Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Effective mucoadhesive liposomal delivery system for risedronate: preparation and in vitro/in vivo characterization
Received 22 January 2014
Accepted for publication 23 March 2014
Published 12 May 2014 Volume 2014:9(1) Pages 2299—2306
DOI https://doi.org/10.2147/IJN.S61181
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Il-Woo Jung, Hyo-Kyung Han
College of Pharmacy, Dongguk University-Seoul, Ilsan-Donggu, Goyang, Republic of Korea
Abstract: In this work, we aimed to develop chitosan-coated mucoadhesive liposomes containing risedronate to improve intestinal drug absorption. Liposomes containing risedronate were prepared with 1,2-distearoryl-sn-glycero-3-phosphocholine and distearoryl-sn-glycero-3-[phospho-rac-(1-glycerol)] using the freeze-drying method, with subsequent coating of the anionic surfaces of the liposomes with chitosan. The in vitro characteristics of the chitosan-coated liposomes were investigated, including their stability, mucoadhesiveness, and Caco-2 cell permeability. This formulation was stable in simulated gastric and intestinal fluids, with the percentage of drug remaining in the liposomes being more than 90% after 24 hours of incubation. Chitosan-coated liposomes also showed strong mucoadhesive properties, implying potential electrostatic interaction with the mucous layer in the gastrointestinal tract. Compared with the untreated drug, chitosan-coated liposomes significantly enhanced the cellular uptake of risedronate, resulting in an approximately 2.1–2.6-fold increase in Caco-2 cells. Further, the chitosan-coated liposomes increased the oral exposure of risedronate by three-fold in rats. Taken together, the results of this study suggest that chitosan-coated liposomes containing risedronate should be effective for improving the bioavailability of risedronate.
Keywords: cellular uptake, bioavailability, mucoadhesiveness, liposome, chitosan
Introduction
Osteoporosis is a progressive bone disease accompanied by porous bones or diminution of bone mass.1 It is a major public health problem for post-menopausal women, many elderly men, and malnourished children, leading to increased mortality and health costs as well as poor quality of life.2 Therefore, prevention and treatment of osteoporosis are very important. Bisphosphonates inhibiting osteoclast resorption are widely used in the treatment and prevention of the disease.3,4 Bisphosphonates are eliminated mainly by urinary excretion of the unchanged drug, and they have very low oral bioavailability.5,6 They also have side effects, including diarrhea, nausea, and vomiting. A particularly serious side effect of oral administration is esophageal inflammation or ulcers.7 Therefore, poor intestinal absorption and mucosal irritation remain major problems with orally administered bisphosphonates.
Risedronate is a potent pyridinyl bisphosphonate used for the treatment of Paget’s disease, osteoporosis, and other bone disorders.8 Risedronate can be taken orally, but to avoid potential development of esophageal ulcers, it should be taken while standing upright and followed by a glass of water.8,9 Moreover, risedronate is poorly absorbed when taken with food. Thus, it is recommended that food and drink other than water should not be taken for 2 hours before and 30 minutes after a dose of risedronate.10 This dosing regimen is very inconvenient for patients, resulting in poor compliance and suboptimal clinical effectiveness of risedronate. In addition, risedronate belongs to Biopharmaceutics Classification System class III (high solubility/low permeability)11 and its oral bioavailability is very low (<1%).8 Therefore, there is a strong need to develop more efficient drug delivery systems for risedronate with less propensity to cause esophageal irritation and better bioavailability.
Recently, Sahana et al12 reported that risedronate-hydroxyapatite nanoparticles administered via intravenous injection were effective for bone-targeted drug delivery. Cationic liposomes and polylactide-co-glycolic-acid microspheres have also been proposed as novel formulations, and are suitable for systemic delivery of risedronate via the pulmonary route.13,14 For oral administration, an adduct of risedronate with titanium dioxide particles and single-unit and multi-unit floating systems of risedronate with Gelucire® 43/01 (Gattefossé, Cedex, France) have been proposed as controlled-release systems to improve the bioavailability of risedronate.15,16 Among the various formulation approaches, lipid-based formulations are considered promising delivery systems because of their low melt viscosity, biocompatibility, and biodegradability.17,18 Liposomal drug delivery systems also have other favorable properties, including their similarity to the biological membrane, the capacity to encapsulate structurally diverse drugs, relatively low toxicity, and an ability to improve undesirable drug properties (eg, low solubility and poor membrane permeability).19,20 Therefore, liposomes have gained a great deal of attention as potential drug delivery systems to maximize drug efficacy.21–23 However, the instability of liposomal vesicles in the gastrointestinal tract often limits the application of liposomal drug delivery systems as oral delivery carriers.24,25 Because stability can be affected by the surface characteristics of liposomes,26,27 coating of the liposomal surface with chitosan may improve the stability of the liposome in the gastrointestinal tract. Furthermore, being a biocompatible, mucoadhesive, and nontoxic polymer,28–31 chitosan can have additional benefits, such as a prolonged residence time in the gastrointestinal tract and enhanced membrane permeability.32–34
Therefore, in this study, chitosan-coated liposomes were prepared to improve the bioavailability of orally administered risedronate. Anionic liposomes of risedronate were prepared with 1,2-distearoryl-sn-glycero-3-phosphocholine (DSPC)/1,2-distearoryl-sn-glycero-3-[phospho-rac-(1-glycerol)] sodium salt (DSPG) and then coated with chitosan. Various in vitro and in vivo characteristics of the obtained liposome were then evaluated.
Materials and methods
Materials
Risedronate sodium was supplied by Hanlim Pharm Co. Ltd. (Seoul, Republic of Korea). DSPC and DSPG were purchased from Avanti (Alabaster, AL, USA). Cholesterol, chitosan, mucin, L-ascorbic acid 2-phosphate sesquimagnesium salt, etidronic acid, and tetrabutylammonium bromide were purchased from Sigma-Aldrich (St Louis, MO, USA). Caco-2 cells were purchased from the Korean Cell Line Bank (Seoul, Republic of Korea). Fetal bovine serum and cell culture media were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Solvents of high-performance liquid chromatography (HPLC) grade were purchased from Merck (Darmstadt, Germany).
Cells
Caco-2 cells were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% nonessential amino acids. The cells were grown in an atmosphere of 5% CO2 and 90% relative humidity at 37°C.
Preparation of liposomes
Risedronate liposomes were prepared using the freeze-drying method. DSPC, DSPG, and cholesterol (6:1:2, molar ratio) were dissolved in tert-butyl alcohol and lyophilized overnight. This lyophilized lipid was rehydrated with risedronate solution and stirred for 2 hours at 55°C or above. After sonication for 80 seconds, the samples were centrifuged at 100,000× g for one hour. The separated liposomes were then resuspended using 0.9% NaCl solution. The last two steps were repeated twice to remove free risedronate.
To prepare the chitosan-coated liposomes, a 0.1% chitosan solution in acetate buffer (pH 4.4) was prepared and mixed with a four-fold volume of liposome suspension. After stirring for 20 minutes, the chitosan-coated liposomes were harvested from the reaction mixture by centrifugation at 25,000× g.
Size, charge, and entrapment efficiency
The liposomal formulation was diluted ten-fold in water, and the size and zeta potential were determined using a zetasizer (Nano-ZS90, Malvern Instruments, Malvern, UK). To estimate the entrapment efficiency (EE%), free risedronate was separated from the liposome counterpart by centrifugation as described above and the supernatant was analyzed by HPLC. EE% was calculated using the following equation:
|
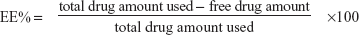
Stability test
The stability of the liposomes in simulated gastric fluid and in simulated intestinal fluid was determined at 37°C. The liposomes were incubated in simulated gastric fluid and in simulated intestinal fluid, and samples were collected at predetermined time points. After centrifugation at 25,000× g for 30 minutes, the supernatant was collected and the amount of drug released was measured by HPLC assay. The effect of different temperatures (4°C, 25°C, and 37°C) on the stability of the liposomes was also examined over a 24-hour period. To determine the stability of the formulation during storage, the amount of drug leakage and the size distribution and zeta potential of the liposomes were evaluated over a 3-month period.
Mucin adsorption study
The adsorption of mucin (extracted from porcine stomach) on the surface of the liposomes was measured as described by Filipovic-Grcic et al35 to evaluate the mucoadhesive properties of the liposomes. First, 1 mL of mucin (1 mg/mL) was stirred with one mL of chitosan-coated liposomes (1 mg/mL and 2 mg/mL) for 2 hours at 37°C. The same procedure was performed for the noncoated liposomes. The suspension was then centrifuged at 25,000× g for one hour. The amount of free mucin was determined in order to assess the amount of mucin adsorbed onto the liposomes according to the difference between total and free mucin.
Uptake study in Caco-2 cells
Cells were seeded into a six-well plate at a density of 5×105 cells per well. Seven days after seeding, the cells were incubated in 25 μg/mL and 100 μg/mL of drug solution (containing untreated drug [no liposome], noncoated liposome, or chitosan-coated liposome). After incubation for 2 hours, the drug solution was removed and the cells were washed three times with ice-cold phosphate-buffered saline. After lysis, the cells were harvested and sonicated for 5 minutes. Acetonitrile was added to the cell lysate, which was then vortexed vigorously and centrifuged for 10 minutes at 5,000× g. The supernatant was collected and the drug concentration in each sample was determined by HPLC assay. The amount of protein in each sample was determined by bicinchoninic acid protein assay.
Pharmacokinetics study
Male Sprague Dawley rats weighing about 250 g were purchased from Nara Biotech Co (Seoul, Republic of Korea). The rats had free access to a normal standard chow diet (Cargill Agri Purina, Kyeonggi-do, Republic of Korea) and tap water. All animal studies were carried out in accordance with the Society of Toxicology Guiding Principles in the Use of Animals in Toxicology. The animals were fasted for 24 hours prior to the experiment. On the day of the study, the rats were divided into three groups and orally administered each drug solution (untreated drug [no liposome], noncoated liposome, or chitosan-coated liposome) at a dose equivalent to 5 mg/kg of risedronate. After oral administration, urine was collected over 48 hours for specified time periods (hours 0–4, 4–12, 12–24, 24–36, and 36–48), and the urine samples obtained were stored at −80°C until analyzed by HPLC.
HPLC assay
In vitro samples
Drug concentration was measured using a HPLC assay as reported by Dissette et al.15 The HPLC system (Flexar®, PerkinElmer, MA, USA) consisted of an ultraviolet light detector, an automatic injector, and two solvent delivery pumps. Separation was performed on a column (Gemini 5 μm, C18, 150 mm ×4.6 mm; Phenomenex®, Torrance, CA, USA). The mobile phase consisted of buffer (5 mM of tetrabutylammonium bromide, 11 mM of sodium phosphate, 1 mM of etidronate, and 1.5 mM of EDTA-2 Na) to methanol (88:12, v/v), adjusted to pH 6.75. The flow rate was 1 mL per minute and the ultraviolet light detection wavelength was set at 262 nm.
Urine samples
Samples were prepared for the HPLC assay as described elsewhere,36 and stored in a deep freezer at −80°C. Before processing, the samples were thawed at room temperature, vortexed, and centrifuged at 3,000× g for 10 minutes. Next, 10 μL of 1.25 M calcium chloride and 100 μL of internal standard (L-ascorbic acid 2-phosphate sesquimagnesium salt) were added to 1 mL of each sample. Addition of 15 μL of 7.5 M NaOH induced formation of a white precipitate. After centrifugation for 30 minutes, 10 μL of 1 M HCl were added to the pellet. Next, 1 mL of water and 10 μL of 7.5 M NaOH were added to produce precipitation. After centrifugation for 30 minutes, the collected pellet was dissolved in 0.6 mL of 0.05 M EGTA (adjusted to pH 6.0) and 0.4 mL of water. The samples were sonicated for 5 minutes to dissolve the precipitate, and the drug concentration in each sample was determined by HPLC assay as mentioned above.
Statistical analysis
All the means are presented with their standard deviation. The statistical analysis was conducted using one-way analysis of variance followed by Dunnett’s correction. A P-value <0.05 was considered to be statistically significant.
Results and discussion
Preparation and in vitro stability of the liposomes
DSPC offers higher vesicle stability and a longer half-life than other phospholipids,23 so was used in preparation of the liposomes to enhance the stability of the liposomal membrane. DSPG was added to provide a negative charge on the liposomal surface, which prevented aggregation of the liposomes by electrostatic repulsion and allowed subsequent coating of the liposomal surface with a positively charged polymer. In addition, given that the glass transition temperature of DSPC and DSPG is about 55°C, these phospholipids should help to retain the rigidity of the liposomal membrane at body temperature, potentially enhancing the stability of the liposomes.37 This was supported well by the stability data obtained at the different temperatures. As shown in Figure 1, under all testing conditions, drug release from the noncoated and coated liposomes was negligible.
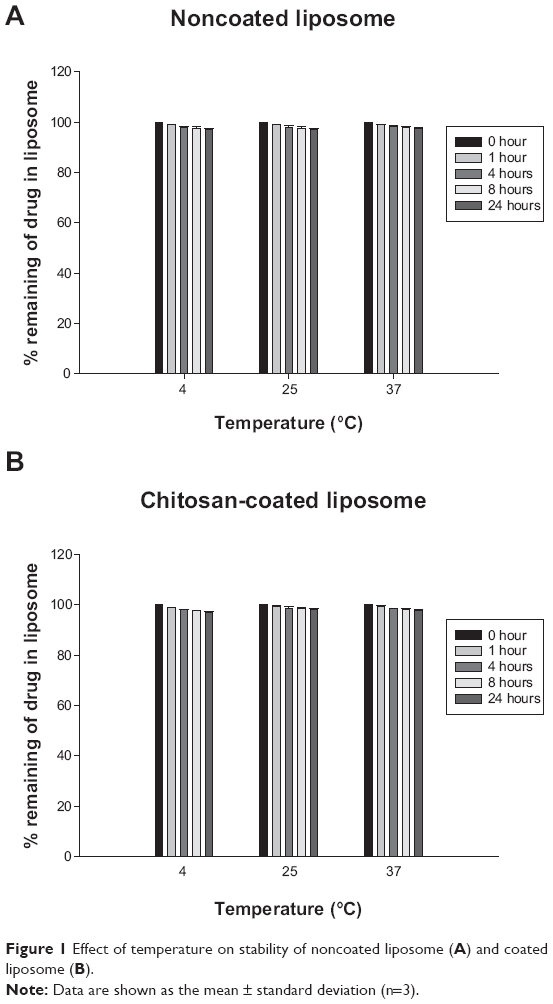 | Figure 1 Effect of temperature on stability of noncoated liposome (A) and coated liposome (B). |
The size and zeta potential values for the obtained liposomes are summarized in Table 1. Negatively charged liposomes were obtained as small vesicles approximately 200 nm in size with a narrow size distribution and an EE of about 32%. After coating the anionic surface of the liposomes with chitosan, the average size of the positively charged liposomes was 955 nm (Table 1).
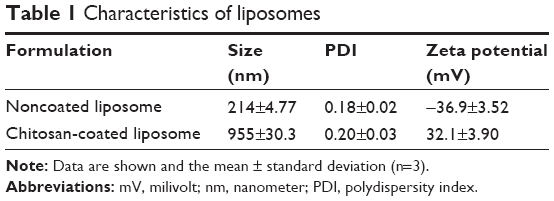 | Table 1 Characteristics of liposomes |
For oral application of a liposomal delivery system to enhance the bioavailability of poorly permeable drugs, the liposomes should be stable in the gastrointestinal tract and remain intact. Therefore, the stability of the obtained liposomes was evaluated in both simulated gastric fluid and simulated intestinal fluid. As shown in Figure 2, both the noncoated liposomes and the chitosan-coated liposomes were stable in simulated gastric fluid and simulated intestinal fluid, with drug leakage from the liposome being minimal during the 24-hour incubation period. Considering that orally administered liposomes are often unstable in the gastrointestinal tract and prone to significant drug leakage because of chemical or enzymatic degradation, the results from the simulated gastric fluid and simulated intestinal fluid studies suggest that both the noncoated liposomes and the chitosan-coated liposomes containing risedronate seem to have good gastrointestinal stability and remain intact in the gastrointestinal tract. This finding indicates that it may be advantageous to keep the drug inside the liposome and thereby away from direct contact with the esophagus to reduce the risk of risedronate-induced esophageal irritation.
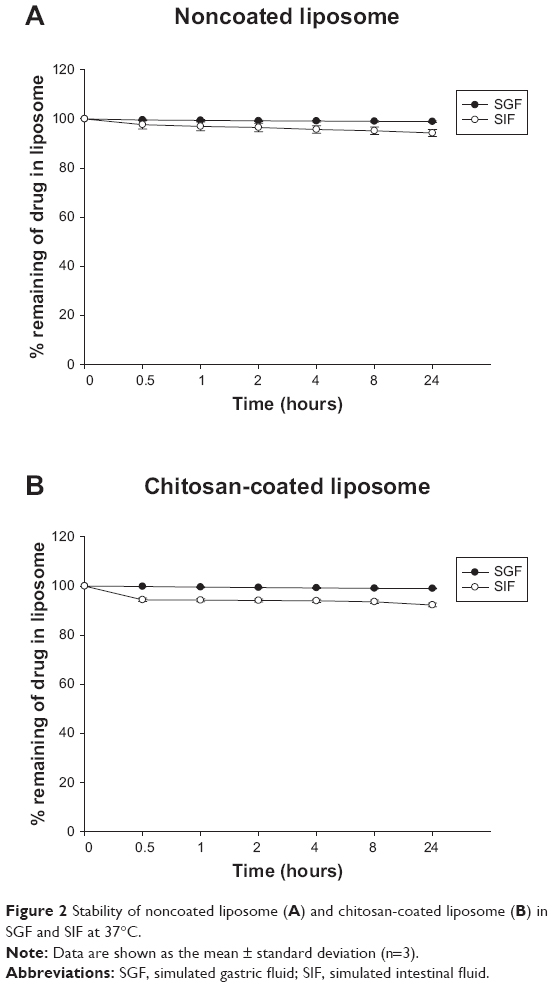 | Figure 2 Stability of noncoated liposome (A) and chitosan-coated liposome (B) in SGF and SIF at 37°C. |
Storage stability was evaluated for 3 months at 4°C and 25°C. Although the surface charge of liposomes can minimize their aggregation via electrostatic repulsion, both noncoated and chitosan-coated liposomes tend to increase in size, possibly due to swelling and aggregation.38 In the present study, drug leakage was minimal (<10%) after storage for 3 months at 4°C and 25°C, implying that the liposomes remained intact. In particular, drug leakage from the chitosan-coated liposomes was negligible, with the coated layer further protecting the liposomal membrane.
Mucin adsorption study
Positively charged chitosan-coated liposomes can interact with negatively charged mucin secreted from intestinal epithelial cells. Therefore, the mucoadhesiveness of the liposomes was estimated by measuring the amount of mucin adsorbed on the surface of the chitosan-coated liposomes. Percent adsorption was determined using the equation below:
|
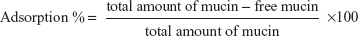
Compared with the noncoated liposomes, the amount of mucin adsorbed on the surface of the chitosan-coated liposomes was 5.4–10.7-fold greater, depending on the liposomal concentration (Figure 3). This result may be explained by the strong electrostatic interaction between the positively charged chitosan and the negatively charged mucin, although hydrogen bonding and hydrophobic interactions may also be involved.39 In general, the mucoadhesive behavior of a polymer is dependent on the chemical groups contributing to the interactions between the polymer and mucus. Strong mucoadhesiveness can be obtained by ionic interactions between positively charged groups and the acidic sialic moieties of mucin.39 It can also be obtained when various chemical groups forming the hydrogen bonds with the mucus gel (eg, hydroxyl, amine, sulfate, and carboxyl groups) are present.39 In that sense, the charge and the chemical groups on chitosan are favorable for mucoadhesion. The strong mucoadhesiveness of chitosan-coated liposomes may increase the residence time of risedronate in the gastrointestinal tract, resulting in prolonged drug absorption.
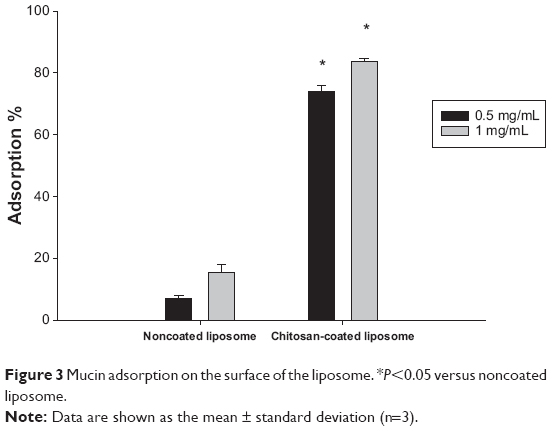 | Figure 3 Mucin adsorption on the surface of the liposome. *P<0.05 versus noncoated liposome. |
Uptake study in Caco-2 cells
Accumulation of risedronate from the different formulations (untreated drug [no liposome], noncoated liposome, chitosan-coated liposome) was measured in Caco-2 cells at drug concentrations of 25 μg/mL and 100 μg/mL. As shown in Figure 4, accumulation of risedronate in cells increased as the drug concentration increased. Compared with the untreated drug (no liposome), cellular uptake of risedronate in the liposomal formulations was significantly enhanced. This enhanced uptake may be due to greater membrane penetration of the hydrophilic drug inside the intact liposome. In addition, the chitosan-coating on the liposomal surface could further enhance the cellular accumulation of risedronate, resulting in an approximately 2.1–2.6-fold increase compared with the untreated drug (Figure 4). Improved cellular uptake via the chitosan-coated liposomes might be explained by several factors. First, chitosan may facilitate the paracellular transport of drugs.40 Chitosan can adhere to the mucosal surface, resulting in transient opening of tight junctions via translocation of the proteins ZO-1 and occludin from the plasma membrane to the cytoplasm and redistribution of F-actin.40 As a result, chitosan may increase the paracellular transport of drugs. Kudsiova and Lawrence41 demonstrated that chitosan-coated phospholipid vesicles reduce the transepithelial electrical resistance of Caco-2 cells and increase paracellular permeability. Second, a chitosan-based delivery system may promote endocytosis of encapsulated drugs by cells.42,43 Previous studies have reported that a chitosan-based formulation could be transported into Caco-2 cells via active endocytosis, with enhanced cellular uptake of the encapsulated molecules.42,43
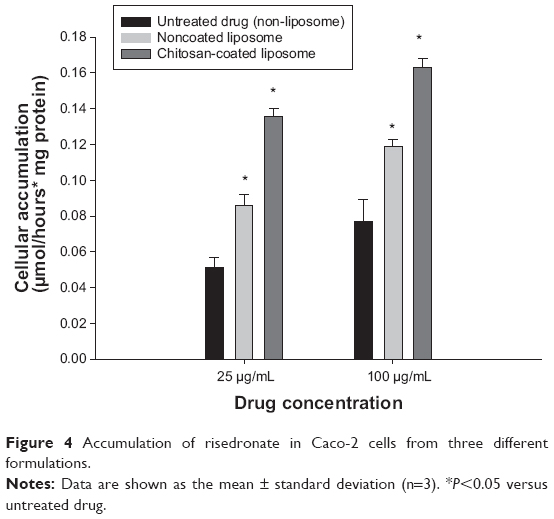 | Figure 4 Accumulation of risedronate in Caco-2 cells from three different formulations. |
Pharmacokinetics study
The major elimination pathway for risedronate is urinary excretion of the unchanged drug.44 Thus, the effect of the formulation on the bioavailability of risedronate was determined by the amount of drug excreted in urine after oral administration of different formulations of the drug to rats.
As summarized in Table 2 and Figure 5, compared with the conventional powder formulation (untreated drug), the liposomal formulation significantly (P<0.05) improved drug absorption in rats. In particular, chitosan-coated liposomes increased the amount of drug excreted in the urine by three-fold in rats compared with the untreated drug, whereas noncoated liposomes increased it by 1.8-fold. This result may be explained by several factors. First, the effect of chitosan on the opening of tight-junction and absorptive endocytosis may contribute, at least in part, to enhanced intestinal absorption of risedronate from the chitosan-coated liposomes.40,42 This is also parallel to the observations regarding uptake of Caco-2 cells. Second, as suggested by the mucin adsorption and vitro stability studies, the chitosan-coated layer might prolong drug residence time at the mucosal sites due to its mucoadhesiveness and also increase the gastrointestinal stability of the liposomal membrane, leading to enhanced membrane transport of the hydrophilic drug inside the intact liposome. Taken together, chitosan-coated liposomes appear to be promising in improving the bioavailability of orally administered risedronate in rats.
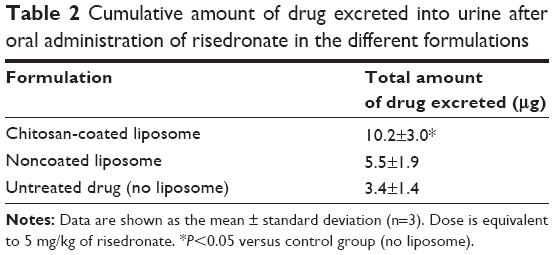 | Table 2 Cumulative amount of drug excreted into urine after oral administration of risedronate in the different formulations |
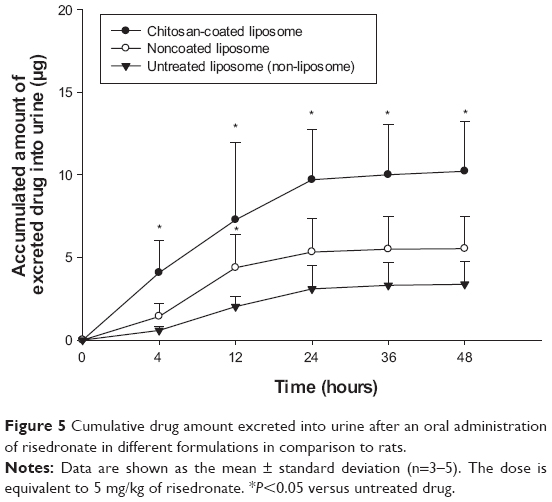 | Figure 5 Cumulative drug amount excreted into urine after an oral administration of risedronate in different formulations in comparison to rats. |
Oral risedronate can form an insoluble complex with calcium and magnesium contained in food. Thus, patients should take it on an empty stomach with water and remain upright for at least 30 minutes after taking the drug.10,45 Use of chitosan-coated liposomes may prevent the interaction with food because risedronate remains inside the liposomes. Further, chitosan-coated liposomes may reduce esophageal irritation by entrapping the drug inside the liposome, keeping it away from direct contact with the esophagus. From this point of view, chitosan-coated liposomes may also improve patient compliance with orally administered risedronate.
Conclusion
In this study, a mucoadhesive liposomal formulation of risedronate was developed with desirable drug delivery properties. The chitosan-coated liposome had good in vitro stability, strong mucoadhesiveness, and enhanced cellular uptake. It also significantly increased the oral absorption of risedronate in rats. Therefore, the chitosan-coated liposomal formulation appears to have the potential to improve the bioavailability of risedronate.
Acknowledgment
This work was supported by the Dongguk University Research Fund of 2010.
Disclosure
The authors report no conflicts of interest in this work.
References
Cummings SR, Melton LJ. Epidemiology and outcomes of osteoporotic fractures. Lancet. 2002;359(9319):1761–1767. | ||
Gullberg B, Johnell O, Kanis JA, Kanis JA. World-wide projections for hip fracture. Osteoporos Int. 1997;7(5):407–413. | ||
Rodan GA. Mechanisms of action of bisphosphonates. Annu Rev Pharmacol Toxicol. 1998;38:375–388. | ||
Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289(5484):1508–1514. | ||
Lin JH. Bisphosphonates: a review of their pharmacokinetic properties. Bone. 1996;18(2):75–85. | ||
Porras A, Holland S, Gertz B. Pharmacokinetics of alendronate. Clin Pharmacokinet. 1999;36(5):315–328. | ||
Sparidans R, Twiss I, Talbot S. Bisphosphonates in bone diseases. Pharm World Sci. 1998;20(5):206–213. | ||
Mitchell DY, St Peter JV, Eusebio RA, et al. Effect of renal function on risedronate pharmacokinetics after a single oral dose. Br J Clin Pharmacol. 2000;49(3):215–222. | ||
Perkins AC, Wilson CG, Frier M, et al. Esophageal transit of risedronate cellulose-coated tablet and gelatin capsule formulations. Int J Pharm. 1999;186(2):169–175. | ||
Lambrinoudaki I, Christodoulakos G, Botsis D. Bisphosphonates. Ann N Y Acad Sci. 2006;1092:397–402. | ||
Takagi T, Ramachandran C, Bermejo M, Yamashita S, Yu LX, Amidon GL. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol Pharm. 2006;3(6):631–643. | ||
Sahana H, Khajuria DK, Razdan R, et al. Improvement in bone properties by using risedronate adsorbed hydroxyapatite novel nanoparticle based formulation in a rat model of osteoporosis. J Biomed Nanotechnol. 2013;9(2):193–201. | ||
Nasr M, Taha I, Hathout RM. Suitability of liposomal carriers for systemic delivery of risedronate using the pulmonary route. Drug Deliv. 2013;20(8):311–318. | ||
Nasr M, Awad GA, Mansour S, Al Shamy A, Mortada ND. Hydrophilic versus hydrophobic porogens for engineering of poly(lactide-co-glycolide) microparticles containing risedronate sodium. Pharm Dev Technol. 2013;18(5):1078–1088. | ||
Dissette V, Bozzi P, Bignozzi CA, et al. Particulate adducts based on sodium risedronate and titanium dioxide for the bioavailability enhancement of oral administered bisphosphonates. Eur J Pharm Sci. 2010;41(2):328–336. | ||
Chauhan B, Shimpi S, Mahadik KR, Paradkar A. Preparation and evaluation of floating risedronate sodium-Gelucire 43/01 formulations. Drug Dev Ind Pharm. 2005;31(9):851–860. | ||
Fricker G, Kromp T, Wendel A, et al. Phospholipids and lipid-based formulations in oral drug delivery. Pharm Res. 2010;27(8):1469–1486. | ||
Porter CJ, Charman WN. In vitro assessment of oral lipid based formulations. Adv Drug Deliv Rev. 2001;50:S127–S147. | ||
Sharma A, Sharma US. Liposomes in drug delivery: progress and limitations. Int J Pharm. 1997;154(2):123–140. | ||
Lian T, Ho RJ. Trends and developments in liposome drug delivery systems. J Pharm Sci. 2001;90(6):667–680. | ||
Law SL, Huang KJ, Chiang CH. Acyclovir-containing liposomes for potential ocular delivery. Corneal penetration and absorption. J Control Release. 2000;63(1–2):135–140. | ||
Flaten GE, Dhanikula AB, Luthman K, Brandl M. Drug permeability across a phospholipid vesicle based barrier: a novel approach for studying passive diffusion. Eur J Pharm Sci. 2006;27(1):80–90. | ||
Epstein H, Gutman D, Cohen-Sela E, et al. Preparation of alendronate liposomes for enhanced stability and bioactivity: in vitro and in vivo characterization. AAPS J. 2008;10(4):505–515. | ||
Anderson M, Omri A. The effect of different lipid components on the in vitro stability and release kinetics of liposome formulations. Drug Deliv. 2004;11(1):33–39. | ||
Rogers JA, Anderson KE. The potential of liposomes in oral drug delivery. Crit Rev Ther Drug Carrier Syst. 1998;15(5):421–480. | ||
Juliano RL, Stamp D. The effect of particle size and charge on the clearance rates of liposomes and liposome encapsulated drugs. Biochem Biophys Res Commun. 1975;63(3):651–658. | ||
Senior JH. Fate and behavior of liposomes in vivo: a review of controlling factors. Crit Rev Ther Drug Carrier Syst. 1987;3(2):123–193. | ||
Chen XG, Lee CM, Park HJ. O/W emulsification for the self-aggregation and nanoparticle formation of linoleic acid-modified chitosan in the aqueous system. J Agric Food Chem. 2003;51(10):3135–3139. | ||
Qi L, Xu Z, Jiang X, Li Y, Wang M. Cytotoxic activities of chitosan nanoparticles and copper-loaded nanoparticles. Bioorg Med Chem Lett. 2005;15(5):1397–1399. | ||
Wu Y, Yang W, Wang C, Hu J, Fu S. Chitosan nanoparticles as a novel delivery system for ammonium glycyrrhizinate. Int J Pharm. 2005;295(1–2):235–245. | ||
Hyung Park J, Kwon S, Lee M, et al. Self-assembled nanoparticles based on glycol chitosan bearing hydrophobic moieties as carriers for doxorubicin: in vivo biodistribution and anti-tumor activity. Biomaterials. 2006;27(1):119–126. | ||
Aspden TJ, Mason JD, Jones NS, Lowe J, Skaugrud O, Illum L. Chitosan as a nasal delivery system: the effect of chitosan solutions on in vitro and in vivo mucociliary transport rates in human turbinates and volunteers. J Pharm Sci. 1997;86(4):509–513. | ||
Takeuchi H, Yamamoto H, Niwa T, Hino T, Kawashima Y. Enteral absorption of insulin in rats from mucoadhesive chitosan-coated liposomes. Pharm Res. 1996;13(6):896–901. | ||
Chornet E, Dumitriu S. Inclusion and release of proteins from polysaccharide-based polyion complexes. Adv Drug Deliv Rev. 1998;31(3):223–246. | ||
Filipovic-Grcic J, Skalko-Basnet N, Jalsenjak I. Mucoadhesive chitosan-coated liposomes: characteristics and stability. J Microencapsul. 2001;18(1):3–12. | ||
Perez-Ruiz T, Martinez-Lozano C, Garcia-Martinez MD. A sensitive post-column photochemical derivatization/fluorimetric detection system for HPLC determination of bisphosphonates. J Chromatogr A. 2009;1216(9):1312–1318. | ||
Mabrey S, Sturtevant JM. Investigation of phase transitions of lipids and lipid mixtures by sensitivity differential scanning calorimetry. Proc Natl Acad Sci U S A. 1976;73(11):3862–3866. | ||
Muppidi K, Pumerantz AS, Wang J, Betageri G. Development and stability studies of novel liposomal vancomycin formulations. ISRN Pharm. 2012;2012:636743. | ||
Sogias IA, Williams AC, Khutoryanskiy VV. Why is chitosan mucoadhesive? Biomacromolecule. 2008;9(7):1837–1842. | ||
Liu N, Park HJ. Chitosan-coated nanoliposome as vitamin E carrier. J Microencapsul. 2009;26(3):235–242. | ||
Kudsiova L, Lawrence MJ. A comparison of the effect of chitosan and chitosan-coated vesicles on monolayer integrity and permeability across Caco-2 and 16HBE14o-cells. J Pharm Sci. 2008;97(9):3998–4010. | ||
Amidi M, Mastrobattista E, Jiskoot W, Hennink WE. Chitosan-based delivery systems for protein therapeutics and antigens. Adv Drug Deliv Rev. 2010;62(1):59–82. | ||
Feng C, Sun G, Wang Z, et al. Transport mechanism of doxorubicin loaded chitosan based nanogels across intestinal epithelium. Eur J Pharm Biopharm. December 4, 2013. [Epub ahead of print]. | ||
Christopher JD, Karen LG. Risedronate: a review of its pharmacological properties and clinical use in resorptive bone disease. Drugs. 2001;61(5):685–712. | ||
Kinov P, Boyanov M. Clinical utility of risedronate in postmenopausal osteoporosis: patient considerations with delayed-release formulation. Int J Womens Health. 2012;4:164–174. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.