Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 8
Economic evaluations of personalized medicine: existing challenges and current developments
Authors Shabaruddin F, Fleeman N, Payne K
Received 30 April 2014
Accepted for publication 11 March 2015
Published 24 June 2015 Volume 2015:8 Pages 115—126
DOI https://doi.org/10.2147/PGPM.S35063
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Fatiha H Shabaruddin,1 Nigel D Fleeman,2 Katherine Payne3
1Department of Pharmacy, University of Malaya, Kuala Lumpur, Malaysia; 2Liverpool Reviews and Implementation Group (LRiG), University of Liverpool, Liverpool, UK; 3Institute of Population Health, The University of Manchester, Manchester, UK
Abstract: Personalized medicine, with the aim of safely, effectively, and cost-effectively targeting treatment to a prespecified patient population, has always been a long-time goal within health care. It is often argued that personalizing treatment will inevitably improve clinical outcomes for patients and help achieve more effective use of health care resources. Demand is increasing for demonstrable evidence of clinical and cost-effectiveness to support the use of personalized medicine in health care. This paper begins with an overview of the existing challenges in conducting economic evaluations of genetics- and genomics-targeted technologies, as an example of personalized medicine. Our paper illustrates the complexity of the challenges faced by these technologies by highlighting the variations in the issues faced by diagnostic tests for somatic variations, generally referring to genetic variation in a tumor, and germline variations, generally referring to inherited genetic variation in enzymes involved in drug metabolic pathways. These tests are typically aimed at stratifying patient populations into subgroups on the basis of clinical effectiveness (response) or safety (avoidance of adverse events). The paper summarizes the data requirements for economic evaluations of genetics and genomics-based technologies while outlining that the main challenges relating to data requirements revolve around the availability and quality of existing data. We conclude by discussing current developments aimed to address the challenges of assessing the cost-effectiveness of genetics and genomics-based technologies, which revolve around two central issues that are interlinked: the need to adapt available evaluation methods and identifying who is responsible for generating evidence for these technologies.
Keywords: pharmacogenetics, pharmacogenomics, cost-effectiveness, economic evaluation, somatic variations, germline variations
Introduction
Personalized medicine, with the aim of safely, effectively, and cost-effectively targeting treatment to a prespecified patient population, has always been a long-time goal within health care. While there are many definitions of the term, the concept of personalized medicine revolves around a central theme relating to the use of combined knowledge (genetics or otherwise) to predict disease susceptibility, disease prognosis, or treatment response of a person to improve the person’s health.1 Progress made in the development of personalized medicine in recent decades has coincided with health care systems placing greater emphasis on evidence-based clinical practice, particularly as they are operating within an increasingly budget-scarce environment. It is often argued that personalizing treatment will inevitably improve clinical outcomes for patients and help achieve more effective use of health care resources. Hence, demand is increasing for demonstrable evidence of clinical and cost-effectiveness to support the use of personalized medicine in health care.
It is increasingly recognized that there is a need to apply the principles and framework of economic evaluation to conduct a comparative analysis of both the costs and outcomes of technologies used to personalize medicine, with the aim of providing sufficiently robust evidence2,3 for decision-makers entrusted with allocating health care budgets.4,5 The demand for such evidence by health care decision-makers has led to widespread publication of economic evaluations of health care technologies in numerous academic journals of assorted medical specialties, including a number of the journals focusing on genetics- and genomics-based technologies used to target medicines. Several systematic reviews of economic evaluations of technologies used to personalize medicine are summarized in Table 1. The economic evaluations identified by these reviews found that many of the technologies assessed were cost-effective, with very few being cost-saving. All reviews highlighted the need to improve the quality of the evidence base, particularly the cost and outcome data used to populate economic evaluations. Nevertheless, the focus of the two most recent reviews6,7 on economic evaluations that utilized quality adjusted life years (QALYs), the preferred outcome for economic evaluations in several jurisdictions,8,9 suggested that the quality of the evidence base may be improving.
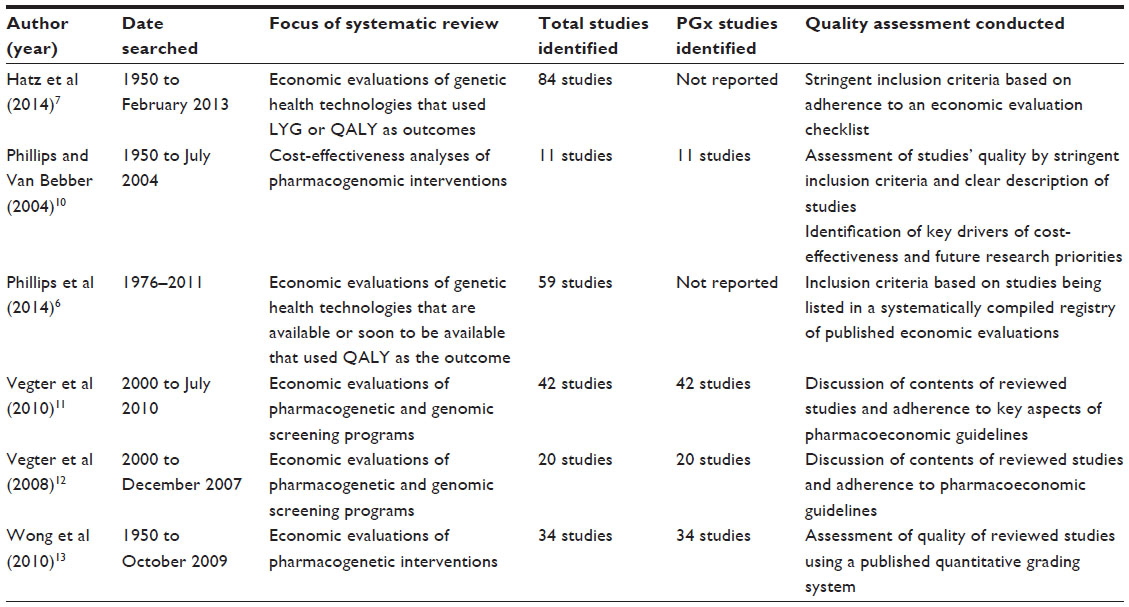 | Table 1 Systematic reviews of economic evaluations of genetics- and genomics-based technologies |
In this paper, we aim to present an overview of the existing challenges and current developments in conducting economic evaluations of genetics- and genomics-targeted technologies (hereafter called pharmacogenetic technologies) as an example of personalized medicine. Several recent reviews have explored different aspects of the challenges affecting the evaluation of pharmacogenetics. Phillips et al14 presented a detailed overview of various aspects relating to economic evaluations of personalized medicine, and highlighted six areas for future research, ranging from applying existing methods to assessments of personalized medicine to developments of new conceptual frameworks and methods specific to these technologies. Godman et al15 discussed current knowledge of the value of personalized medicine and concerns relating to personalized medicine from a payer’s perspective. Annemans et al16 and Buchanan et al17 separately explored methodological challenges of conducting economic evaluations of genomic interventions, and discussed possible solutions to some of these issues. Our paper takes another approach in illustrating the complexity of the challenges faced by pharmacogenetics by highlighting the variations in the issues faced by genetics- and genomics-based companion diagnostic tests for somatic variations, generally referring to genetic variation in a tumor, and germline variations, generally referring to inherited genetic variation in enzymes involved in drug metabolic pathways. Such tests are typically aimed at stratifying patient populations into subgroups on the basis of clinical effectiveness (response) or safety (avoidance of adverse events). Our paper places particular emphasis on technologies used to prevent adverse drug events as they are seldom considered in the literature.18
Pharmacogenetic technologies
Pharmacogenetic technologies can be broadly split into two types of tests with each type using a companion diagnostic to either identify: 1) somatic variations of a disease, for example tumors, or 2) germline variations in individuals, for example, drug metabolizing enzymes. Some examples of companion genetics-based diagnostics currently available for use in practice are tabulated in Table 2. As these examples show, companion diagnostics used to identify somatic variations are generally used to target treatment in terms of clinical response and companion diagnostics used to identify germline variations commonly target medicines administered to specific populations with the aim of minimizing adverse events.
 | Table 2 Selected examples of genetics-based companion diagnostics |
Challenges for economic evaluations of pharmacogenetics
The introduction and use of economic evaluations in health care decision-making historically centered on the assessment of medicines.20 The source of data for an economic evaluation typically comes from either a single trial or a multitude of sources combined within a model-based economic evaluation. A trial-based economic evaluation incorporates data collection of clinical effectiveness, health status, and resource use data into the clinical trial protocol, with the data collected from the trial population. A model-based economic evaluation is based on a systematic compilation of data from many different sources within a single framework, such as evidence of clinical effectiveness, utility and cost data from the literature, practice-based data sources (such as audits and observational studies), and expert opinion. The “gold standard” for the compilation of data for economic models is via systematic reviews of published evidence.21 While the basic framework for economic evaluations of pharmacogenetic technologies is similar to medicines, some specific issues and challenges can be identified and assessed based on economic evaluation checklists.9,22
Defining the study question
An economic evaluation is defined by the study question and the distinct scope of the study. This aspect incorporates the need to clearly describe every characteristic of the intervention being assessed, including descriptions of the intervention, the role and place of the intervention within clinical pathways, the target patient population and the comparator(s). Defining the precise nature of a pharmacogenetic technology can be particularly problematic as the test is not the intervention by itself. Instead, the test is a tool to assist decision-making with regard to future health care, with future clinical pathways largely depending on the results from the test. Testing for a genetic variation of interest can often be conducted using a variety of genotyping methods, with each method having different test characteristics such as test sensitivity, specificity, reproducibility, and precision.23,24 For example, CYP2D6 genotyping can be conducted using a large number of different assays, many of which can be in-house laboratory tests and measuring assorted CYP2D6 variations. This ambiguity as to the exact nature of the intervention (and crucially, therefore, convincing clinical evidence for “the intervention”) actually resulted in a planned economic evaluation being precluded from a health technology assessment of CYP2D6 pharmacogenetic testing.25
Establishing clinical pathways
Identifying the place of a pharmacogenetic test within care pathways is crucial, not only to guide the selection of a relevant comparator (which is usually current standard care in economic evaluations conducted for health technology assessments), but also to guide the use of the companion drug and subsequent treatment pathways to be modeled. Human epidermal growth factor receptor 2 (HER2) testing presents a good example of this. HER2 testing was first developed to assist in the targeting of trastuzumab.26 When first introduced, HER2 testing was conducted only in the metastatic setting27 as clinical evidence only supported the use of trastuzumab in patients with metastatic breast cancer. Testing is now conducted in patients with early stage breast cancer due to the emergence of evidence supporting the use of trastuzumab in both adjuvant and metastatic settings.26,28,29 The exact place along the treatment pathway where testing occurs may change the cost-effectiveness of the intervention because of differences in the type of treatment subsequently received and the costs and outcomes arising from these.
Somatic versus germline testing
HER2 testing is an example of a somatic test. The challenges for establishing an evidence base are arguably greater for germline tests than somatic tests, particularly when germline polymorphisms simultaneously affect the metabolism of various drugs for different diseases; CYP2D6 is one such example. Pharmacogenetic profiles obtained from CYP2D6 tests can be used in multiple patient populations to inform prescribing of various medications for a range of clinical indications.30,31 An economic evaluation needs to be conducted to assess the cost-effectiveness of each specific use of CYP2D6 testing in each specific patient population. The defined scope of an economic evaluation will not consider the added costs and potential benefits that may be incurred from CYP2D6 testing for all other indications that can potentially occur in an individual’s lifetime. It can be argued that one advantage of a test that affects several conditions is that the test need be conducted only once over an individual’s lifetime. However, this fails to consider that tests evolve over time and that clinically important polymorphisms may only become apparent over time, requiring additional, more accurate, genotyping in the future. As tests become more sophisticated so as to be able to concurrently test for multiple genetic polymorphisms,16 defining the study question will become much more complicated, particularly for pharmacogenetic tests of germline polymorphisms.
Data requirements for an economic evaluation
Key data needed for an economic evaluation of a pharmacogenetic test include outcome data on the clinical effectiveness and utility of the technology, changes in health status as well as resource use and related costs of the affected patient population and the uptake of the test. Fundamentally, the challenges relating to the data requirements for the economic analysis of a pharmacogenetic intervention revolve around the availability and quality of existing data.
There is also a need to consider the current lack of agreement on the appropriate methods and parameters to be used when including adverse events in an economic model32 which leaves analysts to decide by themselves on which adverse event parameters to include, with key options being cost, utility, and clinical parameters. These variations in economic modeling practice can directly affect the relative cost-effectiveness of pharmacogenetic technologies that aim to reduce adverse events, where the incremental gains in outcomes are based on reducing adverse events by stratifying patients into subgroups based on their predicted risk. There is a growing need to standardize how adverse event parameters are incorporated in economic models. The main parameters that should be considered for standard inclusion are the frequency of adverse events and their clinical impact on patient outcomes, changes in health status, resource use, and downstream costs due to the adverse events and subsequent changes in clinical pathways.33 Exempting any of these parameters may significantly affect the findings of an economic evaluation, which may subsequently limit the relevance of the analysis for use in health care decision-making.
Clinical effectiveness
There are currently few prospective studies conducted of pharmacogenetics10,11,13 and this paucity has had a large impact on the quality of evidence available to support the use of pharmacogenetics in clinical practice. To understand the reasons for the scarcity of prospective pharmacogenetic studies, it is essential to acknowledge the challenges that are inherent to assessing any health care intervention that relies on stratifying patients into treatment subgroups. Any study involving multiple subgroups needs a sufficiently large sample size to demonstrate significant differences in patient outcomes.34 Conducting multicenter and multinational studies is one potential solution to ensure a trial has sufficient power to show statistically significant differences in patient outcomes but the cost of such studies may be prohibitively high.
These challenges are often more overwhelming when evaluating pharmacogenetics that aim to reduce adverse events compared to those that predict response. A part of the issue relates to the number of subgroups that patients need to be stratified into. Tests for somatic variations that predict response often only need to stratify patients into two groups, consisting of responders and non-responders, usually based on the presence of the polymorphism of interest that predicts the susceptibility of the disease to the companion drug. On the other hand, tests for germline variations to predict adverse events often first require evidence linking genotype to phenotype, which subsequently inform patient stratification and the number of relevant subgroups involved that are usually determined by findings from observational cohort studies linking pharmacogenetic variations and patient outcomes.35–37 The challenges encountered while assessing pharmacogenetics that predict adverse events are also linked to the frequency at which the adverse event of interest occurs.34 While response (or the lack of it) can be measured in every patient, certain adverse events only affect a certain proportion of patients within a study, thus emphasizing the need for studies with large sample sizes.
Ideally, the clinical outcomes of effectiveness should be real clinical end-points instead of surrogate or intermediate outcomes.38 However, particularly in pharmacogenetics, surrogate outcomes may be more common.39 To illustrate this challenge, consider the examples of TPMT and UGT1A1 testing, for which the clinical adverse event of interest is neutropenia represented by hematological laboratory findings and an increased risk of infection. While neutropenia predisposes patients to severe and potentially fatal infection, it has no clinical implication on health status unless it is accompanied by an infection or a clinical sequelae40 and can be classified as a surrogate outcome with clinically relevant events such as febrile and septic neutropenia being the actual clinical outcomes. When surrogate outcomes are used, further evidence on the association between the surrogate outcomes and the real clinical end-points of particular relevance to the patient population should be presented to describe the true value of the intervention.41,42 However, there are scarce data on the actual proportion of neutropenic patients who develop febrile or septic neutropenia in clinical practice43 and assumptions made on the estimated proportion may subsequently weaken the evidence base needed for robust assessment of the cost-effectiveness of TPMT and UGT1A1 tests.37,43
Clinical utility
Challenges faced while determining the clinical utility of a companion diagnostic44 are underpinned by the need to show evidence that patient outcomes are improved by testing. This relies heavily on the pharmacogenetics-guided treatment options that are available once information from a pharmacogenetic test is available to inform prescribing decisions.45 Treatment options for a test that predicts response depend mainly on the alternative treatments available to nonresponders while that for a test reducing adverse events depends primarily on the availability of either an alternative treatment strategy (such as in the case of abacavir and HLA-B*5701 testing) or a pharmacogenetics-based dosing algorithm and evidence linking the alternative dosing strategy with patient outcomes (such as in the case of warfarin and CYP2C9 and VKORC1 tests).35,36
Pharmacogenetics-based dosing algorithms are currently poorly developed. The demand for guidance to utilize pharmacogenetics data in decision-making within clinical practice has led to the development of several pharmacogenetics-based dosing algorithms for warfarin based on the therapeutic range of a drug (a surrogate or intermediate outcome). Clinical data primarily based on various observational studies and some small prospective studies have supported the potential clinical utility of the genotype-guided dosing recommendations.46–49 However, recently published results of two randomized clinical trials of pharmacogenetics-based dosing of warfarin came to divergent conclusions.35,36 Recommendations for genotype-guided dosing that are not supported by robust evidence and which may later be contradicted by results from well-designed randomized controlled trials may discourage the use of pharmacogenetics within clinical practice and subsequently impair the development of the evidence base for the clinical utility of genetics-based diagnostics.
Developing evidence to inform dosing algorithms for tests that reduce adverse events is made more complicated by the lack of timely biomarkers available to assess the efficacy of the reduced doses of treatment, particularly in oncology.50–52 While there may be sufficient evidence to indicate that a pharmacogenetic marker is predictive of adverse events, pharmacogenetics-based evidence guiding the new dosage of a drug and the subsequent impact of altered doses of medicines on patients’ outcome is scarce and demonstrating clinical utility requires evidence linking the alternative dosing strategy with patient outcomes.43,44
Health status
The impact of an intervention on health status is the preferred outcome measure in economic evaluations.53 Methodological issues regarding the valuation of health outcomes for pharmacogenetics, particularly the quality-adjustment (utility) component in QALYs, are similar to those faced by other health care interventions. These issues tend to revolve around the preferred approach to use to value the quality-adjustment and associated preference weights.53 Discussions in the general literature on the challenges of valuing the disutility of adverse events and the use of choice-based stated preference methods to derive utility values of adverse events based on study-specific descriptions of a health condition (vignettes)33 are particularly relevant to the assessment of pharmacogenetic interventions that aim to reduce adverse events. The use of study-specific vignettes often leads to a lack of standardization in the evidence base due to the assorted definitions and vignettes used to describe an adverse event of interest.33 This can cause additional hurdles to the analyst, especially when choosing the utility value that is most suitable for use in an economic model. Despite this fact, questionnaires based on vignettes remain useful in assessing the utility of adverse events. A potential solution to guide the selection of the utility values obtained is to develop vignettes that are based on a clinical definition of an adverse event, instead of a study specific definition.33
Resource use and cost data
Similar to evaluations of other health care interventions, cost data used in evaluations of pharmacogenetics should ideally be based on actual resource use of the patient population of interest, either observed within a clinical trial or based on observational data. Resource use data are subsequently combined with unit cost data to generate the relevant cost data, preferably published national unit costs.53
There are however two key components of costs associated with the use of a pharmacogenetic technology: 1) the short-run downstream costs consisting of the actual testing process, interpretation of the test results, and subsequent change in prescribing, and 2) the long-run downstream costs consisting of the economic impact due to the change in prescribing and subsequent changes to care pathways as well as patient outcomes. Due to the paucity of prospective studies, and challenges in designing and funding them,10,11,13 there is generally limited actual resource use data of the patient population of interest to inform economic evaluations of pharmacogenetics.
There is also limited available information on published acquisition costs for the companion diagnostic component of a pharmacogenetic technology. Unlike medicines, where the costs are published as national price lists for the relevant jurisdictions, there are no national price lists available for genetics- and genomics-based diagnostic tests.54 Furthermore, there is likely to be a marked difference in the unit cost for pharmacogenetic tests developed by a laboratory compared with companion diagnostics that have been through a regulatory approval process and marketed as a standalone test. Large variations in the unit costs of these tests can affect the findings of an economic evaluation and increase uncertainty in the estimated relative cost-effectiveness of a test.
Uptake of pharmacogenetic tests
Potential variations in the uptake of pharmacogenetic tests and in the implementation of the test results within patients’ clinical pathways in economic evaluations are issues that are seldom considered. To date, the majority of economic evaluations have assumed perfect implementation of the test results and perfect uptake of a pharmacogenetic test.14,17 However, a UK-based pragmatic clinical trial (TARGET) assessing the use of TPMT genotyping to inform azathioprine dose selection and reduce the incidence of azathioprine-induced neutropenia37 illustrated how the results of a pharmacogenetic test are not always followed as intended. The trial found that although clinicians followed the trial’s pharmacogenetics-based dosing recommendation for patients with a heterozygous TPMT genotype by starting azathioprine at a low dose, they did not comply to the dosing recommendation for patients with a wild-type TPMT genotype. Instead of immediately starting these patients on the standard maintenance dose, patients with a wild-type TPMT genotype were still prescribed a lower starting dose that is subsequently titrated upwards over time, similar to usual clinical practice. The authors37 suggested that this could be due to the implicit concern that TPMT genotyping does not predict other azathioprine-related adverse events. This concern is likely to apply to most pharmacogenetic interventions that aim to reduce adverse events, as genetics are only one of many factors influencing variability in adverse events profile. The economic evaluation based on the TARGET trial subsequently concluded that TPMT genotyping potentially offers a less expensive alternative than current practice, but it may also have a small but negative effect on health status.55 This example illustrates the need for careful consideration when estimating the uptake of pharmacogenetics within clinical practice, particularly in model-based economic evaluations.
Current developments in the assessment of pharmacogenetics
Current developments to address the challenges of assessing the cost-effectiveness of pharmacogenetic technologies revolve around two central issues that are interlinked: the need to adapt available evaluation methods and identifying who is responsible for generating evidence for pharmacogenetics.
An iterative approach to evaluation
It has been said that the prospects for personalized medicine do not obviate the need for evaluation; they change the kind of evaluation that is needed.56 Current trends observed in the literature illustrate that this is already happening,10,11,13 with most assessments of pharmacogenetics being model-type evaluations in response to the scarcity of trial-based data for pharmacogenetics. The general paucity of outcome, health status, and cost data specific to pharmacogenetic interventions means that it may be necessary in the early developmental stages to design and populate relatively simple economic models as opposed to more complex models.16 An iterative approach could then be employed that systematically and explicitly considers the need for further evidence to reduce decision uncertainty,57 and is consistent with an approach to health technology assessment (HTA) known as constructive technology assessment.58 An iterative approach may enable decision-makers to prioritize both clinical and cost-effectiveness research and allow for timely decision-making with regard to the use of companion diagnostics. Efforts to populate simple economic models in the presence of limited evidence may necessitate the use of nontraditional and alternative sources of data, including methodically documented and reported expert opinion.59 There are assorted elicitation methods relevant to economic modeling, such as the use of surveys60 or Delphi methods,61 to describe patient care pathways for the model structure and mathematical aggregation methods to populate model parameters.62,63 The iterative approach can be accompanied by value of information (Vol) analysis to provide a quantitative estimate of the value of future research to reduce current uncertainty in the evidence used to populate model parameters.64
The need for methodological research
A review by Annemans et al16 presented ten issues that pose specific methodological challenges when designing and conducting model-type economic evaluation of personalized medicine. The review16 emphasized the importance of issues relating to the correct framing of the research question, interpretation of test results, data collection of medical management options after obtaining test results, and expressing the value of tests.
A structured review of the literature by Buchanan et al17 that considered methodological challenges for economic evaluations of genomic technologies identified similar issues to the review by Annemans et al.16 While these issues may also arise in evaluations of most types of health care interventions, it is the sheer quantity and range of challenges that makes it a unique challenge for assessing pharmacogenetics.17 The review by Buchanan et al17 concluded that while new methods may be needed to assess the cost-effectiveness of genomics, there is still insufficient evidence to justify alternative approaches. In view of this and the lack of consensus on economic evaluation methods that can be used for pharmacogenetics, the authors recommend for analysts to conduct extensive and wide-ranging sensitivity analyses.17 Such analyses may consider multiple perspectives, broader ranges, types of costs, and outcomes.17 The analyses could also incorporate various scenario and structural uncertainties, such as variations in the delivery models of pharmacogenetic services.17
However, while expanding scenario and sensitivity analyses is a potential solution for the assessment of pharmacogenetics, this would result in a higher degree of uncertainty of the findings. Variations in the methods used may also hinder the comparison of cost-effectiveness evidence between pharmacogenetic technologies, negating one of the main advantages of utilizing economic evaluations, particularly with the use of QALYs as an outcome measure in health care decision-making. A review of the literature by Beaulieu et al65 noted a high degree of heterogeneity between studies assessing the same pharmacogenetic test and suggested a list of pharmacogenetic-specific parameters that should be consistently included in economic evaluation of pharmacogenetics, such as prevalence of the biomarker, outcome of genetics-based treatment as well as cost of genomic data collection and analysis. The concern that different methodological approaches may lead to conflicting adopting decisions17 also persists and in the long-term, it is still necessary to simultaneously focus on developing methods applicable to pharmacogenetics while ensuring policy makers are clear as to which evaluation methods are preferred to assist decision-making.
There is thus a need for realistic expectations regarding the standards66 and type of clinical evidence needed for reimbursement. To date, a common method employed is retrospective analyses of trial data.67 However, such analyses are only possible where data or samples from trials exist (such as from tissues of patients) and can then be analyzed retrospectively. Clinical information obtained prospectively is usually of a higher quality than retrospective data not least because it is not always feasible to conduct tests retrospectively. In addition, trials need to be sufficiently large, and it should be noted that selection bias may occur where there is missing data. Potential alternative solutions may involve the use of novel trial designs, such as adaptive clinical trials,68,69 evidence from observational studies, and clinical practice.45,66
Generating clinical evidence to populate models
The lack of relevant clinical data to populate model-based economic evaluations of pharmacogenetic tests is cited to be the main barrier to the introduction of personalized medicine into clinical practice.70,71 Adaptive clinical trials have been suggested as a means of speeding up the development of new interventions by reducing the size of the clinical trial by enabling a flexible trial design.68,69 In general, adaptive clinical trial methods are accepted in the exploratory stages of clinical trials.72
The most widely understood adaptive trial design is the group sequential design that allows stopping a trial early if it becomes clear that a treatment is superior or inferior; such a design is accepted as a valid trial design by some regulators, such as the United States Food and Drug Administration (FDA).72 However, many other novel approaches also exist, such as adaptive randomization.68,69 When used properly, it is argued that adaptive clinical trials can result in a more efficient treatment development process, and an increased chance of correctly answering the clinical question of interest. However, improper adaptations can lead to biased studies although it is argued that logistical issues and regulatory concerns, rather than statistical issues, currently limit the use of such trials.68,69 For example, such trials require a different way of thinking about the structure and conduct of Data and Safety Monitoring Boards.68,69
There is also a movement toward using observational studies rather than randomized trials. The Clinical Pharmacogenetics Implementation Consortium (CPIC), based in the United States, develops peer reviewed guidelines on how to use available pharmacogenetic test results in order to optimize treatment based on evidence derived from patients’ medical records. However, the CPIC does not address the crucial question of whether a pharmacogenetic test should or should not be ordered.45 Currently, there are approximately 30 gene-drug pairs with CPIC dosing guidelines.73 The selection of gene-drug pairs is based on several factors, including surveys of experts in the relevant fields, the availability of strong evidence linking drug response or adverse events with genetic variations, clinically actionable prescribing recommendations, alternative therapeutic strategies based on pharmacogenetics, and the actual pharmacogenetic tests for use within clinical practice.45
While the development of CPIC guidelines is a positive step toward moving pharmacogenetics-based individualized treatment into routine clinical practice, it may be argued that they also imply that existing challenges in assessing pharmacogenetics are insurmountable. This is because these guidelines bypass the traditional data requirements needed to support the use of a new intervention and move straight to producing recommendations for its use in clinical practice. Even with the implementation of CPIC guidelines, there may still be insufficient evidence on the clinical and cost-effectiveness of interventions upon which policy makers can make reimbursement decisions.74 The widespread use of any health care intervention depends on its availability within clinical practice. This is typically linked to reimbursement decisions by health care payers. Hence, the CPIC guidelines offer only an alternative in the unusual circumstances where a test’s use is widespread in practice despite lacking a robust evidence base for its effectiveness. This highlights a pressing need to boost the generation of a robust evidence base for pharmacogenetics by alternative means that can satisfy the demands of health care payers.
Responsibility for generating evidence for pharmacogenetics
The derivation of an economic evidence base sufficient to inform health care resource allocation decision has a clear opportunity cost in terms of alternative uses of a research budget.75 It is necessary to be clear who is responsible for generating the evidence required. For medicines, there are clear regulatory and reimbursement systems in place and pharmaceutical companies are aware that they are expected to derive good quality evidence from RCTs to comply with the requirements set by regulators, reimbursement agencies, or national bodies that inform health care funding decisions.76–79 In contrast, there is substantial uncertainty about the level of evidence considered to be sufficient to support the use of pharmacogenetic technologies and in most instances there is also uncertainty as to who is responsible for producing such evidence, particularly the types of evidence on cost-effectiveness that are needed by health care payers and policy makers.80
The task of generating a robust evidence base for pharmacogenetics need not be shouldered by a single sector and can be shared with pharmaceutical companies, which are now increasingly aware of the advantages of developing tests for somatic mutations alongside drug development. A 2007 survey of 16 of the top 20 pharmaceutical companies conducted by McKinsey indicated that on average, up to 50% of drugs in development have an associated biomarker program,81 a finding also found from a separate survey conducted by the Tufts Center for the Study of Drug Development.82 However, the findings by McKinsey also indicated that fewer than 10% of drugs would be launched with a companion diagnostic over the next 5–10 years.81 The authors suggested that this finding largely reflected the benefits and risks faced by pharmaceutical companies when adopting a companion diagnostic strategy.81 Potential benefits cited included increased productivity by decreasing trial sizes, reducing attrition, or increasing speed to market, as well as enhancing commercial performance by boosting market share or supporting higher drug prices.81 Deverka et al83 described how the pharmaceutical company (Roche) that manufactures trastuzumab found that introducing HER2 testing into its pivotal clinical trial reduced development costs (by an estimated US$35 million), trial size (from 2,200 to 470 patients), and the time it took to bring the product to the market (from a projected 10 years to 1.6 years). However, these observations83 from one specific example may not be generalizable to other scenarios. There are potential risks to having an associated personalized medicine program,81 including delays in study development, increased costs, and a decrease in market share caused by the identification of a more specific patient population based on pharmacogenetics.
The generation of an evidence base to inform the reimbursement of pharmacogenetic technologies that involve a companion diagnostic to identify germline variations, such as variations in drug metabolizing enzymes, is likely to be more problematic than for somatic variations. This is because the majority of such pharmacogenetic tests are used to target existing drugs that have been on the market for a number of years and as such, are off patent.73 Another issue is that many of the pharmacogenetic tests for germline variations can be used to inform prescribing decisions of multiple drugs, such as CYP2D6 genotyping.30,31 In such situations, it is not clear who might be responsible (or indeed have the incentive) for generating the required evidence base. The implications of the current paucity of data are showcased by the examples of two HTAs that aimed to conduct model-based economic evaluations of CYP2D6 testing for tamoxifen84 and CYP450 testing of anti-psychotic medications.25 Both HTAs were unable to produce an assessment of cost-effectiveness due to a lack of evidence for clinical effectiveness (which mainly comprised retrospective studies) and consequently, cost-effectiveness.
Current policies of most regulatory and reimbursement systems are not resulting in the compilation of sufficient evidence on the value of identifying responders or patients likely to experience adverse events.66 This situation points to a need to incentivize the generation of evidence for technologies used to personalize medicine.66,85,86 Previous suggestions for economic incentives for evidence generation include value-based price flexibility, intellectual property protection for the evidence generated, realistic expectations for the standards of evidence needed, and public investment to complement the effort of payers and manufacturers.66 Public funding bodies, such as the Medical Research Council in the UK,87 have addressed the lack of evidence by setting up extensive funding streams for stratified medicine, looking specifically at existing medicines. Research funding streams such as these should change the future landscape in terms of the available evidence to support personalized medicine. This increase in available funding must be matched with an associated improvement in the design and conduct of studies to generate an evidence base sufficient to inform whether scarce health care resources should be diverted toward genetics- and genomics-based technologies.
Concluding remarks
Identifying and quantifying the incremental costs and benefits of technologies used to personalize medicine using robust economic evaluations has a key role in the context of health care systems needing to decide how best to allocate scarce health care resources. The discipline of health economics provides methods of economic evaluation to generate evidence of the relative cost-effectiveness of genetics and genomics-based technologies compared with current clinical practice. As these technologies continue to develop, and become more complex (such as with an increasing number of gene variations tested for at a time and able to inform prescribing of multiple drugs), there is a need to constantly develop new evaluative methods and evidence of clinical and cost-effectiveness that is sufficient for timely and informed resource allocation decision-making. However, as the technologies become increasingly complex, the derivation of such evidence also becomes more complex. In this paper, we have highlighted some of the main challenges and presented current developments in addressing these issues.
Decision-makers need to recognize that the development of evidence has substantial implications for the effective use of resources and hence there is a need for economic incentives to support the generation of a robust evidence base for personalized medicine. Resolving methodological issues relating to the derivation of evidence for, and assessment of, genetics and genomics-based technologies and identifying who is responsible for generating evidence are integral to support the reimbursement of genetics and genomics-based tests by health care payers and thus move personalized medicine from bench to bedside.
Disclosure
The authors report no conflicts of interest in this work.
References
Redekop WK, Mladsi D. The faces of personalized medicine: a framework for understanding its meaning and scope. Value Health. 2013; 16(6):S4–S9. | |
Cooper NJ, Sutton AJ, Ades AE, Paisley S, Jones DR. Use of evidence in economic decision models: practical issues and methodological challenges. Health Econ. 2007;16(12):1277–1286. | |
Claxton K. Exploring uncertainty in cost-effectiveness analysis. Pharmacoeconomics. 2008;26(9):781–798. | |
Conti R, Veenstra DL, Armstrong K, Lesko LJ, Grosse SD. Personalized medicine and genomics: challenges and opportunities in assessing effectiveness, cost-effectiveness, and future research priorities. Med Decis Making. 2010;30(3):328–340. | |
Phillips KA, Van Bebber SL. Measuring the value of pharmacogenomics. Nat Rev Drug Discov. 2005;4(6):500–509. | |
Phillips KA, Sakowski JA, Trosman J, Douglas MP, Liang SY, Neumann P. The economic value of personalized medicine tests: what we know and what we need to know. Genet Med. 2014;16(3):251–257. | |
Hatz MH, Schremser K, Rogowski WH. Is individualized medicine more cost-effective? A systematic review. Pharmacoeconomics. 2014; 32(5):443–455. | |
International Society for Pharmacoeconomics and Outcomes Research. Pharmacoeconomic Guidelines Around the World; 2015. Available from: http://www.ispor.org/peguidelines/index.asp. Accessed February 26, 2015. | |
National Institute for Health and Care Excellence (NICE). Guide to the Methods of Technology Appraisal; 2013. Available from: http://www.nice.org.uk/media/D45/1E/GuideToMethodsTechnologyAppraisal 2013.pdf. Accessed May 1, 2014. | |
Phillips KA, Van Bebber SL. A systematic review of cost-effectiveness analyses of pharmacogenomic interventions. Pharmacogenomics. 2004;5(8):1139–1149. | |
Vegter S, Jansen E, Postma MJ, Boersma C. Economic evaluations of pharmacogenetic and genomic screening programs: update of the literature. Drug Dev Res. 2010;71(8):492–501. | |
Vegter S, Boersma C, Rozenbaum M, Wilffert B, Navis G, Postma MJ. Pharmacoeconomic evaluations of pharmacogenetic and genomic screening programmes: a systematic review on content and adherence to guidelines. Pharmacoeconomics. 2008;26(7):569–587. | |
Wong WB, Carlson JJ, Thariani R, Veenstra DL. Cost effectiveness of pharmacogenomics: a critical and systematic review. Pharmacoeconomics. 2010;28(11):1001–1013. | |
Phillips KA, Sakowski JA, Liang SY, Ponce NA. Economic perspectives on personalized health care and prevention. Forum Health Econ Policy. 2013;16(2):30. | |
Godman B, Finlayson AE, Cheema PK, et al. Personalizing health care: feasibility and future implications. BMC Med. 2013;11:179. | |
Annemans L, Redekop K, Payne K. Current methodological issues in the economic assessment of personalized medicine. Value Health. 2013;16(6):S20–S26. | |
Buchanan J, Wordsworth S, Schuh A. Issues surrounding the health economic evaluation of genomic technologies. Pharmacogenomics. 2013;14(15):1833–1847. | |
Craig D, McDaid C, Fonseca T, Stock C, Duffy S, Woolacott N. Are adverse effects incorporated in economic models? A survey of current practice. Int J Technol Assess Health Care. 2010;26(3):323–329. | |
US Food and Drug Administration. Pharmacogenomic Biomarkers in Drug Labeling; 2014. Available from: http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.htm. Accessed May 1, 2014. | |
Williams IM, McIver S, Moore D, Bryan S. The use of economic evaluations in NHS decision-making: a review and empirical investigation. Health Technol Assess. 2008;12(7):1–175. | |
Centre for Reviews and Dissemination. NHS Economic Evaluation Database Handbook; 2007. Available from: http://www.york.ac.uk/inst/crd/pdf/nhseed-handbook2007.pdf. Accessed May 1, 2014. | |
Husereau D, Drummond M, Petrou S, et al; CHEERS Task Force. Consolidated health economic evaluation reporting standards (CHEERS) statement. BMJ. 2013;346:f1049. | |
Isler JA, Vesterqvist OE, Burczynski ME. Analytical validation of genotyping assays in the biomarker laboratory. Pharmacogenomics. 2007;8(4):353–368. | |
Weiss ST, McLeod HL, Flockhart DA, et al. Creating and evaluating genetic tests predictive of drug response. Nat Rev Drug Discov. 2008;7(7):568–574. | |
Fleeman N, Martin Saborido C, Payne K, et al. The clinical effectiveness and cost-effectiveness of genotyping for CYP2D6 for the management of women with breast cancer treated with tamoxifen: a systematic review. Health Technol Assess. 2011;15(33):1–102. | |
Ferrusi IL, Marshall DA, Kulin NA, Leighl NB, Phillips KA. Looking back at 10 years of trastuzumab therapy: what is the role of HER2 testing? A systematic review of health economic analyses. Per Med. 2009;6(2):193–215. | |
Elkin EB, Weinstein MC, Winer EP, Kuntz KM, Schnitt SJ, Weeks JC. HER-2 testing and trastuzumab therapy for metastatic breast cancer: a cost-effectiveness analysis. J Clin Oncol. 2004;22(5):854–863. | |
Blank PR, Schwenkglenks M, Moch H, Szucs TD. Human epidermal growth factor receptor 2 expression in early breast cancer patients: a Swiss cost-effectiveness analysis of different predictive assay strategies. Breast Cancer Res Treat. 2010;124(2):497–507. | |
Parkinson B, Pearson SA, Viney R. Economic evaluations of trastuzumab in HER2-positive metastatic breast cancer: a systematic review and critique. Eur J Health Econ. 2014;15(1):93–112. | |
Hicks JK, Swen JJ, Thorn CF, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin Pharmacol Ther. 2013;93(5):402–408. | |
Crews KR, Gaedigk A, Dunnenberger HM, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for codeine therapy in the context of cytochrome P450 2D6 (CYP2D6) genotype. Clin Pharmacol Ther. 2012;91(2):321–326. | |
Craig D, McDaid C, Fonseca T, Stock C, Duffy S, Woolacott N. Are adverse effects incorporated in economic models? An initial review of current practice. Health Technol Assess. 2009;13(62):1–71, 97–181. | |
Shabaruddin FH, Chen LC, Elliott RA, Payne K. A systematic review of utility values for chemotherapy-related adverse events. Pharmacoeconomics. 2013;31(4):277–288. | |
Noordzij M, Tripepi G, Dekker FW, Zoccali C, Tanck MW, Jager KJ. Sample size calculations: basic principles and common pitfalls. Nephrol Dial Transplant. 2010;25(5):1388–1393. | |
Pirmohamed M, Burnside G, Eriksson N, et al; EU-PACT Group. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med. 2013;369(24):2294–2303. | |
Kimmel SE, French B, Kasner SE, et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med. 2013;369(24):2283–2293. | |
Newman WG, Payne K, Tricker K, et al; TARGET Study Recruitment Team. A pragmatic randomized controlled trial of thiopurine methyltransferase genotyping prior to azathioprine treatment: the TARGET study. Pharmacogenomics. 2011;12(6):815–826. | |
Svensson S, Menkes DB, Lexchin J. Surrogate outcomes in clinical trials: a cautionary tale. JAMA Intern Med. 2013;173(8):611–612. | |
Sorich MJ, Rowland A, Wiese MD. Pharmacogenomic substudies of randomized controlled trials: consideration of safety outcomes. Ther Adv Drug Saf. 2014;5(2):62–66. | |
US National Institutes of Health; National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE); 2013. Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm. Accessed February 25, 2015. | |
Fleming TR, Powers JH. Biomarkers and surrogate endpoints in clinical trials. Stat Med. 2012;31(25):2973–2984. | |
Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996;125(7):605–613. | |
Shabaruddin FH. Economic Evaluation of the UGT1A1 Pharmacogenetic Test [dissertation]. Manchester, UK: The University of Manchester; 2011. | |
Sorich MJ, Coory M. Interpreting the clinical utility of a pharmacogenomic marker based on observational association studies. Pharmacogenomics J. 2014;14(1):1–5. | |
Caudle KE, Klein TE, Hoffman JM, et al. Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr Drug Metab. 2014;15(2):209–217. | |
Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol. 2010;55(25):2804–2812. | |
Johnson JA, Gong L, Whirl-Carrillo M, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther. 2011;90(4):625–629. | |
Anderson JL, Horne BD, Stevens SM, et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II). Circulation. 2012;125(16):1997–2005. | |
Burmester JK, Berg RL, Yale SH, et al. A randomized controlled trial of genotype-based Coumadin initiation. Genet Med. 2011;13(6):509–518. | |
Mayers C, Panzarella T, Tannock IF. Analysis of the prognostic effects of inclusion in a clinical trial and of myelosuppression on survival after adjuvant chemotherapy for breast carcinoma. Cancer. 2001;91(12):2246–2257. | |
Cameron DA, Massie C, Kerr G, Leonard RC. Moderate neutropenia with adjuvant CMF confers improved survival in early breast cancer. Br J Cancer. 2003;89(10):1837–1842. | |
Tinker AV, Speers C, Barnett J, Olivotto IA, Chia S. Impact of a reduced dose intensity of adjuvant anthracycline based chemotherapy in a population-based cohort of stage I-II breast cancers. Ecancermedicalscience. 2008;2:63. | |
Drummond MF, Sculpher M, Torrance G, O’Brien BJ, Stoddart G. Methods for the Economic Evaluation of Health Care Programmes. Oxford, UK: Oxford University Press; 2005. | |
Payne K. Fish and chips all round? Regulation of DNA-based genetic diagnostics. Health Econ. 2009;18(11):1233–1236. | |
Thompson AJ, Newman WG, Elliott RA, Roberts SA, Tricker K, Payne K. The cost-effectiveness of a pharmacogenetic test: a trial-based evaluation of TPMT genotyping for azathioprine. Value Health. 2014;17(1):22–33. | |
Garber AM, Tunis SR. Does comparative-effectiveness research threaten personalized medicine? N Engl J Med. 2009;360(19):1925–1927. | |
Sculpher M, Drummond M, Buxton M. The iterative use of economic evaluation as part of the process of health technology assessment. J Health Serv Res Policy. 1997;2(1):26–30. | |
Douma KF, Karsenberg K, Hummel MJ, Bueno-de-Mesquita JM, van Harten WH. Methodology of constructive technology assessment in health care. Int J Technol Assess Health Care. 2007;23(2):162–168. | |
Philips Z, Bojke L, Sculpher M, Claxton K, Golder S. Good practice guidelines for decision-analytic modelling in health technology assessment: a review and consolidation of quality assessment. Pharmacoeconomics. 2006;24(4):355–371. | |
Shabaruddin FH, Elliott RA, Valle JW, Newman WG, Payne K. Understanding chemotherapy treatment pathways of advanced colorectal cancer patients to inform an economic evaluation in the United Kingdom. Br J Cancer. 2010;103(3):315–323. | |
Sullivan W, Payne K. The appropriate elicitation of expert opinion in economic models: making expert data fit for purpose. Pharmacoeconomics. 2011;29(6):455–459. | |
Leal J, Wordsworth S, Legood R, Blair E. Eliciting expert opinion for economic models: an applied example. Value Health. 2007;10(3):195–203. | |
O’Hagan A, Buck CE, Daneshkhah A, et al. Uncertain Judgements: Eliciting Experts’ Probabilities (Statistics in Practice). Chichester: Wiley; 2006. | |
Sculpher M, Claxton K. Establishing the cost-effectiveness of new pharmaceuticals under conditions of uncertainty – when is there sufficient evidence? Value Health. 2005;8(4):433–446. | |
Beaulieu M, de Denus S, Lachaine J. Systematic review of pharmacoeconomic studies of pharmacogenomic tests. Pharmacogenomics. 2010;11(11):1573–1590. | |
Towse A, Garrison LP Jr. Economic incentives for evidence generation: promoting an efficient path to personalized medicine. Value Health. 2013;16(6 Suppl):S39–S43. | |
European Medicines Agency. Reflection Paper on Methodological Issues Associated with Pharmacogenomic Biomarkers in Relation to Clinical Development and Patient Selection; 2011. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/07/WC500108672.pdf. Accessed February 23, 2015. EMA/446337/2011. | |
Coffey CS, Levin B, Clark C, et al. Overview, hurdles, and future work in adaptive designs: perspectives from a National Institutes of Health-funded workshop. Clin Trials. 2012;9(6):671–680. | |
Kairalla JA, Coffey CS, Thomann MA, Muller KE. Adaptive trial designs: a review of barriers and opportunities. Trials. 2012;13:145. | |
Newman W, Payne K. Removing barriers to a clinical pharmacogenetics service. Per Med. 2008;5(5):471–480. | |
McKinnon RA, Ward MB, Sorich MJ. A critical analysis of barriers to the clinical implementation of pharmacogenomics. Ther Clin Risk Manag. 2007;3(5):751–759. | |
US Food and Drug Administration. Draft Guidance for Industry: Adaptive Design Clinical Trials for Drugs and Biologics; 2011. Available from: http://www.fda.gov/downloads/DrugsGuidance ComplianceRegulatoryInformation/Guidances/UCM201790.pdf. Accessed February 23, 2015. | |
PharmGKB. Dosing Guidelines – CPIC; 2015. Available from: http://www.pharmgkb.org/view/dosing-guidelines.do?source=CPIC#. Accessed February 25, 2015. | |
Byron SK, Crabb N, George E, Marlow M, Newland A. The Health Technology Assessment of companion diagnostics: experience of NICE. Clin Cancer Res. 2014;20(6):1469–1476. | |
Claxton KP, Sculpher MJ. Using value of information analysis to prioritise health research: some lessons from recent UK experience. Pharmacoeconomics. 2006;24(11):1055–1068. | |
US Food and Drug Administration. FDA Basics. What is the Approval Process for a New Prescription Drug? 2014. Available from: http://www.fda.gov/AboutFDA/Transparency/Basics/ucm194949.htm. Accessed February 25, 2015. | |
Medicines and Healthcare Products Regulatory Agency. Apply for a Licence to Market a Medicine in the UK Detailed Guidance; 2014. Available from: https://www.gov.uk/apply-for-a-licence-to-market-a-medicine-in-the-uk. Accessed February 25, 2015. | |
European Medicines Agency. Human Medicines: Regulatory Information; 2015. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/landing/human_medicines_regulatory.jsp&mid=. Accessed February 25, 2015. | |
National Institute for Health and Care Excellence (NICE). Specification for Manufacturer/Sponsor Submission of Evidence; 2014. Available from: https://www.nice.org.uk/proxy/?sourceurl?http://www.nice.org.uk/aboutnice/howwework/devnicetech/specificationformanufacturersponsorsubmissionofevidence.jsp. Accessed February 25, 2015. | |
Payne K, Annemans L. Reflections on market access for personalized medicine: recommendations for Europe. Value Health. 2013;16(6): S32–S38. | |
Davis JC, Furstenthal L, Desai AA, et al. The microeconomics of personalized medicine: today’s challenge and tomorrow’s promise. Nat Rev Drug Discov. 2009;8(4):279–286. | |
Fugel HJ, Nuijten M, Postma M. Stratified medicine and reimbursement issues. Front Pharmacol. 2012;3:181. | |
Deverka PA, Vernon J, McLeod HL. Economic opportunities and challenges for pharmacogenomics. Annu Rev Pharmacol Toxicol. 2010;50:423–437. | |
Fleeman N, McLeod C, Bagust A, et al. The clinical effectiveness and cost-effectiveness of testing for cytochrome P450 polymorphisms in patients with schizophrenia treated with antipsychotics: a systematic review and economic evaluation. Health Technol Assess. 2010;14(3):1–157. | |
Willke RJ, Crown W, Del Aguila M, Cziraky MJ, Khan ZM, Migliori R. Melding regulatory, pharmaceutical industry, and US payer perspectives on improving approaches to heterogeneity of treatment effect in research and practice. Value Health. 2013;16(6 Suppl):S10–S15. | |
Frueh FW. Regulation, reimbursement, and the long road of implementation of personalized medicine – a perspective from the United States. Value Health. 2013;16(6):S27–S31. | |
Medical Research Council. Stratified Medicine; 2015. Available from: http://www.mrc.ac.uk/research/initiatives/stratified-medicine/. Accessed February 26, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.