Back to Journals » Clinical Interventions in Aging » Volume 14
Effect of genetic polymorphisms on Alzheimer’s disease treatment outcomes: an update
Authors Sumirtanurdin R ![]() , Thalib AY, Cantona K, Abdulah R
, Thalib AY, Cantona K, Abdulah R
Received 2 January 2019
Accepted for publication 14 February 2019
Published 29 March 2019 Volume 2019:14 Pages 631—642
DOI https://doi.org/10.2147/CIA.S200109
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Walker
Riyadi Sumirtanurdin,1 Amirah Y Thalib,1 Kelvin Cantona,1 Rizky Abdulah1,2
1Department of Pharmacology and Clinical Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Jatinangor, Indonesia; 2Center of Excellence in Higher Education for Pharmaceutical Care Innovation, Universitas Padjadjaran, Jatinangor, Indonesia
Abstract: Genetic variations in individuals may cause differences in the response to cholinesterase inhibitor drugs used in the treatment of Alzheimer’s disease (AD). Through this review, we aimed to understand the potential relationship between genetic polymorphisms and treatment response in AD. We conducted a systematic review of the studies published from 2006 to 2018 that assessed the relationship between genetic polymorphisms and the pharmacotherapeutic outcomes of patients with AD. Via several possible mechanisms, genetic polymorphisms of many genes, including ABCA1, ApoE3, CYP2D6, CHAT, CHRNA7, and ESR1, appear to have strong correlations with the treatment response of patients with AD. Indeed, these genetic polymorphisms, either in the form of single nucleotide polymorphisms or direct changes to one or more amino acids, have been shown to cause differences in the therapeutic response. In summary, our findings indicate that genetic polymorphisms should be considered in the management of AD to achieve both effective and efficient treatment outcomes in terms of cost and prognosis.
Keywords: Alzheimer’s disease, genetic polymorphisms, treatment response
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive dementia. AD suggests distinctive neuropathology and/or neurochemical deficiency in selective brain regions. It is considered the most common cause of dementia and accounts for 60%–70% of all cases of dementia. Approximately 4.6 million cases of dementia have been reported annually worldwide, and by 2050, 100 million people are projected to have dementia. On the contrary, ~20% of the world’s population aged >80 years has AD.1
AD is characterized by a reduced memory capacity (or dementia), decreased numbers of cholinergic neurons, accumulation of amyloid-β, and neurofibrillary neuronal formation.2 The pathogenesis of AD is considered complex. Although there are several hypotheses involving genetic factors, the precise etiology has not yet been identified.3 AD causes synaptic dysfunction in the early phase of the disease, thereby disrupting the communication of important nerves involved in memory as well as other cognitive functions. Degeneration in AD begins in the medial temporal lobe, particularly in the entorhinal cortex and hippocampus. Damage to these regions may result in memory deficits and is observed as an initial clinical manifestation.4
AD itself is pathologically defined as extracellular accumulation of amyloid-β, intracellular accumulation of tau proteins, loss of neurons and synapses, brain atrophy, and general inflammation. Some genetic mutations have been identified as risk factors and are believed to be involved in the pathological development of AD.3 Several types of genetic mutations or disturbances have been identified in patients with AD, including the presence of dysfunctional mutations, single nucleotide polymorphisms (SNPs),5 mitochondrial mutations, and epigenetic changes.6 A recent study by Jiang et al showed the tendency of genetic variation as a factor responsible for AD development. Furthermore, the same study also suggested that AD genetic variations are significantly enriched in the pathways of the immune system.7
Effective pharmacological treatments of AD are currently lacking. The first-line treatment for mild-to-moderate AD involves acetylcholinesterase inhibitors (AChEI) as well as non-pharmacological therapies, including multidimensional stimulation therapy (MST), to prevent and slow the cognitive impairment often observed in patients with AD. However, the outcomes of those therapies may be influenced by differences in genetic polymorphisms among individuals. For example, polymorphisms of ABCA1 and CYPD26 can affect AChEI therapy, whereas those of ApoE-4 and SNAP-25 can affect MST.3 In this study, we discuss recent findings on the association of genetic polymorphisms with the outcomes of AD treatment. This association has not been comprehensively reviewed, as previous review studies mainly focused on the association of genetic polymorphism with the development of AD.
Methodology
This review included studies published in the PubMed database obtained using the keywords “polymorphism” and “Alzheimer’s therapy.” Reviews, non-English studies, and unrelated studies, such as those reporting the lack of therapeutic outcomes for AD and genetic polymorphisms, were excluded. The flowchart of the literature search is shown in Figure 1.
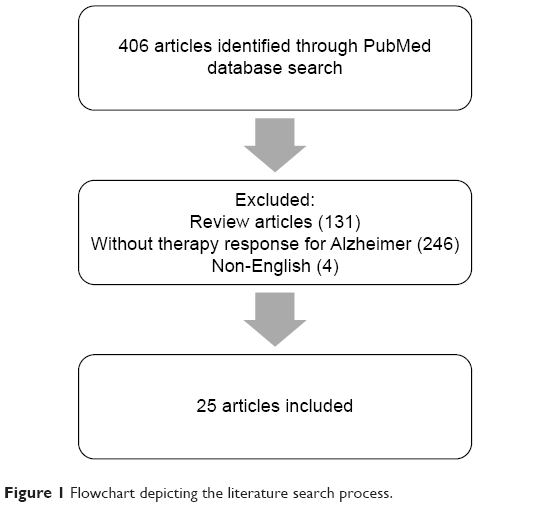 | Figure 1 Flowchart depicting the literature search process. |
Of the total 405 articles obtained in July 2018, we included 24 studies3,5,8–29 that particularly focused on the association between genetic polymorphisms and the outcomes of the treatment of AD (Table 1).
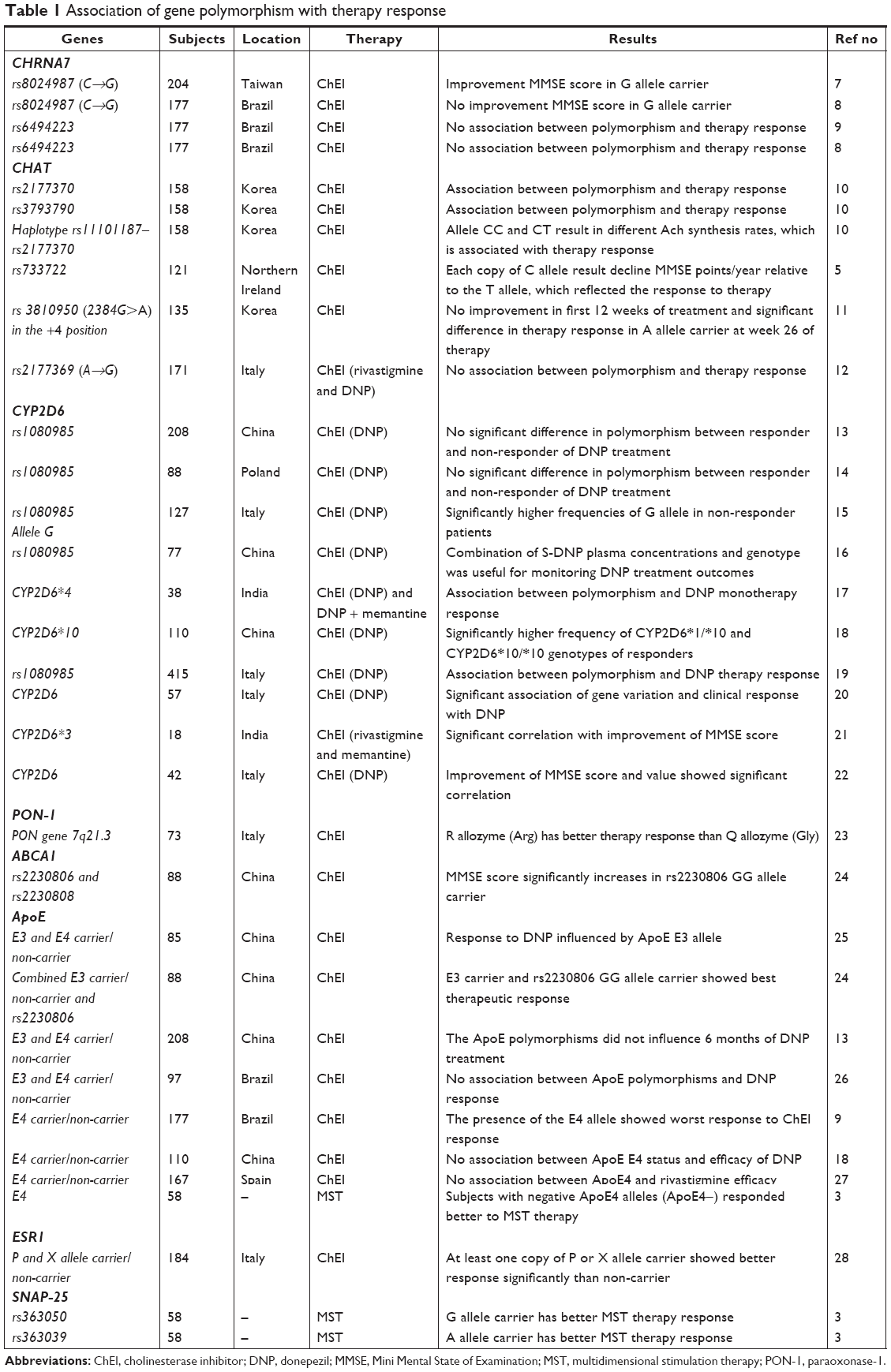 | Table 1 Association of gene polymorphism with therapy response |
Effect of genetic polymorphisms on the pharmacotherapeutic outcomes of AD
Because an effective pharmacotherapeutic management strategy for AD has not yet been established, AChEI therapy is currently used as the first-line drug therapy for AD management. However, genetic polymorphisms reportedly affect the outcomes of AChEI therapy. Indeed, several genes have been studied in this regard, including ABCA1, ApoE, PON-1, CHRNA7, CHAT, ESRI1, and CYP2D6.
ABCA1
ABCA1 is located on chromosome 9 and is reportedly associated with late-onset AD. ATP-binding cassette transporter A1 (ABCA1) is a membrane transporter protein that stimulates cholesterol and phospholipid efflux to apoliproteins.30 In addition, ABCA1 plays a role in cholesterol transport by neutralizing the capacity of Aβ aggregation in an ApoE-dependent manner; this in turn facilitates the elimination of Aβ from the brain, thereby directly transporting it into the blood.31
Lu et al25 reported that patients with AD who have the ABCA1 gene (rs2230806, GG genotype) exhibited considerably better therapeutic outcomes than patients who have the AA or AG genotype. Indeed, patients with the GG genotype were shown to exhibit the best response to donepezil (DNP) therapy (21 of 49 responders) and the lowest frequency as non-responders (4 of 39 non-responders). The authors also tested the ABCA1 gene, rs2230808 base G>A, and reported that it does not considerably affect the therapeutic outcomes.25
ApoE
ApoE is a protein carrier for cholesterol transport in the brain. ApoE is presented as three isoforms as follows: ApoE2, ApoE3, and ApoE4. ApoE2 has been shown to reduce amyloid-β buildup, thus considered a protective factor against AD pathology.26
In 2016, Lu et al26 studied the Han Chinese population and reported that compared with the ApoE3 carriers, the ApoE3 non-carriers in that population responded considerably better to DNP therapy. The authors also reported that compared with ApoE3 carriers, patients with AD who were ApoE3 non-carriers exhibited better therapeutic responses to DNP. In addition, compared with ApoE3 carriers who had the ABCA1 gene rs2230806 AG/AA genotype, patients with AD who were ApoE3 non-carriers and had the ABCA1 gene rs2230806 GG genotype exhibited the best therapeutic responses to DNP.25
Another study reported that compared with ApoE4 carriers, ApoE4 non-carriers exhibited a considerably better response to DNP treatment.10 Another study that observed the association between ApoE4 and AD reported that >80% of the ApoE4-negative patients with AD exhibited marked improvement after 30 weeks as assessed via the AD scores Alzheimer’s Disease Assessment Scale (ADAS), whereas 60% of the ApoE4 carriers had lower ADAS scores compared with baseline. These results indicate that ApoE4 plays a role in AD-related cholinergic dysfunction and may be useful in the prognosis of patients with AD who poorly respond to AChEI therapy.32 In contrast, Miranda et al27 and Zhong et al19 reported no association between ApoE4 polymorphisms and the clinical response to DNP after 6–12 months of follow-up.
Paraoxonase-1 (PON-1)
PON-1 is an arylesterase; it has multiple biological activities, including acetylcholinesterase inhibition. PON-1 can hydrolyze paraoxon, the active metabolite of parathion having toxic acetylcholinesterase properties, to provide protection against exogenous organophosphates.33
The serum level and activity of PON-1 considerably vary in humans and are determined by polymorphisms in related genes.34 The basis of the genetic polymorphism of PON-1 is the change in Gln to Arg at residue 192, which produces the following three possible genotypes: QQ, QR, and RR. The Q allozyme with a Gln at residue 192 has low paraoxon hydrolysis activity, whereas the R allozyme with an Arg at residue 192 exhibits higher activity.35
Pola et al24 reported that compared with patients carrying the Q allele, those carrying the R allele on the PON-1 gene responded better to therapy. This may be due to the mutations that occur, thereby causing differences in the synthesis of PON-1 with different hydrolysis activities. Thus, the R allele is associated with higher enzyme activity. Apart from its role as an endogenous cholinesterase inhibitor, the PON-1 protein has been hypothesized to synergistically interact with drugs that act as AChEIs.24
CHRNA7
The loss of cholinergic neurons and nicotinic acetylcholine receptors (nAChRs) is a major pathological hallmark of AD.36,37 nAChRs are ligand-gated ion channels that mediate the effects of the neurotransmitter acetylcholine. Alpha-7 nAChRs, which are encoded by CHRNA7 on chromosome 15q14, are one of the major nAChR subunits in the central nervous system. Weng et al8 suggested an association between SNPs in CHRNA7 and the response to cholinesterase inhibitors in the treatment of AD. The polymorphism that occurs at rs8024987, which is located in an intron of CHRNA7 in the form of a homozygous or heterozygous C→G transversion, results in better outcomes for cholinesterase inhibitor therapy, with the GG allele providing a better response than the GC allele. However, a further analysis showed that this association was not found in male responders, thereby indicating considerable interaction between gender and SNPs at rs8024987.8 Moreover, Weng et al8 reported that women, particularly those carrying the GG or GC allele, compared with those who are non-carriers or non-galantamine-treated, tend to exhibit better responses to galantamine therapy.
However, conflicting results of the association of CHRNA7 with the outcomes of the treatment of AD have also been reported. In their study, Clarelli et al failed to establish an association between the polymorphism in rs8024987 and cognitive therapy response using cholinesterase inhibitors.9 Studies concerning the polymorphism in rs6494223 also failed to establish an association with the response to AChEI therapy.9,10
Subsequent haplotype analyses have demonstrated that of the four haplotype blocks obtained, one comprising the SNPs at rs885071 (T→G) and rs8024987 (C→G) was associated with therapeutic outcomes. The tendency to respond to the therapy was better when the GG haplotype was observed. However, although the SNPs at rs885071 and rs8024987 are strong in linkage disequilibrium, the pairwise correlations between them are low.8
As described in Figure 2, the correlation between the polymorphisms in CHRNA7 and the cognitive therapy response of ChEI may be associated with multiple pathways. The SNP at rs8024987 occurs in the intron part that might affect the expression of the alpha-7 nAChR via a pre-mRNA splicing mechanism, which leads to changes in protein production. ChEI directly enhances cognition by reducing acetylcholine breakdown, which binds the alpha-7 nAChR, subsequently increasing cholinergic neurotransmission.38 In vitro studies have reported that the alpha-7 nAChR plays a vital role in mediating the neuroprotective effects of ChEI against the toxicity of amyloid-β.39,40 The alpha-7 nAChR can also modulate neurotransmitter release in presynaptic neurons.41 On the contrary, chronic treatment with ChEI also helps increase the alpha-7 nAChR, which induces a positive feedback loop to amplify the effects of ChEI.42 The possible mechanism of polymorphisms in CHRNA7 affects ChEI cognitive response.
 | Figure 2 Possible mechanism of polymorphisms in ChAT and CHRNA7 affecting ChEI cognitive response. |
Choline acetyltransferase (ChAT)
AD is associated with the widespread degeneration of cholinergic neurons; AChEIs have been approved for treating symptoms with the ultimate goal of restoring cholinergic deficits.43 Thus, the cholinergic system is considered a reasonable target in terms of pharmacogenomic studies. Moreover, numerous studies have established an association between genetic polymorphisms in cholinergic genes and the therapeutic effects of AChEIs.5,13 However, the therapeutic response rate of AChEIs varies from 40% to 70%.44
ChAT is an enzyme encoded by ChAT, which plays a role in acetylcholine synthesis by using choline and acetyl-CoA as substrates. ChAT activity is known to be reduced in patients with AD, and this reduction appears to be related to the severity of dementia.13,45
Yoon et al investigated 25 points of SNPs in the ChAT-encoding gene and reported that the two SNPs, rs 2177370 and rs3793790, located in the introns of ChAT, were associated with AChEI drug response. The haplotype analysis demonstrated four haplotype blocks comprising 13 haplotype alleles; in particular, one haplotype block was significantly associated with the AChEI drug response and included two haplotype alleles, with one comprising rs2177370.11
The association of ChAT polymorphisms with AChEI therapy response is associated with the function of AChEIs as an inhibitor of acetylcholinesterase that breaks down acetylcholine. The efficacy of AChEIs depends on the synthesis of acetylcholine itself; thus, when acetylcholine synthesis is impaired owing to the degeneration of cholinergic neurons in patients with AD, the synthesis capacity of the remaining neurons is expected to support the response of the AChEI drug. The CC haplotype itself is reportedly associated with a decrease in the synthesis of ACh, whereas the CT haplotype is associated with a higher rate of ACh synthesis.11
Several studies have investigated SNPs occurring in ChAT. Scacchi et al13 reported that the polymorphism at rs2177369, with G/G with respect to the G/A+A/A genotypes, is considered a risk factor for AD but the authors did not observe an association with the response to ChEI therapy. However, Harold et al5 reported that the occurrence of the C allele results in considerable decline in the Mini Mental State of Examination (MMSE) score, which is associated with the therapeutic response.
In 2015, Lee et al12 reported that they did not observe an association between ChAT A carriers (alleles and non-carriers) and the outcomes of 12 weeks of DNP treatment. However, after 26 weeks of treatment, a considerable difference was observed in the therapeutic outcomes between A ChAT carrier alleles and non-carriers. The mean MMSE score in the Korean version of the Consortium to Establish a Registry for AD assessment battery MMSE-KC increased after 26 weeks of therapy with DNP in the ChAT A carriers.12 The difference in the outcomes of these treatments is associated with the possible efficiency of ChAT translation, which occurs when there is a change (2384 G→A), and the recognition of the initiator codon is improved. Although this is not a rate-limiting enzyme, the substitution of the A allele for G may decrease the production of ChAT, which may be related to the therapeutic effects of DNP.46
ESR1
ESR1 is an estrogen receptor located on chromosome 6q25. ESR1-mediated estrogen activity in the brain occurs via the activation of transmembrane, intracellular, and membrane-bound proteins.47 Scacchi et al29 reported that compared with non-carriers, patients with at least one P and X allele of the two ESR1 SNPs rs2234693 and rs9340799 exhibited a better treatment response. This is likely due to the presence of the P and X alleles, which increase acetylcholine biosynthesis, thereby increasing acetylcholinesterase inhibition.
CYP2D6
CYP2D6 is responsible for the hydroxylation or demethylation of approximately 25% of all clinically important drugs, including antiarrhythmics, antipsychotics, antihistamines, and antidepressants.48 CYP2D6 plays a role in the metabolism of central nervous system agents with narrow therapeutic indexes in which its treatment and accumulation can produce symptoms similar to those of the disease. Substrates and inhibitors metabolized by CYP2D6 have basic and oxidized nitrogen molecules. They also tend to have flat lipophilic regions and functional groups that are capable of electrostatic interactions or forming hydrogen bonds.48
Pilotto et al16 conducted a study during a 6-month follow-up period after DNP therapy in 115 patients (60% responders and 40% non-responders) and reported considerably higher frequencies of patients with G allele at rs1080985 in non-responders than in responders. Further analysis showed that patients with the G allele had a higher risk accompanied with poor response to DNP treatment. This study also suggested that the SNP at rs1080985 in CYP2D6 may influence the clinical efficacy of DNP therapy in patients with mild-to-moderate AD; furthermore, CYP2D6 genotype analysis may be useful in identifying subgroups of patients with AD who exhibit different clinical responses to DNP.16 The results of this study were later confirmed via a follow-up study conducted by Albani et al in 2012.20
In 2013, Zhong et al19 reported a higher frequency of the CYP2D6*1/*10 and CYP2D6*10/*10 genotypes in responders than in non-responders after 6 months of DNP therapy. CYP2D6*1 is a wild-type form of CYP2D6, whereas CYP2D6*10 is mutant allele and the last one exhibits reduction in catalytic activity thus retard metabolism. Furthermore, those with the genotypes CYP2D6*1/*10 and CYP2D6*10/*10 also had higher DNP plasma concentrations and better cognitive scores than those with the CYP2D6*1/*1 genotype. Thus, the authors suggested that patients with AD with mutant alleles (*10) in CYP2D6 may respond better to DNP than those with wild-type alleles (*1).19
Sonali et al22 assessed patients undergoing rivastigmine therapy and reported a considerable allele frequency of CYP2D6*3 polymorphisms, where there is a frameshift mutation in the fifth exon and resulted an inactive enzyme. In addition, a study conducted by the same group in 2014 reported that the frequency of the CYP2D6*3 alleles was considerably associated with the results of DNP monotherapy. The authors also suggested that the CYP2D6 polymorphism may play a role in regulating the plasma concentrations of AD drugs.18 These results were supported by the findings of Lu et al17 who attempted to determine the steady-state plasma concentrations of S-DNP in patients by combining their CYP2D6 genotypes and reported it to be useful to monitor the effectiveness of clinical DNP therapy in patients with AD.
Other studies have also reported that the CYP2D6 genotype analysis is useful for identifying subgroups of patients with AD who had different clinical responses to DNP therapy.21,23 Conflicting results, however, were also reported. Klimkowicz-Mrowiec et al15 and Liu et al14 reported that the CYP2D6 polymorphism did not influence the response to DNP therapy in either the Polish or Chinese population, respectively.
Association of genetic polymorphisms with the outcome of non-pharmacological therapies of AD
The symptoms of AD, including cognitive impairment, function, and behavior, vary greatly among individuals and may depend on the patient’s age at the onset of AD, location of the affected region of the brain that controls cognition, prevalence of behavioral symptoms, and speed of progression.49 Genetic factors themselves are known to play an important role in this variability, because disease progression and functional restoration are highly dependent on the neuroplastic capacity of the remaining neural tissue.50 Synaptosomal-associated protein of 25 kDa (SNAP-25) is one protein in addition to ApoE4 that plays a role in regulating the plasticity of neurons in neural tissues.51 Both genes have been reported to affect the outcomes of non-pharmacological treatments of AD, including MST, which aims to prevent and slow cognitive impairment in patients.3
SNAP-25
SNAP-25 is a vesicular protein composed of 206 amino acids encoded by a gene located on chromosome 20 and is a SNARE target molecule located at the terminal of presynaptic neurons.52 Binding occurs in one of the SNARE domains of SNAP-25 with the syntaxin 1A plasma membrane and in another domain with a vesicle-associated membrane protein (VAMP). Interactions also occur between SNAP-25 and synaptotagmin, in which it is essential to promote the fusion of calcium-mediated membranes as well as control the fusion pore at the final stage of exocytosis, which is the release phase of neurotransmitters.53 In addition to regulating the release of neurotransmitters through exocytosis, SNAP-25 also plays a role in modulating various types of voltage-gated calcium channels (VGCCs), including types N, P/Q, and L, in which the overexpression of SNAP-25 considerably inhibits the function of these channels and lowers the response to depolarization.54
Polymorphisms of SNAP-25 itself are known to be associated with the risk of developing AD. The intron with the alleles rs363050 (A) and rs363043 (T) and the haplotype rs363050/rs363043 A→T are more commonly present in individuals with AD and are associated with pathological values of the fMRI parameter. Guerini et al3 reported that a polymorphism (SNP) in the SNAP-25-encoding gene not only increases the risk of AD but also affects the outcomes of MST therapy. MST therapy is a cognitive stimulation therapy related to recreational and psychomotor therapy that is not only capable of improving cognitive disorders but also serves to increase activity in the temporal region of the thalamus and right insular cortex.55 Indeed, test results have shown that subjects carrying the rs363050 (G) and rs363039 (A) alleles are characterized as having a lower Neuropsychiatric Inventory (NPI) score, reflecting an increase in post-therapy behavioral function using the MST method.3
On the contrary, the type of allele carried, either homozygous or heterozygous, also influences the therapeutic outcomes. Patients with the rs363050 homozygous (GG) allele, who represent a minor population of the overall sample patient population, have been shown to exhibit better improvement in therapeutic outcomes than those with the homozygous (AA) allele, who represent the major population. Patients with heterozygous alleles (AG) also showed better therapeutic outcomes compared with homozygous patients. In rs363039, the patients who were homozygous for the minor allele (AA) exhibited improved behavioral function when compared with those who were homozygous for the major allele (GG). Guerini et al3 also suggested a possible link between the genotype dose effects and NPI scores.
The effect of the polymorphisms (SNPs) of the rs363050 allele on the efficacy of therapy is also associated with the SNAP-25 gene transcription function, in which the allele rs363050(A) has a much higher expression of SNAP-25 than the allele rs363050(G) (Figure 3A).56 The overexpression of SNAP-25 encountered in individuals with the rs363050(A) allele considerably inhibits the function of the VGCC channel and decreases the response to depolarization.54 Calcium influx of nerve terminals is negatively regulated by complex formation between SNAP-25 and VGCC.57 On the contrary, the overexpression of SNAP-25 in adulthood generates deficits in the memory formation process via the role of SNAP-25 in glutamate-dependent excitatory transmission (Figure 3B). The core of the fusion molecule (SNARE Complex) consists of synaptobrevin/VAMP2, syntaxin-1, and SNAP-25.57 The expression of SNAP-25 in the adult dorsal hippocampus also leads to the deregulation of memory consolidation in that region of the brain.58 Furthermore, the overexpression of SNAP-25 in hippocampal neuron cultures is also reportedly associated with synaptic transmission disorders.59
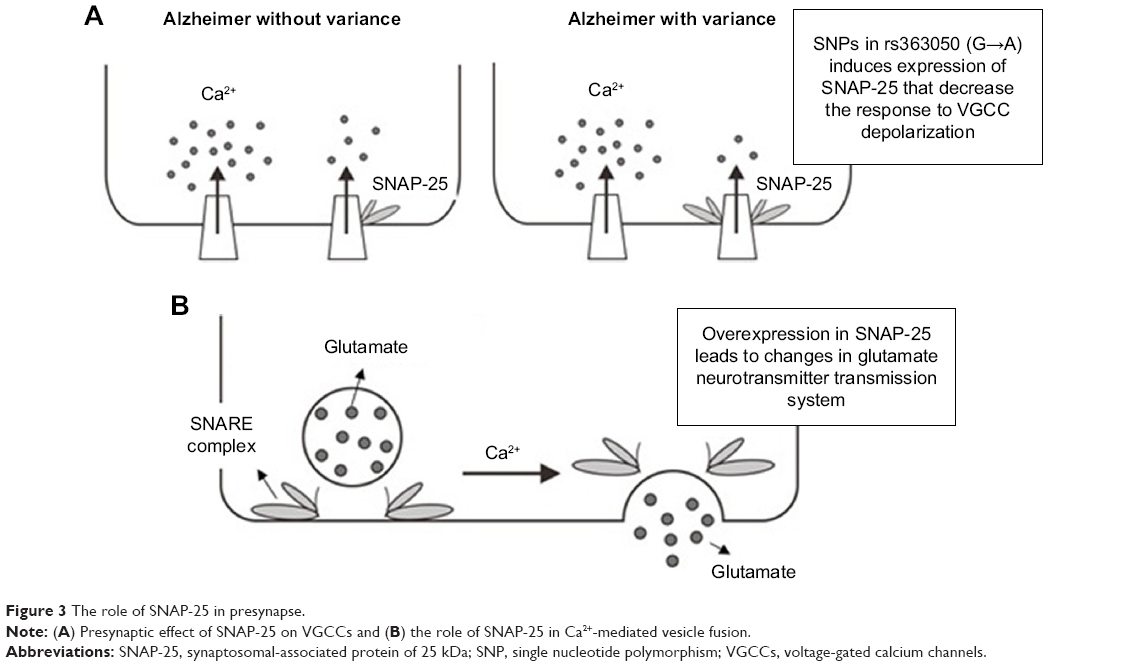 | Figure 3 The role of SNAP-25 in presynapse. |
ApoE
The ApoE protein is a glycoprotein composed of 299 amino acids with varying levels of post-translation stabilization via O-glycosylation of threonine residues.60 Although neurons can produce ApoE under some conditions, even in smaller amounts compared with that produced by astrocytes,61 non-neuron astrocytes and some microglia are the main cell types that express ApoE in the brain.62,63 ApoE4 is one of the isoforms resulting from polymorphisms found in ApoE, with the others being ApoE2 and ApoE3. The three isoforms of ApoE differ by only one or two amino acids at the residues 112 and 158; however, these differences are capable of altering the structure and function of ApoE.64 All three ApoE isoforms promote the occurrence of Aβ42 fibrillation; however, the highest effect is shown by ApoE4.65 On the contrary, ApoE4 increases Aβ40 aggregation more effectively than ApoE3.66 However, some studies have reported that Aβ40 and Aβ42 have opposite effects on the aggregation of Aβ in vivo.67 The aggregation of Aβ40 is known to be more common in patients with AD and exhibits a dose-dependent model with the ApoE4 gene.
Previous studies have shown that patients with AD who carry positive ApoE4 alleles (ApoE4+) exhibit more rapid cognitive impairment; thus, the status of ApoE4 in recent studies may serve to predict the outcomes of cognitive stimulation in visuospatial memory.68 Furthermore, Guerini et al3 also reported that patients with negative ApoE4 alleles (ApoE4−) responded better to MST therapy than those with positive alleles (ApoE4+). The effect of the gene dose on the MMSE score also indicated that compared with patients with the ApoE4+/E4+ allele, those with the ApoE4+/E4− allele exhibited better response to MST therapy, although not better than patients with the E4−/E4− allele did.3 The low response of MST therapy in patients with the E4+/E4+ allele was determined by decreased ApoE lipoprotein function in stimulating synaptic development,69 lipid debris clearance,70 and promotion of growth in the fibers of cellular granules.71
Prevalence of genetic polymorphisms in genes associated with the outcomes of treatment of AD
Interestingly, the phenomenon of genetic polymorphisms that occur in various genes discussed in the review is not rare. As presented in Table 2, various studies have reported on the genotype distribution of these genes.10,11,14,16,19,25–27,29,72–77 Therefore, the consideration of genetic polymorphisms as part of a therapeutic strategy cannot be underestimated and is believed to have a considerable impact. On the contrary, the genotype distribution that occurs in several genes appears to depend on certain geographies and races.
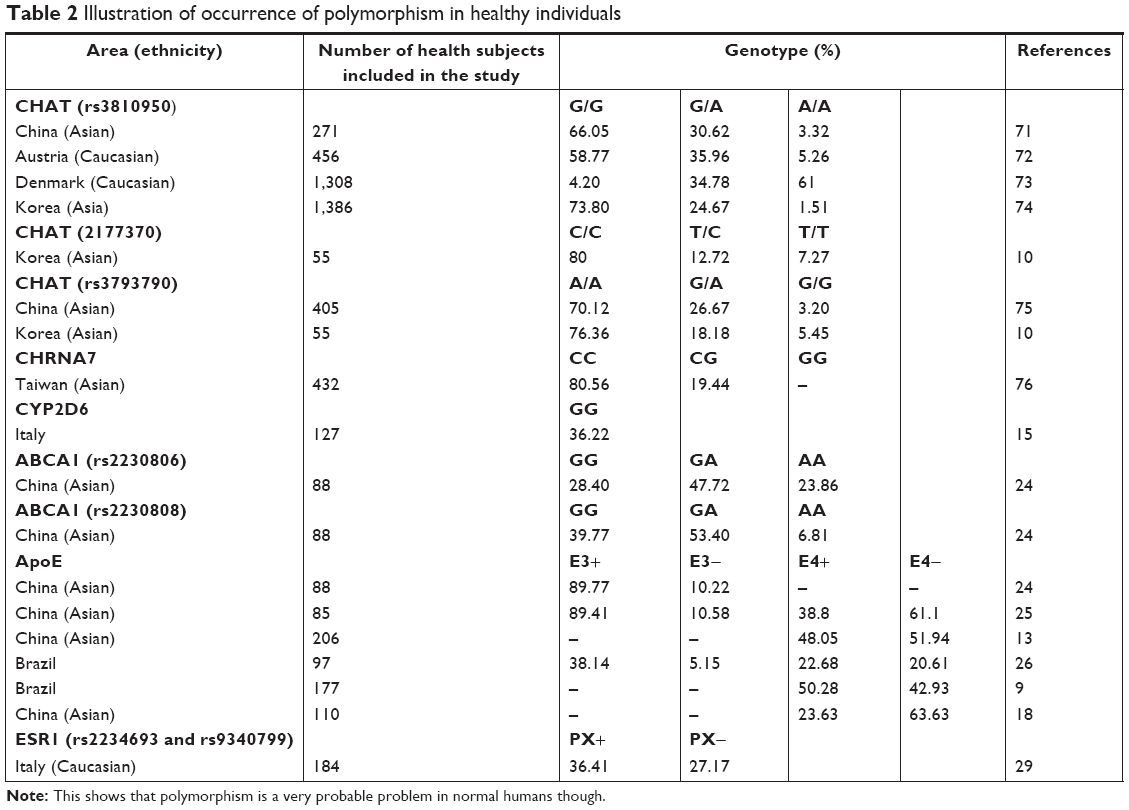 | Table 2 Illustration of occurrence of polymorphism in healthy individuals |
Conclusion and future prospects
Polymorphisms occurring in patients with AD, either in the form of SNPs or direct changes to one or more amino acids in a protein, have been shown to be capable of causing differences in the therapeutic responses in individuals using the same drug. Thus, genetic polymorphisms should be considered to achieve effective and efficient treatment outcomes both in terms of prognosis and cost. A therapeutic guideline based on genetic polymorphisms has actually been established;78–82 therefore, a therapeutic guideline that considers genetic polymorphisms for patients with AD is required to determine effective and efficient therapies for AD.
Acknowledgment
No sources of funding were used to assist in the conduct of this study.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work
References
Mann DM. Pyramidal nerve cell loss in Alzheimer’s disease. Neurodegeneration. 1996;5(4):423–427. | ||
Noor A, Zahid S. A review of the role of synaptosomal-associated protein 25 (SNAP-25) in neurological disorders. Int J Neurosci. 2017;127(9):805–811. | ||
Guerini FR, Farina E, Costa AS, et al. APOE and SNAP-25 polymorphisms predict the outcome of multidimensional stimulation therapy rehabilitation in Alzheimer’s disease. Neurorehabil Neural Repair. 2016;30(9):883–893. | ||
Sheng M, Sabatini BL, Südhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2012;4(5):a005777. | ||
Harold D, Macgregor S, Patterson CE, et al. A single nucleotide polymorphism in ChAT influences response to acetylcholinesterase inhibitors in Alzheimer’s disease. Pharmacogenet Genomics. 2006;16(2):75–77. | ||
Minocherhomji S, Tollefsbol TO, Singh KK. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics. 2012;7(4):326–334. | ||
Jiang Q, Jin S, Jiang Y, et al. Alzheimer’s disease variants with the genome-wide significance are significantly enriched in immune pathways and active in immune cells. Mol Neurobiol. 2017;54(1):594–600. | ||
Weng PH, Chen JH, Chen TF, et al. CHRNA7 polymorphisms and response to cholinesterase inhibitors in Alzheimer’s disease. PLoS One. 2013;8(12):e84059. | ||
Clarelli F, Mascia E, Santangelo R, et al. CHRNA7 gene and response to cholinesterase inhibitors in an Italian cohort of Alzheimer’s disease patients. J Alzheimers Dis. 2016;52(4):1203–1208. | ||
Braga ILS, Silva PN, Furuya TK, et al. Effect of APOE and CHRNA7 genotypes on the cognitive response to cholinesterase inhibitor treatment at different stages of Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2015;30(2):139–144. | ||
Yoon H, Myung W, Lim SW, et al. Association of the choline acetyltransferase gene with responsiveness to acetylcholinesterase inhibitors in Alzheimer’s disease. Pharmacopsychiatry. 2015;48(3):111–117. | ||
Lee KU, Lee JH, Lee DY, et al. The effect of choline acetyltransferase genotype on donepezil treatment response in patients with Alzheimer’s disease. Clin Psychopharmacol Neurosci. 2015;13(2):168–173. | ||
Scacchi R, Gambina G, Moretto G, Corbo RM. Variability of AChE, BCHE, and ChAT genes in the late-onset form of Alzheimer’s disease and relationships with response to treatment with donepezil and rivastigmine. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(4):502–507. | ||
Liu M, Zhang Y, Huo YR, et al. Influence of the rs1080985 single nucleotide polymorphism of the CYP2D6 gene and APOE polymorphism on the response to donepezil treatment in patients with Alzheimer’s disease in China. Dement Geriatr Cogn Dis Extra. 2014;4(3):450–456. | ||
Klimkowicz-Mrowiec A, Wolkow P, Sado M, et al. Influence of rs1080985 single nucleotide polymorphism of the CYP2D6 gene on response to treatment with donepezil in patients with Alzheimer’s disease. Neuropsychiatr Dis Treat. 2013;9:1029–1033. | ||
Pilotto A, Franceschi M, D’Onofrio G, et al. Effect of a CYP2D6 polymorphism on the efficacy of donepezil in patients with Alzheimer disease. Neurology. 2009;73(10):761–767. | ||
Lu J, Wan L, Zhong Y, et al. Stereoselective metabolism of donepezil and steady-state plasma concentrations of S-donepezil based on CYP2D6 polymorphisms in the therapeutic responses of Han Chinese patients with Alzheimer’s disease. J Pharmacol Sci. 2015;129(3):188–195. | ||
Sonali N, Tripathi M, Sagar R, Velpandian T, Subbiah V. Impact of CYP2D6 and CYP3A4 genetic polymorphism on combined cholinesterase inhibitors and memantine treatment in mild to moderate Alzheimer’s disease. Dement Geriatr Cogn Disord. 2014;37(1–2):58–70. | ||
Zhong Y, Zheng X, Miao Y, Wan L, Yan H, Wang B. Effect of cyp2d6*10 and APOE polymorphisms on the efficacy of donepezil in patients with Alzheimer’s disease. Am J Med Sci. 2013;345(3):222–226. | ||
Albani D, Martinelli Boneschi F, Biella G, et al. Replication study to confirm the role of CYP2D6 polymorphism rs1080985 on donepezil efficacy in Alzheimer’s disease patients. J Alzheimers Dis. 2012;30(4):745–749. | ||
Seripa D, Bizzarro A, Pilotto A, et al. Role of cytochrome P4502D6 functional polymorphisms in the efficacy of donepezil in patients with Alzheimer’s disease. Pharmacogenet Genomics. 2011;21(4):225–230. | ||
Sonali N, Tripathi M, Sagar R, Velpandian T, Subbiah V. Clinical effectiveness of rivastigmine monotherapy and combination therapy in Alzheimer’s patients. CNS Neurosci Ther. 2013;19(2):91–97. | ||
Varsaldi F, Miglio G, Scordo MG, et al. Impact of the CYP2D6 polymorphism on steady-state plasma concentrations and clinical outcome of donepezil in Alzheimer’s disease patients. Eur J Clin Pharmacol. 2006;62(9):721–726. | ||
Pola R, Flex A, Ciaburri M, et al. Responsiveness to cholinesterase inhibitors in Alzheimer’s disease: a possible role for the 192 Q/R polymorphism of the PON-1 gene. Neurosci Lett. 2005;382(3):338–341. | ||
Lu J, Fu J, Zhong Y, et al. Association between ABCA1 gene polymorphisms and the therapeutic response to donepezil therapy in Han Chinese patients with Alzheimer’s disease. Brain Res Bull. 2018;140:1–4. | ||
Lu J, Fu J, Zhong Y, et al. The roles of apolipoprotein E3 and CYP2D6 (rs1065852) gene polymorphisms in the predictability of responses to individualized therapy with donepezil in Han Chinese patients with Alzheimer’s disease. Neuroscience Letters. 2016;614:43–48. | ||
Miranda LFJR, Gomes KB, Tito PAL, et al. Clinical response to donepezil in mild and moderate dementia: relationship to drug plasma concentration and CYP2D6 and APOE genetic polymorphisms. J Alzheimers Dis. 2017;55(2):539–549. | ||
Blesa R, Aguilar M, Casanova JP, et al. Relationship between the efficacy of rivastigmine and apolipoprotein E (epsilon4) in patients with mild to moderately severe Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20(4):248–254. | ||
Scacchi R, Gambina G, Broggio E, Corbo RM. Sex and ESR1 genotype may influence the response to treatment with donepezil and rivastigmine in patients with Alzheimer’s disease. Int J Geriatr Psychiatry. 2014;29(6):610–615. | ||
Fitz NF, Carter AY, Tapias V, et al. ABCA1 deficiency affects basal cognitive deficits and dendritic density in mice. J Alzheimers Dis. 2017;56(3):1075–1085. | ||
Lupton MK, Proitsi P, Lin K, et al. The role of ABCA1 gene sequence variants on risk of Alzheimer’s disease. J Alzheimers Dis. 2014;38(4):897–906. | ||
Poirier J, Delisle MC, Quirion R, et al. Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci. 1995;92(26):12260–12264. | ||
Getz GS, Reardon CA. Paraoxonase, a cardioprotective enzyme: continuing issues. Curr Opin Lipidol. 2004;15(3):261–267. | ||
Adkins S, Gan KN, Mody M, La Du BN. Molecular basis for the polymorphic forms of human serum paraoxonase/arylesterase: glutamine or arginine at position 191, for the respective A or B allozymes. Am J Hum Genet. 1993;52(3):598–608. | ||
Humbert R, Adler DA, Disteche CM, Hassett C, Omiecinski CJ, Furlong CE. The molecular basis of the human serum paraoxonase activity polymorphism. Nat Genet. 1993;3(1):73–76. | ||
Coyle JT, Price DL, Delong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219(4589):1184–1190. | ||
London ED, Ball MJ, Waller SB. Nicotinic binding sites in cerebral cortex and hippocampus in Alzheimer’s dementia. Neurochem Res. 1989;14(8):745–750. | ||
Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV. Synaptic potentials mediated via alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci. 1998;18(20):8228–8235. | ||
Takada-Takatori Y, Kume T, Sugimoto M, Katsuki H, Sugimoto H, Akaike A. Acetylcholinesterase inhibitors used in treatment of Alzheimer’s disease prevent glutamate neurotoxicity via nicotinic acetylcholine receptors and phosphatidylinositol 3-kinase cascade. Neuropharmacology. 2006;51(3):474–486. | ||
Akaike A, Takada-Takatori Y, Kume T, Izumi Y. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J Mol Neurosci. 2010;40(1–2):211–216. | ||
Wonnacott S, Barik J, Dickinson J, Jones IW. Nicotinic receptors modulate transmitter cross talk in the CNS: nicotinic modulation of transmitters. J Mol Neurosci. 2006;30(1–2):137–140. | ||
Kume T, Sugimoto M, Takada Y, et al. Up-regulation of nicotinic acetylcholine receptors by central-type acetylcholinesterase inhibitors in rat cortical neurons. Eur J Pharmacol. 2005;527(1–3):77–85. | ||
Ellis JM. Cholinesterase inhibitors in the treatment of dementia. J Am Osteopath Assoc. 2005;105(3):145–158. | ||
Jann MW, Shirley KL, Small GW. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin Pharmacokinet. 2002;41(10):719–739. | ||
Wilcock GK, Esiri MM, Bowen DM, Smith CC. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J Neurol Sci. 1982;57(2–3):407–417. | ||
Kozak M. Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. Embo J. 1997;16(9):2482–2492. | ||
Österlund MK, Hurd YL. Estrogen receptors in the human forebrain and the relation to neuropsychiatric disorders. Prog Neurobiol. 2001;64(3):251–267. | ||
Abraham BK, Adithan C. Genetic polymorphism of CYP2D6. Indian J Pharmacol. 2001;33(3):147–169. | ||
National Institute of Health. Progress Report on Alzheimer’s Disease 2004–2005: New Discoveries, New Insights. Maryland: National Institute of Health; 2005. | ||
Goldberg A, Curtis CL, Kleim JA. Linking genes to neurological clinical practice: the genomic basis for neurorehabilitation. JNPT. 2015;39(1):52–61. | ||
Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–118. | ||
Chang JYH, Stamer WD, Bertrand J, et al. Role of nitric oxide in murine conventional outflow physiology. Am J Physiol Cell Physiol. 2015;309(4):C205–C214. | ||
Brunger AT. Structural insights into the molecular mechanism of Ca(2+)-dependent exocytosis. Curr Opin Neurobiol. 2000;10(3):293–302. | ||
Pozzi D, Condliffe S, Bozzi Y, et al. Activity-dependent phosphorylation of Ser187 is required for SNAP-25-negative modulation of neuronal voltage-gated calcium channels. Proc Natl Acad Sci. 2008;105(1):323–328. | ||
Baglio F, Griffanti L, Saibene FL, et al. Multistimulation group therapy in Alzheimer’s disease promotes changes in brain functioning. Neurorehabilitation and Neural Repair. 2015;29(1):13–24. | ||
Braida D, Guerini FR, Ponzoni L, et al. Association between SNAP-25 gene polymorphisms and cognition in autism: functional consequences and potential therapeutic strategies. Transl Psychiatry. 2015;5(1):e500. | ||
Antonucci F, Corradini I, Fossati G, Tomasoni R, Menna E, Matteoli M. SNAP-25, a known presynaptic protein with emerging postsynaptic functions. Frontiers Synapt Neurosci. 2016;8(89):7. | ||
McKee AG, Loscher JS, O’Sullivan NC, et al. AAV-mediated chronic over-expression of SNAP-25 in adult rat dorsal hippocampus impairs memory-associated synaptic plasticity. J Neurochem. 2010;112(4):991–1004. | ||
Owe-Larsson B, Berglund M, Kristensson K, et al. Perturbation of the synaptic release machinery in hippocampal neurons by overexpression of SNAP-25 with the Semliki Forest virus vector. Eur J Neurosci. 1999;11(6):1981–1987. | ||
Wernette-Hammond ME, Lauer SJ, Corsini A, Walker D, Taylor JM, Rall SC. Glycosylation of human apolipoprotein E. The carbohydrate attachment site is threonine 194. J Biol Chem. 1989;264(15):9094–9101. | ||
Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the APOE locus. J Neurosci. 2006;26(19):4985–4994. | ||
Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 1987;262(29):14352–14360. | ||
Grehan S, Tse E, Taylor JM. Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J Neurosci. 2001;21(3):812–822. | ||
Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci. 2006;103(15):5644–5651. | ||
Ma J, Brewer HB, Potter H. Alzheimer A beta neurotoxicity: promotion by antichymotrypsin, ApoE4; inhibition by a beta-related peptides. Neurobiol Aging. 1996;17(5):773–780. | ||
Castano EM, Prelli F, Wisniewski T, et al. Fibrillogenesis in Alzheimer’s disease of amyloid beta peptides and apolipoprotein E. Biochem J. 1995;306 (Pt 2):599–604. | ||
Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63(3):287–303. | ||
Polito L, Abbondanza S, Vaccaro R, et al. Cognitive stimulation in cognitively impaired individuals and cognitively healthy individuals with a family history of dementia: short-term results from the “Allena-Mente” randomized controlled trial. Int J Geriatr Psychiatry. 2015;30(6):631–638. | ||
Mauch DH, Nägler K, Schumacher S, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294(5545):1354–1357. | ||
White F, Nicoll JA, Roses AD, Horsburgh K. Impaired neuronal plasticity in transgenic mice expressing human apolipoprotein E4 compared to E3 in a model of entorhinal cortex lesion. Neurobiol Dis. 2001;8(4):611–625. | ||
Teter B, Xu PT, Gilbert JR, Roses AD, Galasko D, Cole GM. Defective neuronal sprouting by human apolipoprotein E4 is a gain-of-negative function. J Neurosci Res. 2002;68(3):331–336. | ||
Tang M, Rao D, Ma C, et al. Evaluation of choline acetyltransferase gene polymorphism (2384 G/A) in Alzheimer’s disease and mild cognitive impairment. Dement Geriatr Cogn Disord. 2008;26(1):9–14. | ||
Grünblatt E, Zehetmayer S, Bartl J, et al. Genetic risk factors and markers for Alzheimer’s disease and/or depression in the vita study. J Psychiatr Res. 2009;43(3):298–308. | ||
Mengel-From J, Christensen K, Thinggaard M, McGue M, Christiansen L. Genetic variants in the choline acetyltransferase (ChAT) gene are modestly associated with normal cognitive function in the elderly. Genes Brain Behav. 2011;10(8):876–882. | ||
Lee JJ, Jo SA, Park JH, et al. Choline acetyltransferase 2384G>a polymorphism and the risk of Alzheimer disease. Alzheimer Dis Assoc Disord. 2012;26(1):81–87. | ||
Yang X, Liu W, Yi M, et al. Choline acetyltransferase may contribute to the risk of Tourette syndrome: combination of family-based analysis and case-control study. World J Biol Psychiatry. 2017:1–6. | ||
Weng PH, Chen JH, Chen TF, et al. CHRNA7 polymorphisms and dementia risk: interactions with apolipoprotein ε4 and cigarette smoking. Sci Rep. 2016;6(1):27231. | ||
Amstutz U, Henricks LM, Offer SM, et al. Clinical pharmacogenetics implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103(2):210–216. | ||
Goetz MP, Sangkuhl K, Guchelaar HJ, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and Tamoxifen Therapy. Clin Pharmacol Ther. 2018;103(5):770–777. | ||
Johnson JA, Caudle KE, Gong L, et al. Clinical pharmacogenetics implementation Consortium (CPIC) guideline for Pharmacogenetics-Guided warfarin dosing: 2017 update. Clin Pharmacol Ther. 2017;102(3):397–404. | ||
Bell GC, Caudle KE, Whirl-Carrillo M, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 genotype and use of ondansetron and tropisetron. Clin Pharmacol Ther. 2017;102(2):213–218. | ||
Hicks JK, Sangkuhl K, Swen JJ, et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin Pharmacol Ther. 2017;102(1):37–44. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.