Back to Journals » Clinical Ophthalmology » Volume 12
Effect of clinical parameters on the ocular surface microbiome in children and adults
Authors Cavuoto KM, Mendez R, Miller D ![]() , Galor A
, Galor A ![]() , Banerjee S
, Banerjee S ![]()
Received 25 February 2018
Accepted for publication 1 May 2018
Published 2 July 2018 Volume 2018:12 Pages 1189—1197
DOI https://doi.org/10.2147/OPTH.S166547
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Kara M Cavuoto,1,* Roberto Mendez,2,* Darlene Miller,1 Anat Galor,1,3 Santanu Banerjee2
1Department of Ophthalmology, Bascom Palmer Eye Institute, University of Miami, Miami, FL, 2Department of Surgery, Miller School of Medicine, University of Miami, Miami, FL, 3Department of Ophthalmology, Miami Veterans Administration Medical Center, Miami, FL, USA
*These authors contributed equally to this work
Purpose: To perform a pilot study to characterize the effect of clinical parameters on the ocular surface microbiome (OSM) in children and adults using 16s ribosomal RNA sequencing.
Methods: Prospective, cross-sectional study using 16s sequencing to evaluate the OSM. Comparisons were made in bacterial composition by 1) age, 2) gender, 3) sampling location of the ocular and periocular surfaces, and 4) topical drop use. 16s sequencing was performed using Illumina MiSeq 250 and analyzed using Qiime.
Results: Thirty patients (15 children [mean 3.7 years], 15 adults [mean 60.4 years]) were sampled. Both principal coordinate analysis and unifrac distance analysis showed significant differences in the composition between the pediatric and adult OSMs (both p=0.001). The eyelid margin microbiota did not show any distinct clustering compared to conjunctiva within the pediatric samples but tended to show a distinction between anatomic sites in adult samples. No differences in OSM were noted by topical drop use.
Conclusion: 16s sequencing is a useful tool in evaluating the OSM in patients of all ages, showing a distinct difference between pediatric and adult microbiomes.
Keywords: ocular microbiome, conjunctival microbiome, eyelid margin microbiome, skin microbiome, 16s rRNA, paucibacterial, Qiime
Introduction
A multitude of bacteria inhabit various mucosal surfaces in the human body, with the majority residing in the gastrointestinal surface.1 Although mucosal surface colonization was first thought to occur after birth, it is now understood that colonization begins in utero.2–4 Specific host site factors such as oxygen levels, moisture, pH, and immunologic factors lead to unique bacterial compositions at each anatomic site.5 The microbiome is further shaped by external factors such as environmental changes, aging, and antibiotic use.6
The ocular surface (OS) was initially believed to have little bacterial influence, as several proteins produced by tears have antimicrobial properties.7 Culture-dependent methods have been used in the past to identify the bacteria present on the OS. Overall, coagulase-negative Staphylococci are the most common bacteria isolated from the conjunctiva, lids, or tears.8 However, cultures only demonstrate growth ~50% of the time.9–13 In addition, culture-dependent methods fail to demonstrate that the diverse number of species compose the ocular surface microbiome (OSM).14
16s sequencing is a technique used to identify bacteria based on the sequence of ribosomal RNA (16s rRNA). This methodology is rapid, accurate, and allows for a moderate- to high-resolution understanding of the microbial composition.15 Analysis of conjunctival samples from healthy volunteers has shown that bacteria can be identified from every specimen, roughly corresponding to over 50 distinct bacterial genera, although it is considered paucibacterial when compared to other mucosal surfaces.14,16,17 Of the bacteria present, Corynebacteria, Propionibacteria, and coagulase-negative Staphylococci are consistently the highest contributors to the composition of the OSM, with somewhat variable contributions from other genera.5,8,16
Despite this knowledge, many questions remain regarding the effect of clinical parameters on the composition of the OSM. In this study, our goals were 1) to evaluate differences in OSM composition by age as shown before;6 2) to evaluate whether the OS has a unique microbiome compared to surrounding structures (ie, skin); and 3) to identify the effect of patient-specific characteristics (ie, topical drops) on the OSM, as demonstrated in other studies involving external stimuli and ocular diseases.19,20 Based on prior studies, we hypothesized that the OSM would have a unique composition as compared to the surrounding skin and that older adults would have greater bacterial diversity than younger adults. The novelty of this study lies in confirming the differences between adult and pediatric ocular microbiome and, more importantly, providing comparative microbial profiles of the microbiota on the OS, compared to surrounding areas using a new study population. A comprehensive study of OSM in health and disease and its interaction with the microbiome of surrounding tissue will provide a unique perspective on its role in ocular health. In light of the demonstrated important role of microbiome in disparate mucosal surfaces, a lot of effort is warranted in defining the role of OSM in OS homeostasis and metabolism.
Methods
Ethics statement
This prospective, cross-sectional, observational study was approved by the University of Miami Institutional (IRB approval no 20140717) and Miami Veteran’s Hospital (IRB approval no 3011.03) Institutional Review Boards. The study was conducted at Bascom Palmer Eye Institute and the Miami Veteran’s Administration Hospital in accordance with the principles of the Declaration of Helsinki and also Health Insurance Portability and Accountability Act compliant.
Sample collection
During a 2-month period, all children (<18 years of age) scheduled for a visit in the pediatric ophthalmology clinic at Bascom Palmer Eye Institute and all adults scheduled for a visit in the oculofacial pain clinic at the Miami Veteran’s Hospital were invited to participate. A written informed consent was obtained from patient or parent/guardian, and a written informed assent was obtained from children 7–17 years old. A review of medical records was performed to record pertinent background information including age, sex, birth history, ocular history, past medical history, and surgical history. Any use of ocular and/or systemic medication over the past year was documented. Exclusion criteria included active infection, skin disease, concurrent contact lens use, or administration of oral or topical antibiotics within the prior 90 days.
All specimens were collected by the study personnel in a standardized fashion using sterile gloves. Samples were collected using sterile applicators in the following sites: right periocular skin, right eyelid margin, right conjunctival fornix, and left conjunctival fornix. Special care was taken to avoid touching other ocular structures to avoid contamination. In children, the samples were collected under general anesthesia prior to sterilization of the conjunctiva and periocular tissues for a planned strabismus or glaucoma procedure. In adults, the samples were collected during a scheduled clinic visit for routine care. The collected specimens were then immediately transported by study personnel directly to the microbiology laboratory and stored in a −80°C freezer. The maximum time between storage and DNA extraction for all samples were <2 weeks. In general, the recruitment frequency was 10% of all eligible donors. Prospective donors who declined, in spite of meeting the inclusion/exclusion criteria, did so due to the sole reason of perceived discomfort during and/or after sample collection.
DNA extraction, 16s sequencing, and analysis
Swab heads were aseptically transferred into power-soil sample collection tubes and lysed using MagnaLyser tissue disruptor (Roche, Indianapolis, IN, USA) and total DNA isolated using Power-soil/fecal DNA isolation kit (Mo-Bio, Germantown, MD, USA) as per manufacturer’s specifications. All samples were quantified using the Qubit® Quant-iT dsDNA Broad-Range Kit (Life Technologies, Grand Island, NY, USA) to ensure that they met minimum concentration and mass of DNA and submitted to University of Minnesota Genomic Center for 16s sequencing as described earlier.17 Briefly, to enrich the sample for the bacterial 16S V4 rDNA region, DNA was amplified using fusion primers designed against the surrounding conserved regions that are tailed with sequences to incorporate flow cell adapters and indexing barcodes (Illumina, San Diego, CA, USA). Each sample was polymerase chain reaction (PCR) amplified with two differently barcoded V4–V5 fusion primers and were advanced for pooling and sequencing. For each sample, amplified products were concentrated using a solid-phase reversible immobilization method for the purification of PCR products and quantified by electrophoresis using an (Agilent, Santa Clara, CA, USA) 2100 Bioanalyzer. The pooled 16S V4–V5-enriched, amplified, barcoded samples were loaded into the MiSeq cartridge (Illumina Inc, San Diego, CA, USA), and then onto the instrument along with the flow cell. After cluster formation on the MiSeq Instrument (Illumina, San Diego, CA, USA), the amplicons were sequenced for 250 cycles with custom primers designed for paired-end sequencing.
In addition to patient samples, unused swabs and reagent controls were supplied in triplicate as background. Samples producing amplicons at later cycles compared to majority of samples were concentrated using Agencourt AMPureXP beads (Beckman Coulter, Indianapolis, IN, USA). All samples were sequenced together after barcode normalization subsequent to a preliminary sequencing run.
Using QIIME 1.9.2,18 sequences were quality filtered and demultiplexed using exact matches to the supplied DNA barcodes and primers. Resulting sequences were then searched against the Greengenes database of 16s sequences, clustered at 97% by uclust (closed-reference Operational Taxonomic Unit [OTU] picking) to obtain phylogenetic identities. Analysis for alpha- and beta diversity was done with standardized QIIME workflow as we have shown before.17
Contaminant removal and stringency
DNA extraction reagents and blank sampling tools acted as negative controls and these were carried along with the ocular samples through the same pipeline of DNA extraction to QIIME analysis. The OTU file revealed several unique counts for each blank tool and a few with minor overlap with ocular samples. To discount the possibility of implement-induced artifacts, OTUs with comparable representation in the blank samples were eliminated from further analysis to derive a highly conservative OTU list. Although some authors recommend a method for contaminant filtering involving statistical regression to normalize OTUs based on those present in the control samples,19 this is prone to introduce artifactual elimination of real bacterial species. Hence, this study followed the conventional methods for contaminant removal based on seminal work of de Paiva et al20 and Banerjee et al.21
Statistical analysis
Microbiome analysis with QIIME: OTU tables were rarefied to the sample containing the lowest number of sequences in each analysis. QIIME 1.9.218 was used to calculate alpha diversity (alpha_rarefaction.py) and to summarize taxa (summarize_taxa_through_plots.py). Principal coordinate analysis (pCoA) was done within this program using observation ID level. The Adonis test was utilized for finding significant whole-microbiome differences among discrete categorical or continuous variables with randomization/Monte Carlo permutation test (with Bonferroni correction). The fraction of permutations with greater distinction among categories (larger cross-category differences) than that observed with the nonpermuted data was reported as the p-value. All other data forms were analyzed for significance by the Mann–Whitney U-test (GraphPad Prism).
Results
The patient characteristics are provided in Table 1. Proteobacteria was shown to be one of the dominant phyla on the OSM, composing more than 60% of all phyla. The next most common phyla were Firmicutes, Bacteroidetes, and Actinobacteria. Pediatric samples had a consistent 4.6-fold abundance over the adult samples in terms of total OTU counts at the phylum level. Adult microbiomes exhibited consistent depletion of Proteobacteria (4.15-fold), Fusobacteria (8.81-fold), Firmicutes (9.10-fold), and Bacteroidetes (34.4-fold) compared to the pediatric population. On the other hand, there was a 2-fold increase in OTUs associated with phylum Actinobacteria in adults, whereas phylum Cyanobacteria did not exhibit any measurable changes between the two groups (Figure 1A and Figure S1). At the mapped genus level, we found a core reduction in Streptococcus (3.5-fold), Staphylococcus (2-fold), and Brachybacterium (2-fold) in adults compared to pediatric samples. Genera seen to be relatively more abundant in adults were Corynebacterium (3.9-fold), Paracoccus (2.9-fold), and Propionibacterium (not found in pediatric samples). This observation is in alignment with a similar study by Zhou et al16 however, we could not detect some of the genera identified in this study, possibly due to the geographical and ethnic differences or due to the region of 16s rRNA sequenced (V1 vs V4–V5 in this study).
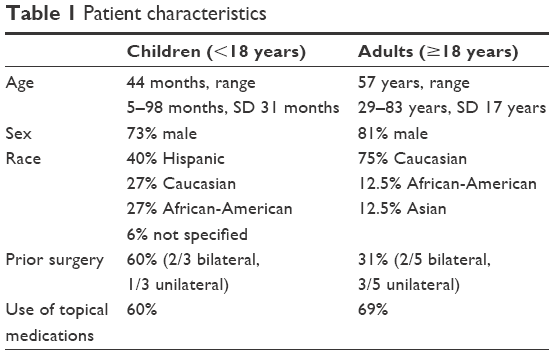 | Table 1 Patient characteristics |
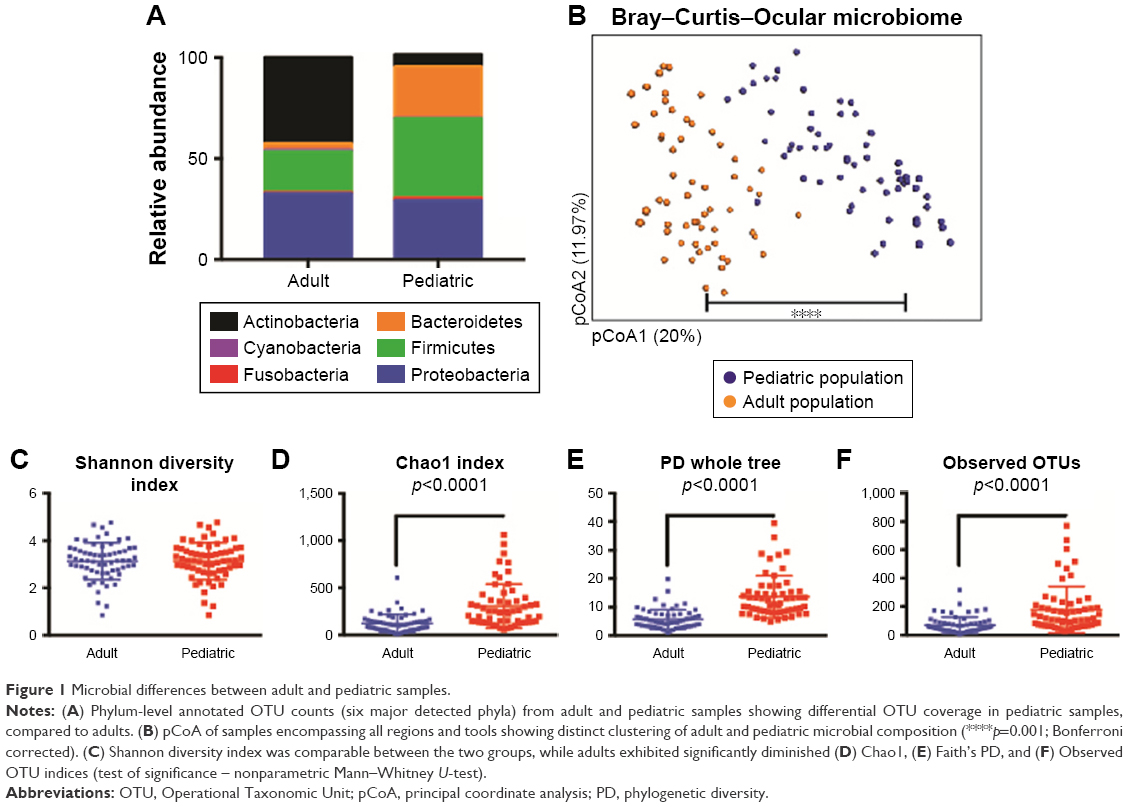 | Figure 1 Microbial differences between adult and pediatric samples. |
Bray–Curtis pCoA (beta diversity) was used to qualitatively examine differences in microbial composition. Pediatric and adult samples clustered separately with significant differences (Figure 1B). Shannon’s H index showed the numbers of different OTUs encountered (diversity), as well as the instances these unique OTUs were sampled (evenness) were similar between pediatric and adult samples, despite significant differences in absolute OTU counts (Figure 1C). As Shannon’s index is blinded to the identities of the OTUs, we also included Chao1 diversity22 to account for the rarity or abundance of individual OTUs and provide a measure of species richness for individual groups (Figure 1D). This analysis demonstrated significantly diminished richness in adults compared to the pediatric population. Faith’s phylogenetic diversity measurement23 also showed that a significant reduction in diversity was observed in adult samples compared to pediatric samples (Figure 1E). Pediatric samples, however, had significantly higher unique OTUs compared to adult samples (Figure 1F).
In overall comparison, adult and pediatric samples clustered separately with significant differences in a principle coordinate plot (Figure 2A). In the same plot, when we compared the periocular skin, conjunctivae, and eyelid margin microbiota, a clear and statistically significant distinction was seen between adult periocular skin when compared to the eyelid margin and conjunctiva. In contrast, there were no significant differences between the pediatric periocular skin and OSM (Figure 2B). This is furthermore clear, when we reanalyzed the adult samples alone (Figure 2C). These data suggest that the paucibacterial nature of OSM extends to the eyelid margin as well. In adults, significant differences were found between periocular microbiome and OSM compared to pediatric samples, despite an opposite trend in species richness/evenness. This implies that while the microbial composition is always different between the periocular skin, eyelid margin, and conjunctivae between the two age groups (Figures S2A–C, respectively), with aging, genera-level changes become more pronounced between periocular skin and OSM/eyelid margin. This is further evident when examining the classwise spread of the microbes, in which there was a distinct expansion of classes from phylum Firmicutes (Bacilli and Clostridia) in the adult periocular skin, at the expense of alpha- and beta-proteobacteria (Figure S2D).
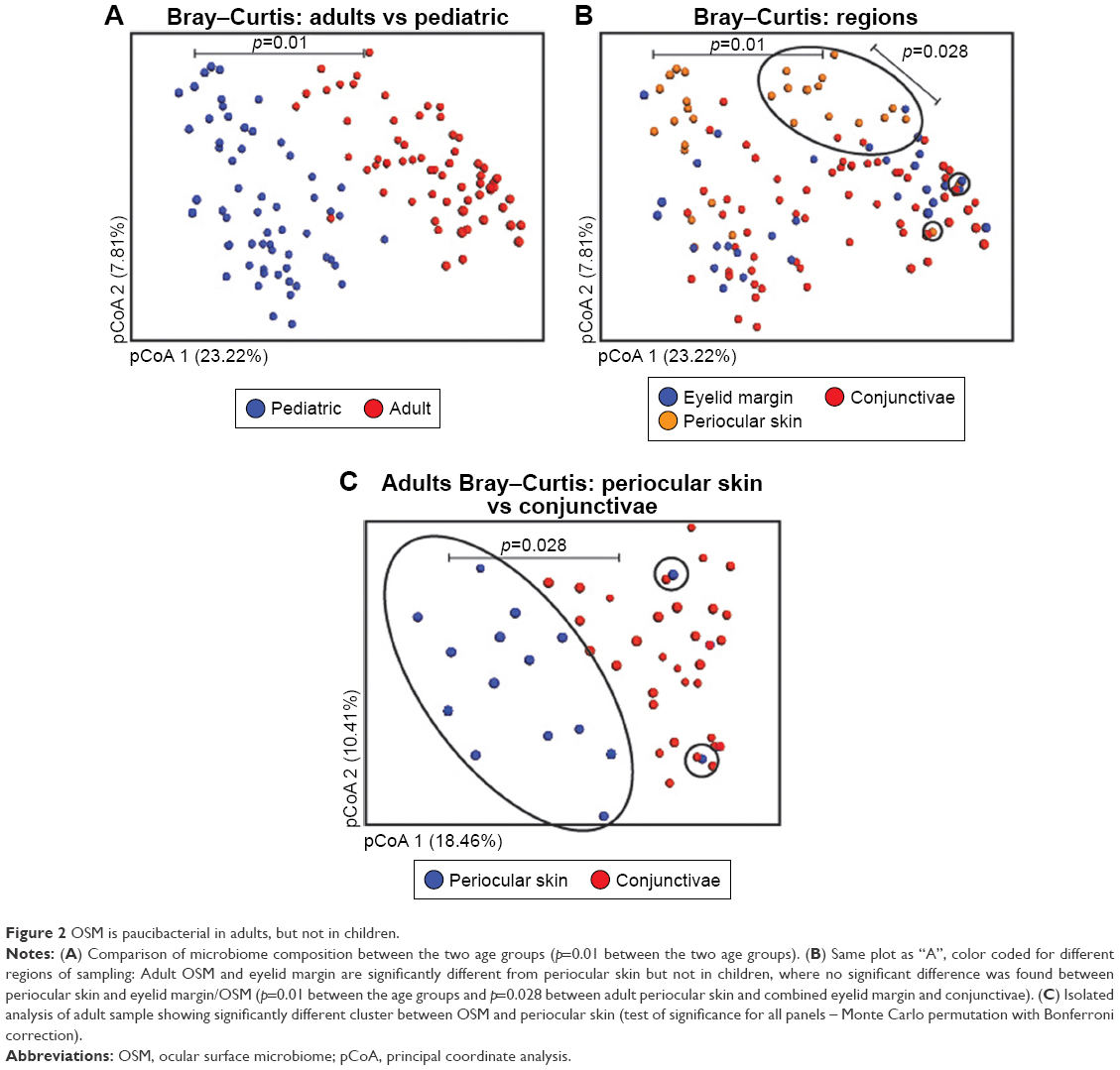 | Figure 2 OSM is paucibacterial in adults, but not in children. |
Although age and sampling location were significant factors, there were no differences between other clinical parameters that were examined. When comparing the right and left eyes, even distribution was seen between the two age clusters, implying that microbial homeostasis is fairly preserved between the two eyes, irrespective of age (Figure 3A). In a similar fashion, no significant differences were seen between the genders (male vs female; Figure 3B), whether surgery was performed in one eye (Figure 3C) or whether the patient was maintained on topical medications (Figure 3D).
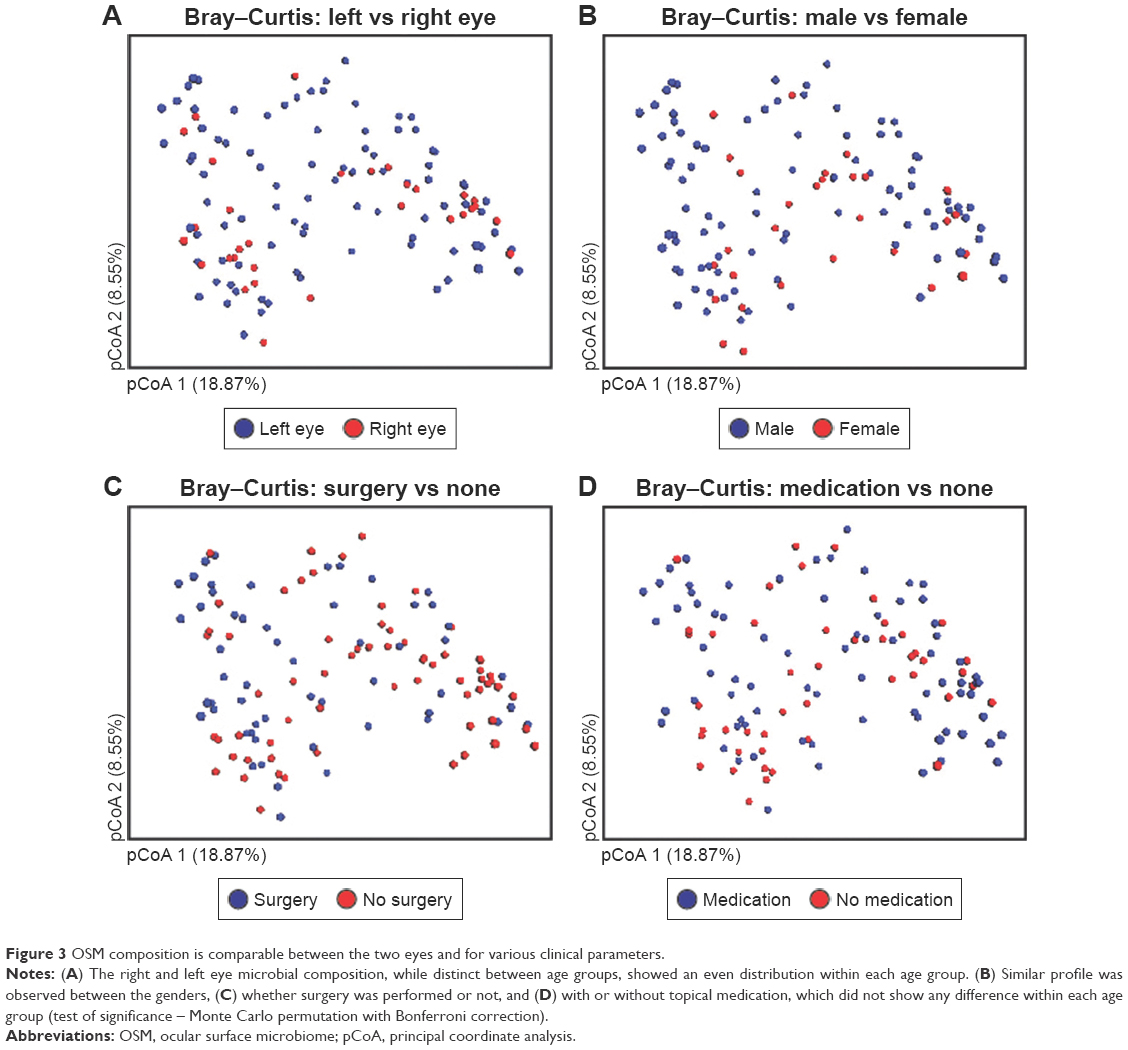 | Figure 3 OSM composition is comparable between the two eyes and for various clinical parameters. |
Discussion
The OSM is thought to be paucibacterial, with an average of 245 different genera per sample as demonstrated in prior studies.21 This is dramatically lower than the gastrointestinal microbiome, in which an average of more than 1,000 genera are present.1 The paucibacterial nature of the OSM is confirmed by our data, in which both the adult and pediatric OTU counts were much lower than the gastrointestinal tract. Interestingly, we also saw a greater number of OTUs in the pediatric population compared to the adult population, which would imply that the paucibacterial nature of OSM is acquired over aging of the individual.
One of the major observations in our study was the significant overall difference between adult and pediatric OSM, which is aligned with the study by Zhou et al.16 The relative abundance of major phyla from adult OSM agrees with the reported composition from other groups in terms of sequencing depth, overall OTU coverage, and phylogenetic identities.16,22 The overall OTU counts were significantly higher in the pediatric samples, with no difference in the evenness of distribution (Shannon index). There were, however, unique compensatory differences between the two age groups in terms of Bacteroidetes, Actinobacteria, and Firmicutes as captured by taxa analysis and other diversity indices.
Additionally, we demonstrated significant differences between the periocular skin microbiome and OSM in adults. The compositional changes in adult periocular skin are shown to be a combined effect of specific expansion of phylum Firmicutes in periocular skin and its reduction in OSM. As an additional unique and novel finding, we show that pediatric microbiome composition of the eyelid margin closely resembles the OSM.
Our study findings need to be considered in light of several limitations. Although 16s sequencing identifies a wider variety of microbes than culture-based techniques, it is more susceptible to noise, sampling errors, and contamination. Second, we do not discuss microbiome to the level of specific genera in this report, which is a limitation of the 16s sequencing method in general. Third, our population included a small number of patients at opposite ends of the age spectrum, and thus they do not likely reflect the true composition of the OSM of the general population across various ages, ethnicities, and exposures. Although our cohort of patients did not demonstrate a difference in the microbial composition between individuals using topical medications, other studies do suggest that long-term use of topical medications does affect the OSM.24 It is possible that the individuals had not used the medications for a sufficient length of time to induce change and/or that our cohort was not large enough to fully evaluate this finding. Additionally, our patients often had ocular morbidities and/or topical ocular medication use, which is also not reflective of the general population. Encouragingly, we did not find significant differences in the composition of the OSM by these comorbidities (eye drop use, history of surgery). Finally, the ethnic distribution of adult and pediatric patients was different. This is due to the recruitment location. Each group reflects the general population of the site from which they were recruited. The pediatric patients recruited from Bascom Palmer Eye Institute reflect the surrounding community of Miami-Dade County, whereas the adults recruited from the Veterans Hospital reflect a broader patient population, typical for the site. Further studies with larger patient populations would be needed to explore these differences, as the current study was a pilot study and not designed to investigate these differences.
Conclusion
Our pilot study provides a foundation for understanding the age-related and site-specific differences of the OSM. Future studies can be directed toward the underlying mechanisms for these differences and their impact on OS immunity and metabolism.
Disclosure
The authors report no conflicts of interest in this work.
References
Power SE, O’Toole PW, Stanton C, Ross RP, Fitzgerald GF. Intestinal microbiota, diet and health. Br J Nutr. 2014;111(3):387–402. | ||
Wassenaar TM, Panigrahi P. Is a foetus developing in a sterile environment? Lett Appl Microbiol. 2014;59(6):572–579. | ||
Eder M, Farina N, Sanabria RR, et al. Normal ocular flora in newborns delivered in two hospital centers in Argentina and Paraguay. Graefes Arch Clin Exp Ophthalmol. 2005;243(11):1098–1107. | ||
Isenberg SJ, Apt L, Yoshimori R, McCarty JW, Alvarez SR. Source of the conjunctival bacterial flora at birth and implications for ophthalmia neonatorum prophylaxis. Am J Ophthalmol. 1988;106(4):458–462. | ||
Huang YJ. The respiratory microbiome and innate immunity in asthma. Curr Opin Pulm Med. 2015;21(1):27–32. | ||
Banerjee R. Introduction to the thematic minireview series: host-microbiome metabolic interplay. J Biol Chem. 2017;292(21):8544–8545. | ||
Azkargorta M, Soria J, Ojeda C, et al. Human basal tear peptidome characterization by CID, HCD, and ETD followed by in silico and in vitro analyses for antimicrobial peptide identification. J Proteome Res. 2015;14(6):2649–2658. | ||
Willcox MD. Characterization of the normal microbiota of the ocular surface. Exp Eye Res. 2013;117:99–105. | ||
Fernandez-Rubio ME, Rebolledo-Lara L, Martinez-Garcia M, Alarcon-Tomas M, Cortes-Valdes C. The conjunctival bacterial pattern of diabetics undergoing cataract surgery. Eye (Lond). 2010;24(5):825–834. | ||
Pramhus C, Runyan TE, Lindberg RB. Ocular flora in the severely burned patient. Arch Ophthalmol. 1978;96(8):1421–1424. | ||
Chern KC, Shrestha SK, Cevallos V, et al. Alterations in the conjunctival bacterial flora following a single dose of azithromycin in a trachoma endemic area. Br J Ophthalmol. 1999;83(12):1332–1335. | ||
Hori Y, Maeda N, Sakamoto M, Koh S, Inoue T, Tano Y. Bacteriologic profile of the conjunctiva in the patients with dry eye. Am J Ophthalmol. 2008;146(5):729–734. | ||
Huang Y, Yang B, Li W. Defining the normal core microbiome of conjunctival microbial communities. Clin Microbiol Infect. 2016;22(7):643.e7–643.e12. | ||
Dong Q, Brulc JM, Iovieno A, et al. Diversity of bacteria at healthy human conjunctiva. Invest Ophthalmol Vis Sci. 2011;52(8):5408–5413. | ||
Sharon I, Banfield JF. Microbiology. Genomes from metagenomics. Science. 2013;342(6162):1057–1058. | ||
Zhou Y, Holland MJ, Makalo P, et al. The conjunctival microbiome in health and trachomatous disease: a case control study. Genome Med. 2014;6(11):99. | ||
Kugadas A, Gadjeva M. Impact of microbiome on ocular health. Ocul Surf. 2016;14(3):342–349. | ||
Lu LJ, Liu J. Human microbiota and ophthalmic disease. Yale J Biol Med. 2016;89(3):325–330. | ||
Zhang H, Zhao F, Hutchinson DS, et al. Conjunctival microbiome changes associated with soft contact lens and orthokeratology lens wearing. Invest Ophthalmol Vis Sci. 2017;58(1):128–136. | ||
de Paiva CS, Jones DB, Stern ME, et al. Altered mucosal microbiome diversity and disease severity in sjogren syndrome. Sci Rep. 2016;6:23561. | ||
Banerjee S, Sindberg G, Wang F, et al. Opioid-induced gut microbial disruption and bile dysregulation leads to gut barrier compromise and sustained systemic inflammation. Mucosal Immunol. 2016;9(6):1418–1428. | ||
Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. | ||
Ozkan J, Nielsen S, Diez-Vives C, Coroneo M, Thomas T, Willcox M. Temporal stability and composition of the ocular surface microbiome. Sci Rep. 2017;7(1):9880. | ||
Ohtani S, Shimizu K, Nejima R, et al. Conjunctival bacteria flora of glaucoma patients during long-term administration of prostaglandin analog drops. Invest Ophthalmol Vis Sci. 2017;58(10):3991–3996. |
Supplementary materials
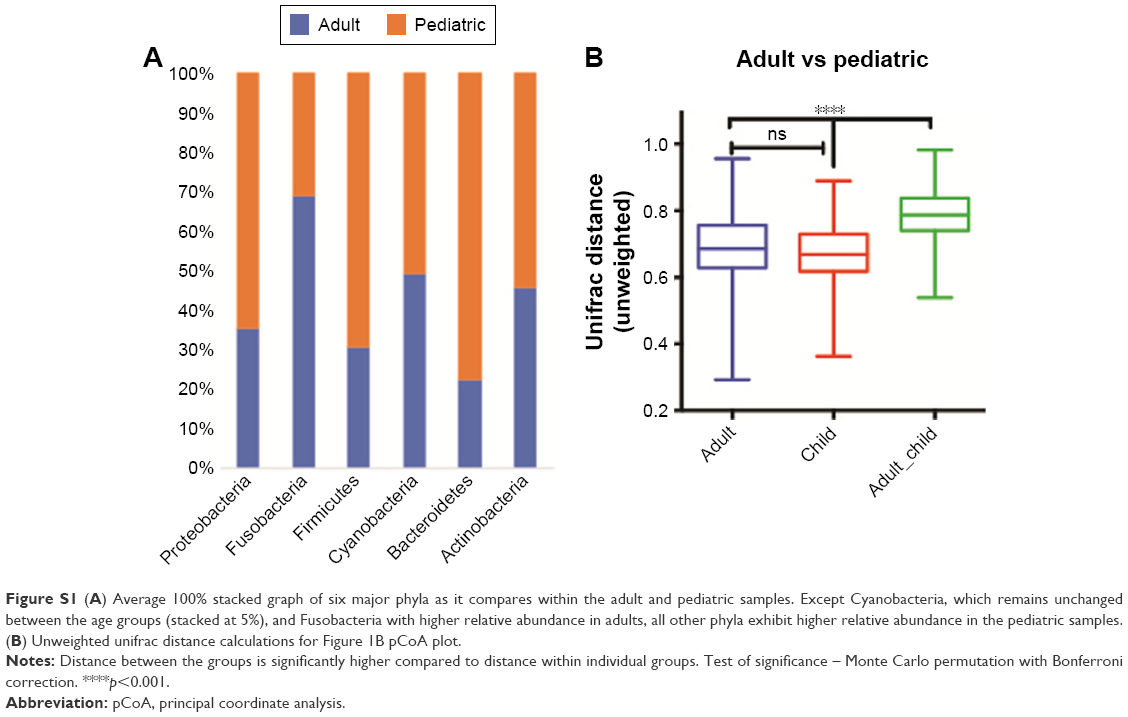 | Figure S1 (A) Average 100% stacked graph of six major phyla as it compares within the adult and pediatric samples. Except Cyanobacteria, which remains unchanged between the age groups (stacked at 5%), and Fusobacteria with higher relative abundance in adults, all other phyla exhibit higher relative abundance in the pediatric samples. (B) Unweighted unifrac distance calculations for Figure 1B pCoA plot. |
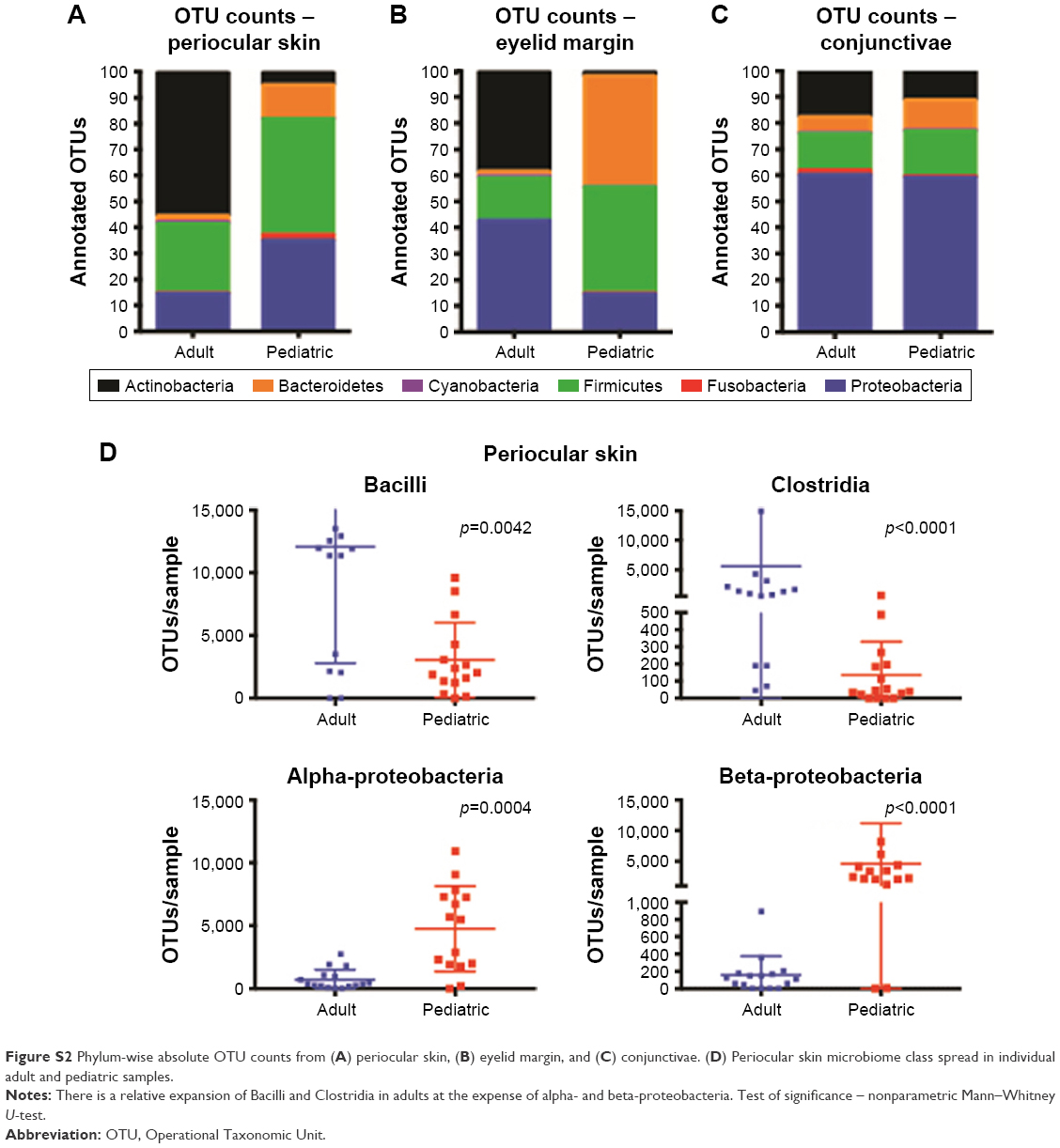 | Figure S2 Phylum-wise absolute OTU counts from (A) periocular skin, (B) eyelid margin, and (C) conjunctivae. (D) Periocular skin microbiome class spread in individual adult and pediatric samples. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.