension, PATENT-2. They concluded that sGCs are well tolerated and improved outcomes, including 6MWD, PVR, functional class, and N-terminal pro–B-type natriuretic peptide.68 Thus, riociguat is a valid alternative to PDE-5i in the future, but data on ES are lacking. Moreover, as yet, there are no head-to-head comparisons between sGCs and PDE-5i; therefore, its use in patients with CHD remains limited.
Prostacyclin analogs and prostacyclin receptor agonists
Prostacyclin is produced predominantly in endothelial cells and induces potent vasodilatation of all vascular beds by stimulating the production of cAMP and inhibiting growth of smooth muscle cells. It is an endogenous inhibitor of platelet aggregation and is cytoprotective and antiproliferative.69 A dysregulation of the prostacyclin pathway has been implicated in PAH, thus, it is a target for pharmacotherapy. Clinical use of prostacyclin has expanded thanks to the production of stable analogs that have different pharmacokinetic properties but share similar pharmacodynamic effects. Infusion of prostacyclin was the first therapy shown to reduce mortality in a controlled study of patients with severe iPAH. However, prostacyclin can only be administered through an intravenous, subcutaneous, or nebulized (inhaled) route, which can influence patients’ compliance, tolerance, and increase the risk of infection when tunneled catheters are used. Intravenous infusions, particularly in the setting of a right-to-left shunt, may increase the risk of thrombotic and infective complications related to the line, including endocarditis or cerebral abscesses. Therefore, subcutaneous or nebulized administration is more appealing for this cohort. Moreover, in patients with right-to-left shunting systemic side effects are common, reducing tolerance and the ability to uptitrate the dose, while an abrupt interruption may result in rebound PH, deterioration, and even death.
Beraprost is an orally active prostacyclin analog, currently in use in Japan. In an RCT in 130 patients, improvements in exercise capacity at 6 months were observed, but there was no hemodynamic improvement or long-term outcome benefits.70 Its use in ES is limited and is associated with frequent adverse events such as headache, flushing, jaw pain, and diarrhea. Since 2013, oral treprostinil has been available in the USA. In the initial primary efficacy study, oral treprostinil in combination with an ERA and/or a PDE-5i for the treatment of PAH trial (FREEDOM-M), patients demonstrated an improvement in their 6MWD when compared to those receiving placebo. In further phase-3 studies, FREEDOM-C and FREEDOM-C2, no significant benefit was seen in exercise with median 6MWD. Furthermore, all patients with PAH-CHD had repaired defects.71,72
Epoprostenol, a synthetic prostacyclin, has a short half-life (3–5 minutes) and requires continuous administration by means of an infusion pump and permanent tunneled catheter. Epoprostenol has been well studied in patients with iPAH and has shown improvements in exercise capacity, quality of life, and hemodynamics.73 Similar improvements have been reported in small cohorts of patients with ES.74 A case series of eight ES patients on intravenous epoprostenol showed improved oxygenation and 6MWDs after treatment.75 However, data are limited and epoprostenol is currently mainly used as a third-line agent in patients with ES. Fears of a high risk of endocarditis or systemic embolic events with intravenous epoprostenol in ES are not supported by available data.
Iloprost is a chemically stable prostacyclin analog available as intravenous, oral, or aerosol administration. The inhaled form is felt to be more pulmonary selective and, therefore, minimizes systemic side effects.76 In this form, it requires 6–8 inhalations a day due to its 25 minutes half-life. In PAH and chronic thromboembolic PH, an RCT evaluating repeated iloprost inhalations when compared to placebo showed an increase in exercise capacity and improvement in symptoms, PVR, and clinical events.77 Intravenous iloprost is thought to be as effective as epoprostenol. The effects of oral iloprost have not been assessed in PAH.
Treprostinil is another stable prostacyclin, with a longer half-life of 3 hours. It is currently available for subcutaneous and intravenous administration. Data on its use in the congenital population is extrapolated from a large RCT of various forms of PAH, of which 24% had PAH-CHD. There were benefits in exercise capacity, hemodynamics, and clinical events, but a high frequency of site pain limits subcutaneous administration.78 Evidence on the intravenous administration of treprostinil in PAH-CHD is very limited.79 Nonetheless, long-term use of treprostinil is felt to have similar survival benefits as intravenous epoprostenol.80
Selexipag is one of the latest pulmonary therapies to be tested in PAH. It is an orally available selective prostacyclin IP receptor agonist. Although, its mode of action is similar to that of endogenous prostacyclin, by means of IP receptor agonism, it is pharmacologically distinct from prostacyclin. An initial phase-2 proof of concept pilot RCT examining the safety and efficacy of selexipag in PAH patients showed a reduction in PVR after 17 weeks of treatment. Safety was evaluated in relation to the frequency of treatment-emergent adverse events, premature study drug discontinuation, changes from baseline measurements of vital signs, ECG, and laboratory parameters.81 An event-driven phase-3 RCT of selexipag versus placebo followed 923 patients, 10% of which had PAH associated with a corrected congenital shunt. This trial showed that selexipag alone or in combination with an ERA and/or a PDE-5i reduced composite morbidity and mortality by 40% (hazard ratio 0.6, P<0.001).82 Currently, there are no data on the use of selexipag in patients with ES; however, this drug is a likely contender for investigation in this group.
Combination therapy
There is limited information on the efficacy of combination therapy in ES, even though most experts feel that combining therapies and targeting different pathomechanistic pathways improve results. Increasingly, the evidenced-based trend in iPAH is to treat patients aggressively with upfront or early sequential double or triple therapy, as early initiation of combination therapy is believed to delay disease progression and improve survival.83,84 This is reflected in the 2015 European Respiratory Society/European Society of Cardiology consensus guidelines on the management PH, whereby the treatment algorithm now suggests upfront combination therapy, even in those with less severe disease.62 As yet there are no studies to test upfront combination therapy against aggressive sequential therapy, thus, the application of either option remains at the discretion of the clinician.
Conversely, adult patients with ES often have prevalent established disease and are stable for long periods. Sequentially adding second- and third-line agents appear acceptable for those failing to meet clinical targets or with evidence of deterioration. One randomized double-blind crossover trial evaluated the effect of combination therapy with sildenafil and bosentan in 21 patients with ES. They found that bosentan alone significantly improved 6MWD, PVR, and pulmonary blood flow. When combined with sildenafil, there was no further improvement in walk distance; however, there was an increase in oxygen saturations at rest.54 In a prospective open-label study, D’Alto et al also studied the combined effect of bosentan and sildenafil in ES patients and reported an improvement in functional class.85 Interestingly, Diller et al showed no survival benefit on direct comparison of patients with combination therapy versus monotherapy.24 However, patients on combination therapy likely represented a sicker population with more advanced symptoms, hence making direct comparison unreliable. These results should not preclude escalation of therapy in ES, whereby alleviation of symptoms and exercise capacity maybe associated with clinical stabilization and may indeed have an impact on survival over time.24 ES is a progressive condition, which often requires sequential treatment escalation, whereas upfront combination therapy is currently not routinely used.
ES in patients with Down syndrome
Down syndrome (DS) is a well-recognized genetic condition associated with several medical morbidities, intellectual disability, and characteristic physical features. In patients with DS, there is a high prevalence of congenital heart defects in the order of 40%–60% and is a major cause of both morbidity and mortality in this group. An atrioventricular septal defect is the most common structural abnormality, while other abnormalities such as mitral valve prolapse may develop later in life.86 Children with DS and CHD develop progressive pulmonary vascular changes earlier than those without DS with similar CHD, possibly due to intrinsic endothelial dysfunction of the pulmonary vasculature and elevation of plasma levels of asymmetric dimethyl arginine, which might contribute to the development of PH.87,88 Despite the significant disease burden in this population, data are very limited on the use of PAH therapies and there are no randomized clinical trials in this population. In an attempt to reflect the success of bosentan in patients with ES, an open-label study of 28 patients with DS were followed up for 11.5 months confirming that this drug was well tolerated with no serious adverse events and significant improvement in walk distance but no changes in quality of life questionnaires.89 However, concerns on the validity of 6MWD tests, reliability of quality of life questionnaires, patient compliance to treatment or regular blood monitoring, and the risks related to invasive assessments such as the need for general anesthesia make the evaluation and management challenging in patients with DS. In the United States, hospitalizations for patients with DS/CHD are decreasing but those who are hospitalized are more likely to die during admission but less likely to undergo a cardiac procedure.90 A multidisciplinary approach in specialist centers is advocated to enable the management of ES in patients with DS.91
Pregnancy in patients with ES
Maternal mortality in the presence of ES is extremely high and has not improved sufficiently in the last 2 decades, despite advances in treatment.92–94 Mortality rates remain worryingly high, at 30%–50% for the expectant mother and, as a result, patients are strongly advised to avoid pregnancy. Many women with ES are also often treated with pulmonary therapies, which are teratogenic (ERAs), while cyanosis makes spontaneous abortions likely. Also, the expectant mother with ES should be advised on the risk of transmission of CHD to her offspring, varying from 3% to 5% compared with 1% risk for the general population.95
Unfortunately, some women who are fully informed and understand the maternal and fetal risk and complications may still become pregnant, while others who are lost to specialist follow-up may present pregnant without having received appropriate preconception counseling. If a patient decides to continue her pregnancy, careful follow-up in centers with appropriate experience in ES, CHD, high-risk obstetrics, anesthesia, and intensive care, so as to manage complications promptly and appropriately. Patients on warfarin are at risk of fetal embryopathy. In a study by Vitale et al, they found no cases of embryopathy in women taking a dose of <5 mg/day.96 However, these figures have not been replicated in other studies, with other groups reporting a 5% risk of embryopathy in women taking <5 mg/day and 7% for those taking >5 mg/day.97 It is recommended that women who must remain on some form of anticoagulation throughout pregnancy are prescribed either unfractionated heparin or low-molecular-weight heparin in their first and third trimesters and warfarin in the second trimester, as optimum anticoagulation particularly for mechanical valves is best achieved with warfarin. ERAs are teratogenic and should be discontinued. ERAs can be replaced by PDE-5i and prostanoids that pose less of a fetal risk. Inhaled NO or intravenous prostanoids may also be used in the intensive care unit if a pulmonary hypertensive crisis or right ventricular failure occurs, which can occur up to several weeks postpartum.61
A discussion about contraceptive methods is imperative, especially if patients are on bosentan. Progesterone-only formulations, such as depot injections and subdermal implants, are a reasonable option; however, progesterone-only pills alone are not optimal because of unacceptably low efficacy rates. Contraceptive pills containing estrogen are contraindicated due to an increased risk of thromboembolism.95 Double contraception (with barrier methods) is recommended, especially in patients on bosentan, due to the high teratogenicity and an interaction, which reduces the contraceptive efficacy.
Intrauterine devices are effective, but may convey a certain risk of infection, with the greatest fear being endocarditis. Possible vasovagal events during insertion can be poorly tolerated by the patient, hence should be performed in hospital. Sterilization is an option, but a laparoscopic procedure carries a high intraoperative risk: beyond the risks relating to the anesthetic and reduced cardiac output from positive pressure ventilation, insufflation of the abdomen with carbon dioxide, arrhythmias, bleeding, and air embolism are also of concern.
Conclusion
In the modern era of more sophisticated medical and surgical management of CHD, the number of patients who develop ES is dropping rapidly; thanks to timely repair of the defect. However, a small proportion of CHD patients in developed countries and many more in developing countries still develop ES, a severe and debilitating condition, which affects the individual’s capacity for exertion, reproduction, and quality of life. ES patients have benefitted greatly from recent advances in the management of this condition, especially the introduction of PAH therapies. Further work needs to be done to improve risk stratification and identify treatment targets that can guide treatment initiation and escalation in ES patients.
Disclosure
Michael A Gatzoulis and the Royal Brompton Hospital Adult Congenital Heart Centre and Centre for Pulmonary Hypertension have received support from the British Heart Foundation. Michael A Gatzoulis and Konstantinos Dimopoulos have acted as consultants for Actelion, Pfizer, Bayer and GSK and received unrestricted educational grants from Actelion, Pfizer and GSK. Rafael Alonso-Gonzalez has acted as a consultant for Lilly Spain and Pfizer Spain. Stephen J Wort has received unrestricted educational and research grants from Bayer UK, Pfizer UK, Actelion UK and GSK UK as well as the Pulmonary Hypertension Association (UK). Heba Nashat has received an educational grant from Actelion UK. Carl Harries has received educational grants from Actelion UK, Bayer UK and MSD UK. Laura C Price has received educational funding from Actelion UK and a research grant from GSK UK. The authors report no other conflicts of interest in this work.
References
Van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58(21):2241–2247. | ||
Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 Guidelines for the management of adults with congenital heart disease: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines (writing committee to develop guidelines on the management of adults with congenital heart disease). Circulation. 2008;118(23):e714–e833. | ||
Engelfriet PM, Duffels MG, Moller T, et al. Pulmonary arterial hypertension in adults born with a heart septal defect: the Euro Heart Survey on adult congenital heart disease. Heart. 2007;93(6):682–687. | ||
Wood P. The Eisenmenger syndrome or pulmonary hypertension with reversed central shunt. I. Br Med J. 1958;2(5098):701–709. | ||
Diller GP, Dimopoulos K, Broberg CS, et al. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: a combined retrospective and case-control study. Eur Heart J. 2006;27(14):1737–1742. | ||
Young D, Mark H. Fate of the patient with the Eisenmenger syndrome. Am J Cardiol. 1971;28(6):658–669. | ||
Duffels MG, Engelfriet PM, Berger RM, et al. Pulmonary arterial hypertension in congenital heart disease: an epidemiologic perspective from a Dutch registry. Int J Cardiol. 2007;120(2):198–204. | ||
Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34–D41. | ||
Saha A, Balakrishnan KG, Jaiswal PK, Venkitachalam CG, Tharakan J, Titus T, Kutty R. Prognosis for patients with Eisenmenger syndrome of various aetiology. Int J Cardiol. 1994;45(3):199–207. | ||
Moceri P, Kempny A, Liodakis E, et al. Physiological differences between various types of Eisenmenger syndrome and relation to outcome. Int J Cardiol. 2015;179:455–460. | ||
Alonso-Gonzalez R, Lopez-Guarch CJ, Subirana-Domenech MT, et al. Pulmonary hypertension and congenital heart disease: an insight from the REHAP National Registry. Int J Cardiol. 2015;184:717–723. | ||
Dimopoulos K, Okonko DO, Diller GP, et al. Abnormal ventilatory response to exercise in adults with congenital heart disease relates to cyanosis and predicts survival. Circulation. 2006;113(24):2796–2802. | ||
Dimopoulos K, Diller GP, Koltsida E, et al. Prevalence, predictors, and prognostic value of renal dysfunction in adults with congenital heart disease. Circulation. 2008;117(18):2320–2328. | ||
Dimopoulos K, Diller GP, Petraco R, et al. Hyponatraemia: A strong predictor of mortality in adults with congenital heart disease. Eur Heart J. 2010;31(5):595–601. | ||
Dimopoulos K, Wort SJ, Gatzoulis MA. Pulmonary hypertension related to congenital heart disease: a call for action. Eur Heart J. 2014;35(11):691–700. | ||
Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC Guidelines for the management of infective endocarditis: the Task Force for the management of infective endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J. 2015;36(44):3075–3128. | ||
Peacock A, Ross K. Pulmonary hypertension: a contraindication to the use of {beta}-adrenoceptor blocking agents. Thorax. 2010;65(5):454–455. | ||
Bogaard HJ, Natarajan R, Mizuno S, et al. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010;182(5):652–660. | ||
Grinnan D, Bogaard HJ, Grizzard J, et al. Treatment of group I pulmonary arterial hypertension with carvedilol is safe. Am J Respir Crit Care Med. 2014;189(12):1562–1564. | ||
Diller GP, Kempny A, Inuzuka R, et al. Survival prospects of treatment naive patients with Eisenmenger: a systematic review of the literature and report of own experience. Heart. 2014;100(17):1366–1372. | ||
Bolger AP, Sharma R, Li W, et al. Neurohormonal activation and the chronic heart failure syndrome in adults with congenital heart disease. Circulation. 2002;106(1):92–99. | ||
Trojnarska O. Heart failure in the adult patient with congenital heart disease. Cardiol J. 2007;14(2):127–136. | ||
Hopkins WE, Kelly DP. Angiotensin-converting enzyme inhibitors in adults with cyanotic congenital heart disease. Am J Cardiol. 1996;77(5):439–440. | ||
Diller GP, Korten MA, Bauer UM, et al; Competence Network for Congenital Heart Defects Investigators. Current therapy and outcome of Eisenmenger syndrome: data of the German National Register for congenital heart defects. Eur Heart J. 2016;37(18):1449–1455. | ||
Oya H, Nagaya N, Satoh T, et al. Haemodynamic correlates and prognostic significance of serum uric acid in adult patients with Eisenmenger syndrome. Heart. 2000;84(1):53–58. | ||
Rosove MH, Perloff JK, Hocking WG, Child JS, Canobbio MM, Skorton DJ. Chronic hypoxaemia and decompensated erythrocytosis in cyanotic congenital heart disease. Lancet. 1986;2(8502):313–315. | ||
Oechslin E. Hematological management of the cyanotic adult with congenital heart disease. Int J Cardiol. 2004;97 Suppl 1:109–115. | ||
Silversides CK, Salehian O, Oechslin E, et al. Canadian Cardiovascular Society 2009 Consensus Conference on the management of adults with congenital heart disease: complex congenital cardiac lesions. Can J Cardiol. 2010;26(3):e98–e117. | ||
Broberg CS, Bax BE, Okonko DO, et al. Blood viscosity and its relationship to iron deficiency, symptoms, and exercise capacity in adults with cyanotic congenital heart disease. J Am Coll Cardiol. 2006;48(2):356–365. | ||
Ammash N, Warnes CA. Cerebrovascular events in adult patients with cyanotic congenital heart disease. J Am Coll Cardiol. 1996;28(3):768–772. | ||
Tay EL, Peset A, Papaphylactou M, et al. Replacement therapy for iron deficiency improves exercise capacity and quality of life in patients with cyanotic congenital heart disease and/or the Eisenmenger syndrome. Int J Cardiol. 2011;151(3):307–312. | ||
Oechslin E, Mebus S, Schulze-Neick I, et al. The adult patient with Eisenmenger syndrome: a medical update after dana point part III: specific management and surgical aspects. Curr Cardiol Rev. 2010;6(4):363–372. | ||
Rosenthal A, Nathan DG, Marty AT, Button LN, Miettinen OS, Nadas AS. Acute hemodynamic effects of red cell volume reduction in polycythemia of cyanotic congenital heart disease. Circulation. 1970;42(2):297–308. | ||
Van De Bruaene A, Delcroix M, Pasquet A, et al. Iron deficiency is associated with adverse outcome in Eisenmenger patients. Eur Heart J. 2011;32(22):2790–2799. | ||
Sandoval J, Aguirre JS, Pulido T, et al. Nocturnal oxygen therapy in patients with the Eisenmenger syndrome. Am J Respir Crit Care Med. 2001;164(9):1682–1687. | ||
Harinck E, Hutter PA, Hoorntje TM, et al. Air travel and adults with cyanotic congenital heart disease. Circulation. 1996;93(2):272–276. | ||
Silversides CK, Granton JT, Konen E, Hart MA, Webb GD, Therrien J. Pulmonary thrombosis in adults with Eisenmenger syndrome. J Am Coll Cardiol. 2003;42(11):1982–1987. | ||
Pollack CV, Jr. Evidence supporting idarucizumab for the reversal of dabigatran. Am J Med. 2016;129(11S):S73–S79. | ||
Dimopoulos K, Diller GP, Piepoli MF, Gatzoulis MA. Exercise intolerance in adults with congenital heart disease. Cardiol Clin. 2006;24(4):641–660, vii. | ||
Hambrecht R, Fiehn E, Weigl C, et al. Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation. 1998;98(24):2709–2715. | ||
Goret L, Reboul C, Tanguy S, Dauzat M, Obert P. Training does not affect the alteration in pulmonary artery vasoreactivity in pulmonary hypertensive rats. Eur J Pharmacol. 2005;527(1–3):121–128. | ||
Sandberg C, Thilen U, Wadell K, Johansson B. Adults with complex congenital heart disease have impaired skeletal muscle function and reduced confidence in performing exercise training. Eur J Prev Cardiol. 2015;22(12):1523–1530. | ||
Kaemmerer H, Mebus S, Schulze-Neick I, et al. The adult patient with eisenmenger syndrome: a medical update after dana point part I: epidemiology, clinical aspects and diagnostic options. Curr Cardiol Rev. 2010;6(4):343–355. | ||
McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–2294. | ||
Ammash NM, Connolly HM, Abel MD, Warnes CA. Noncardiac surgery in Eisenmenger syndrome. J Am Coll Cardiol. 1999;33(1):222–227. | ||
Bennett JM, Ehrenfeld JM, Markham L, Eagle SS. Anesthetic management and outcomes for patients with pulmonary hypertension and intracardiac shunts and Eisenmenger syndrome: a review of institutional experience. J Clin Anesth. 2014;26(4):286–293. | ||
Waddell TK, Bennett L, Kennedy R, Todd TR, Keshavjee SH. Heart-lung or lung transplantation for Eisenmenger syndrome. J Heart Lung Transplant. 2002;21(7):731–737. | ||
Stoica SC, McNeil KD, Perreas K, et al. Heart-lung transplantation for Eisenmenger syndrome: early and long-term results. Ann Thorac Surg. 2001;72(6):1887–1891. | ||
Christie JD, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult lung and heart-lung transplant report – 2010. J Heart Lung Transplant. 2010;29(10):1104–1118. | ||
Mebus S, Schulze-Neick I, Oechslin E, et al. The adult patient with Eisenmenger syndrome: a medical update after dana point part II: medical treatment – study results. Curr Cardiol Rev. 2010;6(4):356–362. | ||
D’Alto M, Diller GP. Pulmonary hypertension in adults with congenital heart disease and Eisenmenger syndrome: current advanced management strategies. Heart. 2014;100(17):1322–1328. | ||
Galie N, Beghetti M, Gatzoulis MA, et al; Bosentan Randomized Trial of Endothelin Antagonist Therapy-5 (BREATHE-5) Investigators. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation. 2006;114(1):48–54. | ||
Mukhopadhyay S, Nathani S, Yusuf J, Shrimal D, Tyagi S. Clinical efficacy of phosphodiesterase-5 inhibitor tadalafil in Eisenmenger syndrome – a randomized, placebo-controlled, double-blind crossover study. Congenit Heart Dis. 2011;6(5):424–431. | ||
Iversen K, Jensen AS, Jensen TV, Vejlstrup NG, Sondergaard L. Combination therapy with bosentan and sildenafil in Eisenmenger syndrome: a randomized, placebo-controlled, double-blinded trial. Eur Heart J. 2010;31(9):1124–1131. | ||
Dimopoulos K, Inuzuka R, Goletto S, Giannakoulas G, Swan L, Wort SJ, Gatzoulis MA. Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation. 2010;121(1):20–25. | ||
Cacoub P, Dorent R, Maistre G, et al. Endothelin-1 in primary pulmonary hypertension and the Eisenmenger syndrome. Am J Cardiol. 1993;71(5):448–450. | ||
Gatzoulis MA, Beghetti M, Galie N, et al. Longer-term bosentan therapy improves functional capacity in Eisenmenger syndrome: results of the BREATHE-5 open-label extension study. Int J Cardiol. 2008;127(1):27–32. | ||
Zuckerman WA, Leaderer D, Rowan CA, Mituniewicz JD, Rosenzweig EB. Ambrisentan for pulmonary arterial hypertension due to congenital heart disease. Am J Cardiol. 2011;107(9):1381–1385. | ||
Pulido T, Adzerikho I, Channick RN, et al; SERAPHIN Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. | ||
ClinicalTrials.gov. Clinical Study to Evaluate the Effects of Macitentan on Exercise Capacity in Subjects With Eisenmenger Syndrome; 2016 [cited Nov 21, 2016]. Available from: https://clinicaltrials.gov/ct2/show/NCT01743001. Accessed November 25, 2016. | ||
Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351(14):1425–1436. | ||
Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by:Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplanation (ISHLT). Eur Heart J. 2016;37(1):67–119. | ||
Singh TP, Rohit M, Grover A, Malhotra S, Vijayvergiya R. A randomized, placebo-controlled, double-blind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertension. Am Heart J. 2006;151(4):e851–e855. | ||
Chau EM, Fan KY, Chow WH. Effects of chronic sildenafil in patients with Eisenmenger syndrome versus idiopathic pulmonary arterial hypertension. Int J Cardiol. 2007;120(3):301–305. | ||
Garg N, Sharma MK, Sinha N. Role of oral sildenafil in severe pulmonary arterial hypertension: clinical efficacy and dose response relationship. Int J Cardiol. 2007;120(3):306–313. | ||
Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–340. | ||
Galie N, Muller K, Scalise AV, Grunig E. PATENT PLUS: a blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. Eur Respir J. 2015;45(5):1314–1322. | ||
Rosenkranz S, Ghofrani HA, Beghetti M, et al. Riociguat for pulmonary arterial hypertension associated with congenital heart disease. Heart. 2015;101(22):1792–1799. | ||
Jones DA, Benjamin CW, Linseman DA. Activation of thromboxane and prostacyclin receptors elicits opposing effects on vascular smooth muscle cell growth and mitogen-activated protein kinase signaling cascades. Mol Pharmacol. 1995;48(5):890–896. | ||
Galie N, Humbert M, Vachiery JL, et al; Arterial Pulmonary Hypertension and Beraprost European (ALPHABET) Study Group. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39(9):1496–1502. | ||
Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(5):624–633. | ||
Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142(6):1383–1390. | ||
Galie N, Seeger W, Naeije R, Simonneau G, Rubin LJ. Comparative analysis of clinical trials and evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):81S–88S. | ||
Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation. 1999;99(14):1858–1865. | ||
Fernandes SM, Newburger JW, Lang P, Pearson DD, Feinstein JA, Gauvreau K, Landzberg MJ. Usefulness of epoprostenol therapy in the severely ill adolescent/adult with Eisenmenger physiology. Am J Cardiol. 2003;91(5):632–635. | ||
Badesch DB, McLaughlin VV, Delcroix M, Vizza CD, Olschewski H, Sitbon O, Barst RJ. Prostanoid therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):56S–61S. | ||
Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322–329. | ||
Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800–804. | ||
Tapson VF, Gomberg-Maitland M, McLaughlin VV, Benza RL, Widlitz AC, Krichman A, Barst RJ. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: a prospective, multicenter, open-label, 12-week trial. Chest. 2006;129(3):683–688. | ||
Lang I, Gomez-Sanchez M, Kneussl M, Naeije R, Escribano P, Skoro-Sajer N, Vachiery JL. Efficacy of long-term subcutaneous treprostinil sodium therapy in pulmonary hypertension. Chest. 2006;129(6):1636–1643. | ||
Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40(4):874–880. | ||
Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522–2533. | ||
Galie N, Rubin L, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008;371(9630):2093–2100. | ||
Sitbon O, Sattler C, Bertoletti L, et al. Initial dual oral combination therapy in pulmonary arterial hypertension. Eur Respir J. 2016;47(6):1727–1736. | ||
D’Alto M, Romeo E, Argiento P, et al. Bosentan-sildenafil association in patients with congenital heart disease-related pulmonary arterial hypertension and Eisenmenger physiology. Int J Cardiol. 2012;155(3):378–382. | ||
Freeman SB, Bean LH, Allen EG, et al. Ethnicity, sex, and the incidence of congenital heart defects: a report from the National Down Syndrome Project. Genet Med. 2008;10(3):173–180. | ||
Cua CL, Rogers LK, Chicoine LG, Augustine M, Jin Y, Nash PL, Nelin LD. Down syndrome patients with pulmonary hypertension have elevated plasma levels of asymmetric dimethylarginine. Eur J Pediatr. 2011;170(7):859–863. | ||
Cappelli-Bigazzi M, Santoro G, Battaglia C, et al. Endothelial cell function in patients with Down’s syndrome. Am J Cardiol. 2004;94(3):392–395. | ||
Duffels MG, Vis JC, van Loon RL, et al. Down patients with Eisenmenger syndrome: is bosentan treatment an option? Int J Cardiol. 2009;134(3):378–383. | ||
Baraona F, Gurvitz M, Landzberg MJ, Opotowsky AR. Hospitalizations and mortality in the United States for adults with Down syndrome and congenital heart disease. Am J Cardiol. 2013;111(7):1046–1051. | ||
Dimopoulos K, Kempny A. Patients with Down syndrome and congenital heart disease: survival is improving, but challenges remain. Heart. 2016;102(19):1515–1517. | ||
Gleicher N, Midwall J, Hochberger D, Jaffin H. Eisenmenger’s syndrome and pregnancy. Obstet Gynecol Surv. 1979;34(10):721–741. | ||
Weiss BM, Zemp L, Seifert B, Hess OM. Outcome of pulmonary vascular disease in pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol. 1998;31(7):1650–1657. | ||
Bedard E, Dimopoulos K, Gatzoulis MA. Has there been any progress made on pregnancy outcomes among women with pulmonary arterial hypertension? Eur Heart J. 2009;30(3):256–265. | ||
Thorne S, MacGregor A, Nelson-Piercy C. Risks of contraception and pregnancy in heart disease. Heart. 2006;92(10):1520–1525. | ||
Vitale N, De Feo M, De Santo LS, Pollice A, Tedesco N, Cotrufo M. Dose-dependent fetal complications of warfarin in pregnant women with mechanical heart valves. J Am Coll Cardiol. 1999;33(6):1637–1641. | ||
Soma-Pillay P, Nene Z, Mathivha TM, Macdonald AP. The effect of warfarin dosage on maternal and fetal outcomes in pregnant women with prosthetic heart valves. Obstet Med. 2011;4(1):24–27. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.