Back to Journals » Clinical Interventions in Aging » Volume 9
Effects of hypercapnia on peripheral vascular reactivity in elderly patients with acute exacerbation of chronic obstructive pulmonary disease
Authors de Matthaeis A, Greco A ![]() , Dagostino MP, Paroni G, Fontana A
, Dagostino MP, Paroni G, Fontana A ![]() , Vinciguerra M, Mazzoccoli G, Seripa D, Vendemiale G
, Vinciguerra M, Mazzoccoli G, Seripa D, Vendemiale G
Received 14 November 2013
Accepted for publication 24 January 2014
Published 29 May 2014 Volume 2014:9 Pages 871—878
DOI https://doi.org/10.2147/CIA.S57548
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Angela de Matthaeis,1 Antonio Greco,2,* Mariangela Pia Dagostino,2 Giulia Paroni,2 Andrea Fontana,3 Manlio Vinciguerra,1,4,5 Gianluigi Mazzoccoli,1,* Davide Seripa,2 Gianluigi Vendemiale6
1Division of Internal Medicine and Chronobiology Unit, 2Geriatrics Unit and Gerontology, Geriatrics Research Laboratory, Department of Medical Sciences, 3Unit of Biostatistics, IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Foggia, 4Euro-Mediterranean Institute of Sciences and Technology, Palermo, Italy; 5University College London, Institute for Liver and Digestive Health, Division of Medicine, Royal Free Campus, London, UK; 6Geriatrics Unit, University of Foggia, Foggia, Italy
*These authors contributed equally to this work
Abstract: Blood acid-base imbalance has important effects on vascular reactivity, which can be related to nitric oxide (NO) concentration and increased during hypercapnia. Release of NO seems to be linked to H+ and CO2 concentration and to exacerbation of chronic obstructive pulmonary disease (COPD), a common medical condition in the elderly. Flow-mediated dilation (FMD), a valuable cardiovascular risk indicator, allows assessment of endothelial-dependent vasodilation, which is to a certain extent mediated by NO. We investigated the effects of hypercapnia and acid-base imbalance on endothelial-dependent vasodilation by measurement of FMD in 96 elderly patients with acute exacerbation of COPD. Patients underwent complete arterial blood gas analysis and FMD measurement before (phase 1) and after (phase 2) standard therapy for acute exacerbation of COPD and recovery. Significant differences between phase 1 and phase 2 were observed in the mean values of pH (7.38±0.03 versus 7.40±0.02, P<0.001), pO2 (59.6±4.9 mmHg versus 59.7±3.6 mmHg, P<0.001), pCO2 (59.3±8.63 mmHg versus 46.7±5.82 mmHg, P<0.001), FMD (10.0%±2.8% versus 8.28%±2.01%, P<0.001) and blood flow rate (1.5±0.3 m/s versus 1.5±0.3 m/s, P=0.001). FMD values were positively correlated with pCO2 values (r=0.294, P=0.004) at baseline. A significant correlation was also found between relative changes in FMD and pCO2 levels, passing from phase 1 to phase 2 (r=0.23, P=0.023). Patients with higher baseline endothelium-dependent vasodilation as evaluated by FMD showed greater modification with regard to pCO2 changes (2.6±1.39 versus 1.59±1.4, P=0.012). In conclusion, endothelium-dependent vasodilation as evaluated by FMD was elevated during hypercapnia, and varied significantly according to pCO2 changes in patients with higher baseline levels, suggesting that vascular reactivity in acute COPD exacerbations in the elderly depends on integrity of the vascular endothelium.
Keywords: hypercapnia, elderly, chronic obstructive pulmonary disease, vascular reactivity, flow-mediated dilation
Introduction
Hypercapnia and the accompanying acidosis are considered to induce strong vasodilating effects on the coronary and cerebral circulation in mammals, and previous data have implicated nitric oxide (NO) in the vasodilatory response of vascular endothelium to hypercapnia.1–3 Although the role of NO in the response of the cerebral circulation to hypercapnia has been extensively investigated,4 few studies have addressed the role of NO in the peripheral circulation under conditions of elevated blood carbon dioxide (CO2) tension in the elderly.5 This issue is important considering that the vasculature of the brain is more sensitive to changes in arterial pCO2 when compared with the peripheral circulation and in particular with regard to the vasculature of the forearm.6
Endothelium-derived NO has been shown to play an important contributory role in the coronary vasodilatory response to a variety of physiological stimuli, including hypoxia, hypercapnia, and reactive hyperemia subsequent to transient occlusion.7 However, the effect of acid-base homeostasis and NO on vascular reactivity has been mainly studied in animal models.8
Flow-mediated dilation (FMD) measurement at the level of the brachial artery is a broadly available method used to test endothelium-dependent vasodilation, as well as to measure indirectly endothelial NO production induced by increased local blood flow (shear stress).9–11 Flow-induced changes in arterial diameter by forearm compression evoke the endothelial response to reduced blood flow, which is characterized by initial narrowing of the blood vessel reflecting the vascular/endothelial response to resting levels of shear stress. Conversely, in response to a sudden increase in blood flow, arterial dilatation ensues. FMD renders the ability of the endothelium to adjust the biosynthesis and release of mediators to produce vasodilation,12 at least in part mediated by NO, as substantiated by the results of a recent meta-analysis.13 FMD is a valuable and reliable indicator of cardiovascular risk and is an important and useful tool in cardiovascular risk stratification in the elderly.14
It is well known that low pH values induce vascular smooth muscle relaxation and that blood vessels have high sensitivity to changes in pCO2.15–17 Moreover, pCO2 is considered to be an important regulator of the cerebral circulation in mammals.18,19
An essential and intrinsic property of the cerebral vasculature with the aim to maintain metabolic constancy is autoregulation of local blood flow. This physiological process, also known as chemoregulation, is frequently challenged by the CO2 concentration.20,21 The CO2-NO axis is a cardinal pathway in the chemoregulation of cerebral blood flow in humans,19 and impairment of endothelial function associated with decrease of CO2 cerebrovascular reactivity has been demonstrated in patients affected by cardiovascular disease.20
Endothelial dysfunction, defined as a functional and reversible alteration of endothelial cells, leading to a shift of the actions of the endothelium toward reduced vasodilation, proinflammatory state and proliferative and prothrombotic properties, results from functional changes characterized by vasospasm, coagulation abnormalities, and increased vascular proliferation, and has been reported to be the initial step in atherosclerosis.22 Peripheral endothelial function can be noninvasively evaluated by measurement of FMD at the level of the brachial artery by using high-resolution ultrasound methods.23,24
As stated above, an important mediator of FMD is endothelium-derived NO, and in the present study we investigated the role of NO in the peripheral circulation during hypercapnia in patients with exacerbation of chronic obstructive pulmonary disease (COPD) by means of FMD measurement. We assessed the relationship between endothelium-dependent vasodilation and pCO2 changes in the clinical setting of acute exacerbation of COPD after treatment with standard therapy (β2-agonists, anticholinergic drugs, and corticosteroids, as well as oxygen and antibiotic therapy in cases of respiratory failure) as indicated by the international guidelines.25 We demonstrated a significant increase in pH values and a decrease in pCO2 and FMD values, and higher baseline endothelium-dependent vasodilation corresponding to higher pCO2 reactivity. Our findings suggest that endothelium-dependent vasodilation, as evaluated by FMD measurement, increases during hypercapnia and depends on the integrity of the vascular endothelium in elderly patients with acute COPD exacerbation.
Materials and methods
Standard protocols and patient consent
This was a cross-sectional cohort study fulfilling the requirements of the Declaration of Helsinki and the guidelines for Good Clinical Practice, the Strengthening the Reporting of Observational Studies in Epidemiology, and the National Institute for Health and Clinical Excellence guidelines. Approval to conduct experiments using human subjects was obtained from the local ethics committee on human experimentation. Written informed consent for this research was obtained from each patient or from relatives/legal guardians in the case of critically disabled patients.
Patient recruitment
From January to December 2010, 220 consecutive elderly patients with a clinical diagnosis of acute exacerbation of COPD were evaluated for enrollment in the study. The recruitment process was conducted on consecutive subjects hospitalized in the Department of Medical Sciences, specifically in the Division of Internal Medicine and in the Geriatrics Unit. The diagnosis of COPD was made according to Global Initiative for Chronic Obstructive Lung Disease criteria for the diagnosis, classification, and severity of COPD.25
Clinical evaluation and inclusion/exclusion criteria
All patients had smoking habits (>20 pack-years) and irreversible airflow limitation (forced expiratory volume in one second <80% of predicted), and were evaluated by spirometry, arterial blood gas analysis (ABGA), and chest radiography. Exclusion criteria included other coexisting pulmonary diseases, body mass index <20 or >30 kg/m2, metabolic syndrome, diabetes mellitus, arterial hypertension (blood pressure values >130/80 mmHg), arterial hypotension (blood pressure values <90/60 mmHg), blood pressure values in the left and right arm differing by more than 15 mmHg, a history of Raynaud’s phenomenon, a congenital abnormality of the arm or hand, known cardiovascular comorbidities, neoplastic diseases or a radical mastectomy on either side, acute and chronic renal failure, carotid stenosis, normal pCO2 levels, and no recovery after standard therapy for COPD exacerbations (inhaled β2-agonists, anticholinergic drugs, and corticosteroids, and oxygen and antibiotic therapy when respiratory failure was present).
Only those patients affected by acute exacerbation of COPD, who reported pCO2 >45 mmHg as evaluated by ABGA before treatment (phase 1) and who responded to 5 days of standard therapy with at least a 5% reduction of their respective baseline pCO2 after treatment (phase 2) were considered for the study. Patients taking medications that could influence endothelial function (eg, nitrates) were excluded.
The inclusion criteria for the initial study sample were used for the screening stage of 220 subjects with acute exacerbation of COPD; 96 patients satisfied the inclusion criteria and were enrolled. All patients enrolled in the study underwent ABGA and FMD measurement in both phase 1 and phase 2 (Figure 1). In order to avoid the possible influence of hypoxemia on endothelial function, we titrated oxygen administration when necessary to maintain arterial oxyhemoglobin saturation at ≥94%.
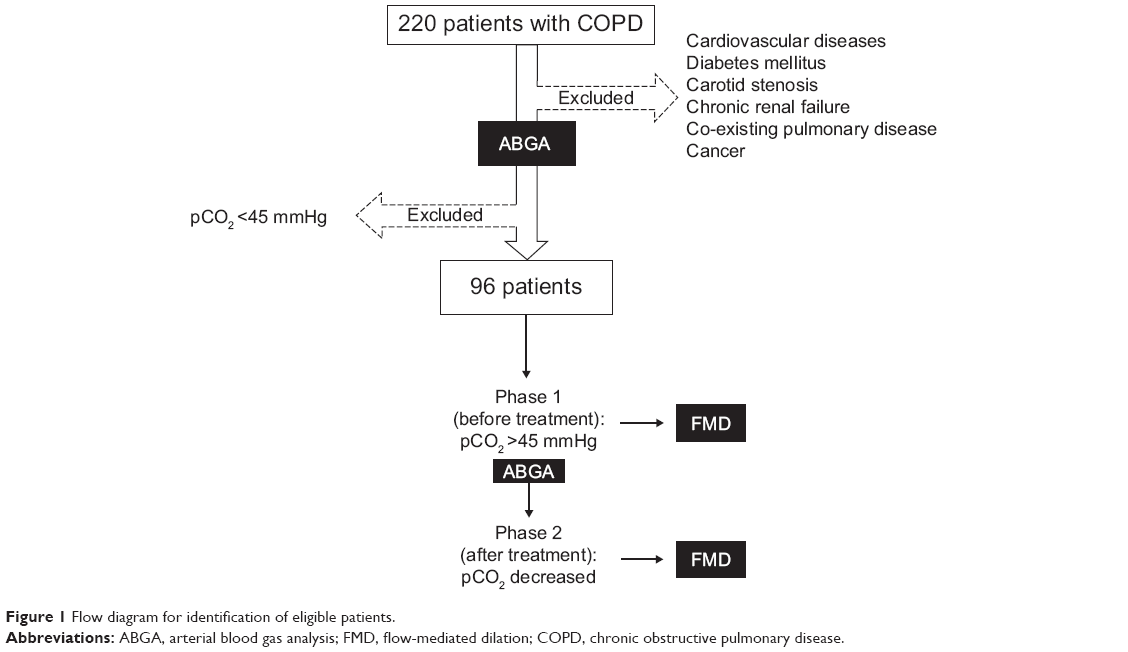 | Figure 1 Flow diagram for identification of eligible patients. |
Assessment of vascular function
Noninvasive evaluation by ultrasonography of the endothelial-dependent vasodilatation was done by measurement of FMD in the brachial artery of the nondominant arm according to international standards.10,26–28 The diameter of the brachial artery was measured by an expert sonographer (AdM) on ultrasound images obtained in B-mode with a high resolution 5–10 mHz multifrequency linear probe connected to a Technos MPX (Esaote, Genova, Italy). All the subjects were fasting and had not been smoking, drinking alcohol or coffee, or taking antioxidant agents for at least 16 hours (last meal at 6 pm on the day before evaluation). During phase 1 and phase 2 of the study, the subjects were examined between 10 am and 11 am and after 15 minutes of rest in the supine position in a quiet, temperature-controlled room (20°C–22°C). We explored the brachial artery at the level of the antecubital fossa on a longitudinal plane, optimizing the depth and the acoustic window pre-sets and keeping them in the same position during the study, using a mechanical probe-holder arm. An automated sphygmomanometer was used to monitor blood pressure and pulse throughout the examination. A standard blood pressure cuff was positioned around the nondominant arm 5 cm below the antecubital fossa, and the artery was imaged 5–9 cm above the antecubital fossa. After basal measurement of vessel diameter and speed of flow in the brachial artery, the sphygmomanometer cuff was inflated around the forearm at a pressure of 50 mmHg higher than the basal systolic pressure and deflated after 5 minutes. Measurements of vessel diameter and speed of flow were taken 30 seconds before deflation and every 30 seconds for 2 minutes after deflation. The mean arterial diameter was measured at the end of diastole, which was determined by simultaneous monitoring of the electrocardiogram (the diastolic diameter of the brachial artery per beat was synchronized with the electrocardiographic R-wave). The measurement was registered at the media-adventitia interface of the anterior (near) and posterior (far) vessel walls. For each scan, the diameters were measured in four heart cycles and the average was calculated by dividing the difference between the maximum diameter and the basal one by the maximum diameter. Intraobserver variability for repeated measurements of the resting arterial diameter was 0.023%±0.004%.
Statistical analysis
Baseline patient characteristics were reported as means ± standard deviations and as frequencies and percentages for continuous and categorical variables, respectively. Assumption of a normal distribution was checked by a Q-Q plot and the Shapiro–Wilks and Kolmogorov–Smirnov tests. Baseline comparisons between men and women were performed using the chi-square test and two-sample t-test for categorical and continuous variables, respectively. Pearson’s correlation between FMD and pCO2 values was estimated at baseline (phase 1), as well as at phase 2.
Comparisons between phase 1 and phase 2 for mean pH, pCO2, pO2, FMD, and blood flow rate were performed using the paired t-test or Wilcoxon signed rank test as appropriate. Moreover, to account for the potential role of confounding factors on the effect of standard therapy a multivariate hierarchical linear model for repeated measurements was performed using FMD values as continuous outcome, and baseline patient characteristics, clinical variables, and pharmacological treatment as covariates. This method borrows strength from correlated measures within each subject over time. Specifically, two separate models were built. The first model (the so-called “fully adjusted” model) included the time variable (ie, an indicator variable which takes the value 1 if referred to phase 2 FMD measurements) and the following baseline covariates: age, sex, smoking habit, pCO2, hyperemic blood flow rate, inhalation therapy (bronchodilators), and treatment with antibiotics, oxygen, calcium channel blockers, aspirin, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and β-blockers. The second one (so-called “selected model”) included a parsimonious set of the mentioned covariates, other than the time variable, using the classical stepwise selection method (retention threshold, P<0.10). A two-sided P-value <0.05 was considered to be statistically significant. Statistical analyses were performed using SAS Release version 9.1 (SAS Institute, Cary, NC, USA).
Results
Sample size and power analysis
A sample size of 96 patients had 90% power to detect a mean of paired differences for FMD values (phase 2 – phase 1) of -0.635, with an estimated standard deviation of differences of 1.44 and with a significance level (type I error) of 0.05 using a two-sided paired t-test.
The study sample included 96 patients, ie, 74 men (77%) and 22 women (33%) with a mean age of 72.4±5.4 (range 65–96) years. Both baseline patient characteristics and the list of medications being taken are reported in Table 1. No statistically significant sex differences were observed.
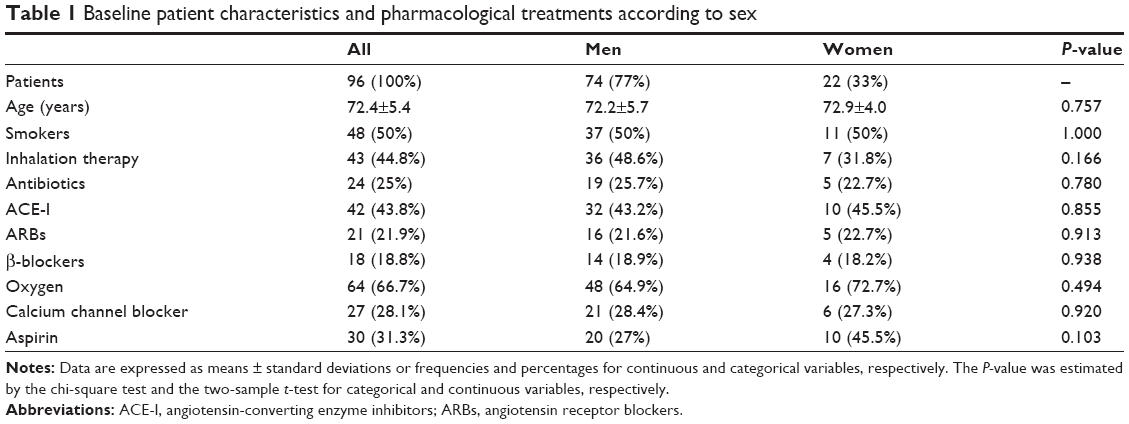 | Table 1 Baseline patient characteristics and pharmacological treatments according to sex |
Baseline clinical variables for the patients in phase 1 and phase 2 are reported in Table 2. Statistically significant differences between phase 1 and phase 2 were observed for all the considered variables (Table 2, all P≤0.001). FMD values were positively correlated with pCO2 values (r=0.294, P=0.004) at baseline. A scatter plot of FMD values against pCO2 values (both measured at phase 1) is reported on Figure 2. Besides, a significant positive correlation was found between relative changes in FMD and pCO2 levels, passing from phase 1 to phase 2 (r=0.23, P=0.023). Patients with higher baseline endothelium-dependent vasodilation as evaluated by FMD measurement (>10%)23 showed greater modification with regard to pCO2 changes (2.6±1.39 versus 1.59±1.4, P=0.012).
 | Table 2 Comparison between clinical variables before (phase 1) and after (phase 2) standard therapy |
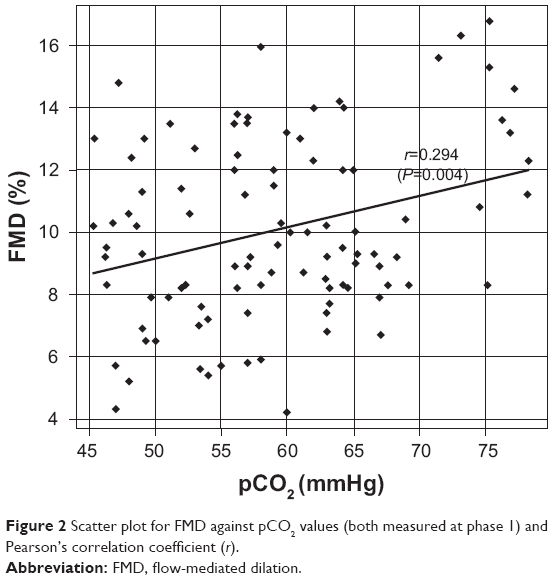 | Figure 2 Scatter plot for FMD against pCO2 values (both measured at phase 1) and Pearson’s correlation coefficient (r). |
FMD and pCO2 were also measured in 76 independent elderly subjects (mean age 69.8±7.3 years, 32.9% males). No significant difference in mean FMD values was found between COPD patients at baseline and controls (10.0%±2.8% versus 9.6%±2.7%, respectively, P=0.344), whereas a marked difference was found between mean pCO2 values (59.3±8.6 mmHg versus 39.6±1.8 mmHg, respectively, P<0.001). No correlation was found between FMD and pCO2 values in the control group (r=0.03, P=0.812).
Regression coefficients with the standard error from a multivariate hierarchical linear model for repeated measurements are reported in Table 3. As shown, after standard therapy, mean FMD values decreased significantly by 1.8%±0.15% (P<0.001) controlled for baseline characteristics of patients, clinical variables, and pharmacological treatments. Only hyperemic blood flow rate (P=0.001) and aspirin therapy (P=0.010) were associated with basal FMD values.
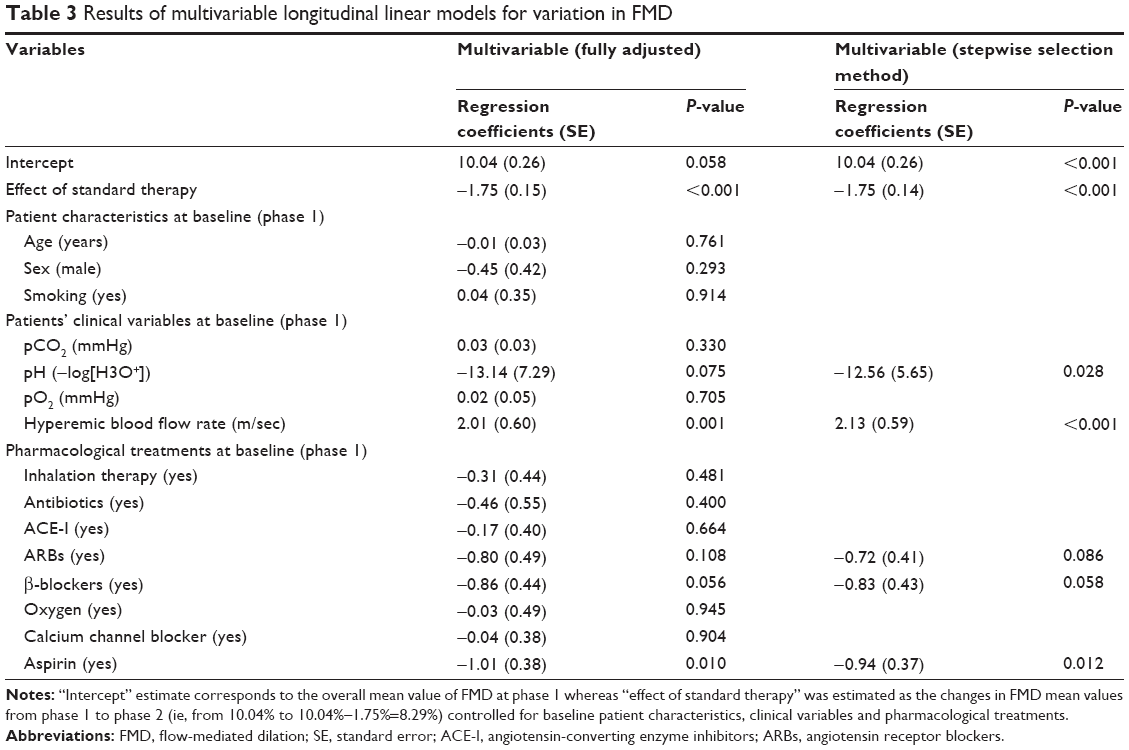 | Table 3 Results of multivariable longitudinal linear models for variation in FMD |
Discussion
The present study found an improvement in endothelial-dependent vasodilation during hypercapnia in patients with acute exacerbation of COPD. Higher vasodilation during hypercapnia is a common finding in the acute phase of COPD, and is reversed after recovery. Our data highlight an enhanced endothelium-dependent vasodilation that might be explained by increased NO production during hypercapnia. Our results are consistent with reports that alterations in NO production or activity may have a major role in endothelium-dependent vasodilation,29 and are corroborated by recent data implicating NO in the vasodilatory responses to hypercapnia.8,30
We observed that higher mean FMD basal values were associated with greater changes between phase 1 and phase 2 FMD mean values, indicating higher endothelium-dependent vasodilation. This evidence suggests that endothelium-dependent vasodilation in response to altered metabolic determinants, such as hypercapnia, requires normal endothelial function, and vascular reactivity in acute COPD exacerbations depends on vascular endothelium integrity in the elderly.
The role of NO in hypercapnia-induced vasodilation is not confined to the central nervous system. Several studies have shown increased coronary blood flow during hypercapnia, and NO has been proposed as a mediator of in situ hypercapnia-induced coronary vasodilation in the dog heart.31,32 Coronary vasodilation during selective intracoronary hypercapnia was attenuated after in situ inhibition of NO synthase with N6-nitro-L-arginine methyl ester or N6-monomethyl-L-arginine in the canine heart. These data suggest that NO plays an important role in coronary vasodilation during hypercapnia. Moreover, studies in isolated aortic strips suggest that vasorelaxation in response to CO2 could be mediated, at least in part, by NO.33 Indeed, most of the studies performed in animal models report that NO synthase activity may be increased, with consequent stimulation of NO production in conditions of low pH.34–36 Overall, although the precise contribution of endothelial NO synthase during hypercapnia is still unclear, previous animal and human studies indicate that endothelial NO synthase is an important, but not exclusive, mediator of hypercapnia-induced vascular modifications.
Our results are in agreement with the involvement of NO in hypercapnia and post-hypercapnia-related vasodilation, and suggest that assessment of endothelium-dependent vasodilation by measurement of FMD is a useful method for evaluating the NO vasodilator system in the setting of aging and COPD in vivo.10,11,13 It has also been reported that endothelial-derived NO may be involved in CO2-dependent systemic blood flow regulation, which in turn depends on the integrity of the vascular endothelium.37,38 Our findings are in agreement with this. Further, we found a significant difference in post-hyperemic flow rate in the hypercapnia and normocapnia phases (Table 2). This suggests that good hypercapnia-induced endothelial-dependent vasodilation is related to efficient endothelial function, as well as increased NO production, the latter likely due to a direct effect of CO2 on the vascular endothelium rather than related to the shear stress induced by an increased peripheral flow rate. The significant positive correlation found between increased endothelium-dependent vasodilation as assessed by measurement of FMD and elevated pCO2 values suggests a functional role for NO in peripheral vascular reactivity to CO2.
Our data are in contrast with a previous study reporting impaired endothelial function in patients affected by stable COPD and a negative correlation between the severity of illness and endothelium-dependent vasodilation.39 Importantly, we have to bear in mind that study considered normocapnic patients with COPD, whereas we focused on FMD changes related to hypercapnia during the acute phase of COPD exacerbation. Notably, acute stress may induce known (eg, hypercapnia) and unknown factors contributing to increased production of endothelial NO. In fact, previous studies have reported significantly increased expression of inducible NO synthase in the neutrophils and macrophages of COPD patients.40 Other studies have also shown higher NO levels in breath condensate and induced sputum in patients with mild or severe COPD, as compared with control subjects.41–43 Persistent stress may result in derangement and dampening of endothelial function, and consequently lower endothelial vasoreactivity. In fact, patients with COPD have an increased risk of cardiovascular disease, probably due to their endothelial dysfunction. COPD is linked to systemic inflammation, which has a propensity to augment in due course and become boosted during acute exacerbations. In turn, systemic inflammation promotes evolution of cardiovascular disease, and an association between systemic inflammation and increased risk of cardiovascular morbidity and mortality has been documented in patients with COPD.39 The link between COPD and cardiovascular disease may be represented by abnormalities in systemic vascular function, and FMD may depict vasomotor function, as well as predict cardiovascular morbidity and mortality during acute exacerbations.
The main limitations of our study are its cross-sectional design and the use of only one method to assess endothelial function, in particular the lack of evaluation of nitroglycerin-mediated dilation, which indicates endothelial-independent dilation. We must also stress that the pO2 levels recorded in our COPD patients were very low, although severe hypoxemia is common in the context of the baseline hypercapnic states frequently seen in COPD patients and is related to the altered response of respiratory drive, especially in the clinical setting of acute functional worsening.44
In conclusion, our data demonstrate that hypercapnia during acute exacerbations of COPD can influence endothelium-dependent vasodilation as assessed by measurement of FMD, and a larger decrease in FMD could point to greater reactivity to pCO2 changes. The modifications in vascular reactivity induced by variations of CO2 tension in peripheral blood could depend on the integrity of the vascular endothelium, could be mediated by the NO synthase/NO pathway, and could be related to cardiovascular risk in this clinical setting, particularly in elderly patients.
Author contributions
AdM performed the FMD measurements, conceived the original idea, and developed the protocol. AG conceived the original idea, developed the protocol, completed the analysis, and wrote the manuscript. MPD helped with data collection, participated in protocol development, and approved the manuscript. GM and MV helped with data collection, completed the analysis, and wrote the manuscript. GP, DS, and GV provided overall supervision and revised the manuscript for intellectual content. AF performed statistical analysis. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Acknowledgment
This work was fully supported by the “5×1,000” voluntary contributions and by the Italian Ministry of Health through IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Foggia, Italy (Research Program 2009–2011 for complex diseases).
Disclosure
The authors report no conflicts of interest in this work.
References
Ely SW, Sawyer DC, Scott JB. Local vasoactivity of oxygen and carbon dioxide in the right coronary circulation of the dog and pig. J Physiol. 1982;332:427–439. | ||
Iadecola C, Zhang F. Nitric oxide-dependent and -independent components of cerebrovasodilation elicited by hypercapnia. Am J Physiol. 1994;266(2 Pt 2):546–552. | ||
Puscas I, Coltau M, Domuta G, Baican M, Puscas C, Pasca R. Carbonic anhydrase I inhibition by nitric oxide: implications for mediation of the hypercapnia-induced vasodilator response. Clin Exp Pharmacol Physiol. 2000;27(1–2):95–99. | ||
Fabricius M, Rubin I, Bundgaard M, Lauritzen M. NOS activity in brain and endothelium: relation to hypercapnic rise of cerebral blood flow in rats. Am J Physiol. 1996;271(5):2035–2044. | ||
Estevez AY, Phillis JW. Hypercapnia-induced increases in cerebral blood flow: roles of adenosine, nitric oxide and cortical arousal. Brain Res. 1997;758(1–2):1–8. | ||
Vantanajal JS, Ashmead JC, Anderson TJ, Hepple RT, Poulin MJ. Differential sensitivities of cerebral and brachial blood flow to hypercapnia in humans. J Appl Physiol (1985). 2007;102(1):87–93. | ||
José Montoya J, Fernández N, Monge L, Diéguez G, Villalón AL. Nitric oxide-mediated relaxation to lactate of coronary circulation in the isolated perfused rat heart. J Cardiovasc Pharmacol. 2011;58(4):392–398. | ||
Celotto AC, Capellini VK, Baldo CF, Dalio MB, Rodrigues AJ, Evora PRB. Effects of acid-base imbalance on vascular reactivity. Braz J Med Biol Res. 2008;41(6):439–445. | ||
Joannides R, Haefeli WE, Linder L, et al. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91(5):1314–1319. | ||
Corretti MC, Anderson TJ, Benjamin EJ, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39(2):257–265. | ||
Green D. Flow-mediated dilation does reflect nitric oxide-mediated endothelial function. J Appl Physiol. 2005;99(3):1233–1234. | ||
Inaba H, Takeshita K, Uchida Y, et al. Recovery of flow-mediated vasodilatation after repetitive measurements is involved in early vascular impairment: comparison with indices of vascular tone. PLoS One. 2014;9(1):e83977. | ||
Green DJ, Dawson EA, Groenewoud HM, Jones H, Thijssen DH. Is flow-mediated dilation nitric oxide mediated? A meta-analysis. Hypertension. 2014;63(2):376–382. | ||
Nagai K, Shibata S, Akishita M, et al. Efficacy of combined use of three non-invasive atherosclerosis tests to predict vascular events in the elderly; carotid intima-media thickness, flow-mediated dilation of brachial artery and pulse wave velocity. Atherosclerosis. 2013;231(2):365–370. | ||
Wolff H, Lennox WG. The cerebral circulation: XII. The effects on pial vessel of variations in the O2 and CO2 content of the blood. Arch Neurol Psychiatry. 1930;23:1097–1120. | ||
Masamoto K, Tanishita K. Oxygen transport in brain tissue. J Biomech Eng. 2009;131(7):074002. | ||
van Bel F, van de Bor M, Baan J, Ruys JH. The influence of abnormal blood gases on cerebral blood flow velocity in the preterm newborn. Neuropediatrics. 1988;19(1):27–32. | ||
Wahl M, Schilling L. Regulation of cerebral blood flow – a brief review. Acta Neurochir Suppl (Wien). 1993;59:3–10. | ||
Willie CK, Tzeng YC, Fisher JA, Ainslie PN. Integrative regulation of human brain blood flow. J Physiol. 2014;592(Pt 5):841–859. | ||
Strandgaard S, Paulson OB. Regulation of cerebral blood flow in health and disease. J Cardiovasc Pharmacol. 1992;19 Suppl 6:S89–S93. | ||
Lavi S, Gaitini D, Milloul V, Jacob G. Impaired cerebral CO2 vasoreactivity: association with endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2006;291(4):H1856–H1861. | ||
Lahera V, Goicoechea M, de Vinuesa SG, et al. Endothelial dysfunction, oxidative stress and inflammation in atherosclerosis: beneficial effects of statins. Curr Med Chem. 2007;14(2):243–248. | ||
Bots ML, Westerink J, Rabelink TJ, de Koning EJ. Assessment of flow-mediated vasodilatation (FMD) of the brachial artery: effects of technical aspects of the FMD measurement on the FMD response. Eur Heart J. 2005;26(4):363–368. | ||
Peretz A, Leotta DF, Sullivan JH, et al. Flow mediated dilation of the brachial artery: an investigation of methods requiring further standardization. BMC Cardiovasc Disord. 2007;7:11–19. | ||
Global Initiative for Chronic Obstructive Lung Disease. The Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease 2013. Available from: http://www.goldcopd.org/uploads/users/files/GOLD_Report_2013_Feb20.pdf. Accessed April 4, 2014. | ||
Clarkson P, Celermajer DS, Powe AJ, Donald AE, Henry RM, Deanfield JE. Endothelium-dependent dilatation is impaired in young healthy subjects with a family history of premature coronary disease. Circulation. 1997;96(10):3378–3383. | ||
Black MA, Cable NT, Thijssen DH, Green DJ. Importance of measuring the time course of flow-mediated dilatation in humans. Hypertension. 2008;51(2):203–210. | ||
Thijssen DH, Black MA, Pyke KE, et al. Assessment of flow-mediated dilation in humans: a methodological and physiological guideline. Am J Physiol Heart Circ Physiol. 2011;300(1):H2–H12. | ||
Meredith IT, Currie KE, Anderson TJ, Roddy MA, Ganz P, Creager MA. Postischemic vasodilation in human forearm is dependent on endothelium-derived nitric oxide. Am J Physiol. 1996;270(4 Pt 2):H1435–H1440. | ||
Gaskell WH. On the tonicity of the heart and blood vessels. J Physiol. 1880;3(1):48–92.16. | ||
Clancy RL, Gonzalez NC. Effect of respiratory and metabolic acidosis on coronary vascular resistance (38528). Proc Soc Exp Biol Med. 1975;148(1):307–311. | ||
Gurevicius J, Salem MR, Metwally AA, Silver JM, Crystal GJ. Contribution of nitric oxide to coronary vasodilation during hypercapnic acidosis. Am J Physiol. 1995;268(1 Pt 2):H39–H47. | ||
Fukuda S, Matsumoto M, Nishimura N, et al. Endothelial modulation of norepinephrine-induced constriction of rat aorta at normal and high CO2 tensions. Am J Physiol. 1990;258(4 Pt 2):H1049–H1054. | ||
Pellegrino DA, Koenig HM, Albreth RF. Nitric oxide synthesis and regional cerebral blood flow responses to hypercapnia and hypoxia in the rat. J Cereb Blood Flow Metab. 1993;13(1):80–87. | ||
Heinzel B, John M, Klatt P, Böhme E, Mayer B. Ca2+/calmodulin dependent formation of hydrogen by brain nitric oxide synthase. Biochem J. 1992;281(Pt 3):627–630. | ||
Niwa K, Lindauer V, Villringer A, Dirnagl U. Blockade of nitric oxide synthesis in rats strongly attenuates the CBF response to extracellular acidosis. J Cereb Blood Flow Metab. 1993;13(3):535–539. | ||
Kelm M, Schrader J. Control of coronary vascular tone by nitric oxide. Circ Res. 1990;66(6):1561–1575. | ||
Krainik A, Hund-Georgiadis M, Zysset S, Von Cramon DY. Regional impairment of cerebrovascular reactivity and BOLD signal in adults after stroke. Stroke. 2005;36(6):1146–1152. | ||
Eickhoff P, Valipour A, Kiss D, et al. Determinants of systemic vascular function in patients with stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178(12):1211–1218. | ||
van der Vliet A, Eiserich JP, Shigenaga MK, Cross CE. Reactive nitrogen species and tyrosine nitration in the respiratory tract: epiphenomena or a pathobiologic mechanism of disease? Am J Respir Crit Care Med. 1999;160(1):1–9. | ||
Corradi M, Montuschi P, Donnelly LE, Pesci A, Kharitonov SA, Barnes PJ. Increased nitrosothiols in exhaled breath condensate in inflammatory airway diseases. Am J Respir Crit Care Med. 2001;163(4):854–858. | ||
Kanazawa H, Shoji S, Yoshikawa T, Hirata K, Yoshikawa J. Increased production of endogenous nitric oxide in patients with bronchial asthma and chronic obstructive pulmonary disease. Clin Exp Allergy. 1998;28(10):1244–1250. | ||
Kanazawa H, Yoshikawa J. Elevated oxidative stress and reciprocal reduction of vascular endothelial growth factor levels with severity of COPD. Chest. 2005;128(5):3191–3197. | ||
Erbland ML, Ebert RV, Snow SL. Interaction of hypoxia and hypercapnia on respiratory drive in patients with COPD. Chest. 1990;97(6):1289–1294. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.