Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 19
Dyskeratosis Congenita: Clinical Phenotype and Genetic Features in a Sibling Pair
Authors Deng X, Guo Z, Chen P, Niu M
Received 26 November 2025
Accepted for publication 17 February 2026
Published 4 March 2026 Volume 2026:19 578757
DOI https://doi.org/10.2147/CCID.S578757
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monica K. Li
Xianhe Deng,1,2,* Ziyu Guo,3,* Pancun Chen,1 Mu Niu1
1Department of Dermatology, The Fifth People’s Hospital of Hainan Province, Haikou, People’s Republic of China; 2Department of Dermatology, Affiliated Dermatology Hospital of Hainan Medical University, Haikou, People’s Republic of China; 3Department of Dermatology, Xiangya Hospital, Central South University, Changsha, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Mu Niu, Department of Dermatology, The Fifth People’s Hospital of Hainan Province, Haikou, People’s Republic of China, Email [email protected] Pancun Chen, Department of Dermatology, The Fifth People’s Hospital of Hainan Province, Haikou, People’s Republic of China, Email [email protected]
Abstract: Dyskeratosis congenita (DC) is a rare, inherited bone marrow failure syndrome resulting from mutations in genes responsible for telomere maintenance. We report a familial case of DC in two brothers, who exhibited the classic diagnostic triad of reticulate skin pigmentation, oral leukoplakia, and nail dystrophy. Genetic analysis identified a rare, hemizygous missense mutation (c.92A>C, p.Gln31Pro) in the DKC1 gene. This case underscores the variable expressivity of DKC1 mutations and reinforces the importance of recognizing the characteristic mucocutaneous features for timely diagnosis and management of this multisystem disorder.
Keywords: dyskeratosis congenita, congenital, mucocutaneous triad, DKC1 gene
Case Presentation
Clinical History
The proband, a 26-year-old male fisherman, presented with a 13-year history of progressive hypopigmented patches on his sun-exposed chest and back. His personal and medical history was otherwise unremarkable. A significant family history revealed that his elder brother had developed similar cutaneous lesions, strongly suggesting a heritable condition.
Physical Examination
The proband’s examination revealed a striking pattern of reticulate, hypopigmented macules and patches with subtle scaling, distributed over his face, upper arms, chest, and back (Figure 1A–D). His fingernails exhibited significant dystrophy, including thinning (platyonychia) and prominent longitudinal ridging (Figure 1F). Oral inspection confirmed the presence of leukoplakia, thereby completing the classic diagnostic triad (Figure 1E). His affected brother presented with a highly similar cutaneous phenotype, characterized by reticulate hyperpigmentation on the face, neck, and trunk (Figure 1G and H).
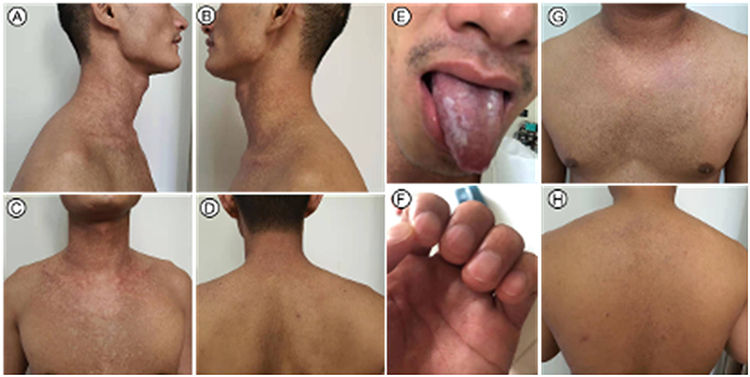 |
Figure 1 Patient’s clinical features (A–F) Reticulate depigmentation on the neck (A and B), chest (C), and back (D), leukoplakia of the oral mucosa, and nail atrophy with longitudinal ridging (E and F) in the proband. (G and H) Reticulate depigmentation on the neck, upper chest (G), and back (H) in the patient’s older brother. |
Investigations
Routine laboratory studies, including complete blood count and serum biochemistry, were within normal limits. A fungal smear from a chest lesion was negative. Histopathological examination of a skin biopsy from the proband revealed hyperkeratosis, mild acanthosis with irregular elongation of the rete ridges, basal layer hyperpigmentation, focal vacuolization of basal cells, pigment incontinence in the papillary dermis, and a mild perivascular lymphocytic infiltrate—findings consistent with DC (Figure 2).
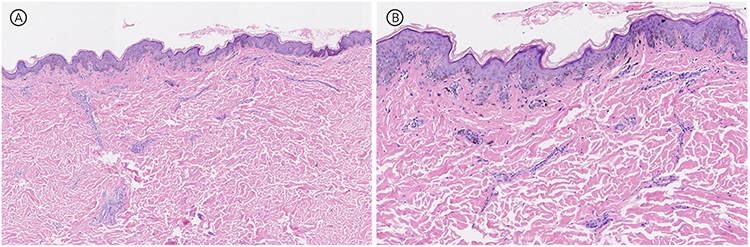 |
Figure 2 Skin biopsy of the anterior chest (Proband): Hyperkeratosis, mild thickening of the epidermal spinous layer, irregular downward extension of the epidermal ridges, and increased pigmentation in the basal layer. Focal basal cell liquefactive degeneration is observed, with pigment incontinence in the superficial dermis. A small number of lymphocytes are infiltrating around the blood vessels. (A) Hematoxylin-eosin, original magnification × 4; (B) Hematoxylin-eosin, original magnification × 10. |
Genetic Analysis
Whole-exome sequencing was performed, which identified a hemizygous c.92A>C (p.Gln31Pro) mutation in exon 3 of the DKC1 gene (NM_001363.5) in the proband (Figure 3). This specific missense mutation, while previously documented in the literature and databases as a pathogenic variant, is rare and its presentation in this familial context provides valuable clinical insights.
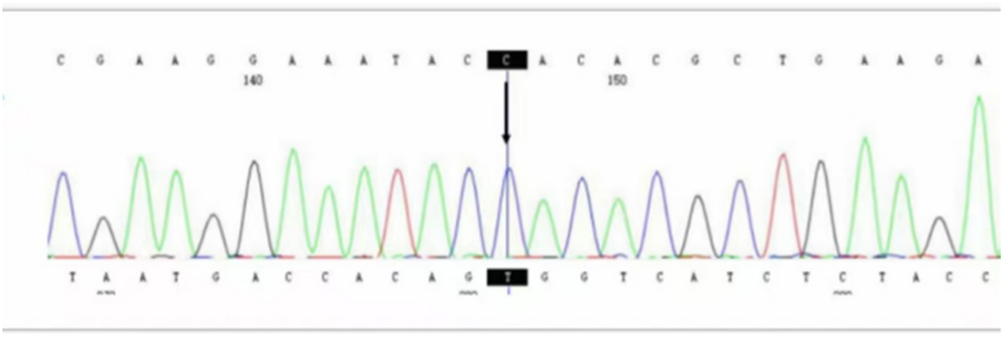 |
Figure 3 Whole exome sequencing and data analysis (Proband): A novel mutation site, C.92A>C (p.Q31P), was identified in exon 3 of the DKC1 gene (NM_001363.5). |
Management and Follow-Up
A definitive diagnosis of DC was established. The current management strategy is centered on symptomatic care, sun protection, and proactive surveillance for the development of bone marrow failure, pulmonary fibrosis, and malignancies. Both patients have, to date, declined invasive procedures such as bone marrow biopsy. Their mucocutaneous symptoms have remained stable under regular follow-up.
Discussion
Dyskeratosis congenita (DC) is a rare genetic disorder of bone marrow failure, with an estimated incidence of 1 in 1,000,000 individuals.1,2 First described in 1906 as Zinsser-Cole-Engman syndrome, DC is characterized by the mucocutaneous triad: dystrophic nails, oral leukoplakia, and reticulate hyperpigmentation of the skin.3 The phenotypic spectrum of DC encompasses a wide range of organ system involvement, including classic DC, Hoyeraal-Hreidarsson syndrome, Revesz syndrome, Coats plus, and isolated cases of aplastic anemia, pulmonary fibrosis, or liver disease.4–6
The disease is caused by mutations in genes associated with telomerase activity, with DKC1 being the most commonly implicated gene, accounting for about 60% of cases.7 Other associated genes include TERC, TERT, NOP10, NHP2, TINF2, WRAP53 (TCAB1), CTC1, RTEL1, ACD, PARN, NAF1, POT1, STN1, PRA1, and ZCCHC8.2,8,9 These genes encode proteins responsible for maintaining telomere function and stability. However, approximately 30% of patients still lack a clearly identified pathogenic gene.
The clinical presentation of DC can vary depending on the mutated gene. Severe forms, such as Hoyeraal-Hreidarsson syndrome and Revesz syndrome, often manifest in early childhood and are associated with mutations in TINF2, RTEL1, and DKC1.10,11 DC with typical skin features usually presents in adolescence or early adulthood, primarily due to DKC1 mutations or homozygous mutations in TERT, TERC, or CTC1.12 In contrast, patients with TERC and TERT heterozygous mutations often experience bone marrow failure and develop complications such as pulmonary fibrosis, liver fibrosis, and osteoporosis in their later years.12 Young adults with moderate to severe disease face the highest risk for malignancies, particularly leukemia.4
DC symptoms commonly begin in childhood, although the onset age can vary.13 Skin pigmentation changes and nail abnormalities often precede other manifestations. Bone marrow failure typically occurs before the age of 30, with 80–90% of patients exhibiting bone marrow abnormalities by their 30s.14,15 In some cases, bone marrow failure appears before mucocutaneous changes, leading to an initial diagnosis of idiopathic aplastic anemia.16 The diverse clinical features of DC make diagnosis based solely on clinical criteria challenging. Bone marrow failure is the primary cause of early mortality and can lead to malignancies and fatal pulmonary complications.4,14
This case of two brothers with dyskeratosis congenita provides multiple insights into the disease. First, the strong family clustering supports the genetic basis of DC. Both patients exhibit a mutation in the DKC1 gene, confirming X-linked recessive inheritance and ruling out de novo mutations. Despite sharing the same mutation and similar genetic backgrounds, the two brothers display different clinical phenotypes. The proband exhibits a more complete triad, while his older brother primarily presents with skin involvement. This illustrates the significant phenotypic heterogeneity seen in DKC1-related DC, which may be influenced by other genetic modifiers or epigenetic mechanisms. Further investigation is needed to understand the underlying factors contributing to this variability. This family case highlights the importance of thorough family history assessment and systematic genetic screening of at-risk family members, particularly male relatives, once DC is diagnosed. Early detection and genetic counseling are essential for timely diagnosis, intervention, and improved prognosis.
Currently, no specific treatments exist for DC, and the prognosis remains unfavorable. Most patients receive symptomatic management. To date, the only potential curative treatment for bone marrow failure associated with telomere biology disorders is hematopoietic cell transplantation.14 Consequently, early diagnosis and timely intervention are crucial for enhancing the quality of life for affected individuals.17
Conclusion
This familial case of DC exemplifies the classic manifestation of the disease and underscores the continued relevance of known DKC1 mutations in clinical practice. It serves as a reminder for clinicians to consider DC in patients presenting with the characteristic mucocutaneous triad, even in adulthood, and highlights the critical role of genetic confirmation in guiding patient management and family counseling.17
Ethics Statement
Informed consent for publication, including the images, was obtained from the patient. Institutional approval for the publication of the case details was obtained from the Ethics Committee of Hainan Fifth People’s Hospital. Additionally, written informed consent for publication was obtained from the patient’s brother.
Funding
This project was supported by Hainan Province Clinical Medical Center.
Disclosure
Xianhe Deng and Ziyu Guo are co-first authors for this study. Pancun Chen and Mu Niu are co-correspondence authors for this study. The authors report no conflicts of interest in this work.
References
1. Mcgrath JA. Dyskeratosis congenita: new clinical and molecular insights into ribosome function. Lancet. 1999;353(9160):1204–5. doi:10.1016/S0140-6736(99)00011-2
2. Alsabbagh MM. Dyskeratosis congenita: a literature review. J Dtsch Dermatol Ges. 2020;18(9):943–967.
3. Dokal I. Dyskeratosis congenita. A disease of premature ageing. Lancet. 2001;358:S27. doi:10.1016/S0140-6736(01)07040-4
4. Alter BP, Giri N, Savage SA, et al. Cancer in dyskeratosis congenita. Blood. 2009;113(26):6549–6557. doi:10.1182/blood-2008-12-192880
5. Mahansaria S, Kumar S, Bharathy KG, et al. Liver transplantation after bone marrow transplantation for end stage liver disease with severe hepatopulmonary syndrome in dyskeratosis congenita: a literature first. J Clin Exp Hepatol. 2015;5(4):344–347. doi:10.1016/j.jceh.2015.10.003
6. Walkup LL, Myers KC, Willmering MM, et al. Modern lung magnetic resonance imaging to screen for pulmonary complications in patients with dyskeratosis congenita. Am J Respir Crit Care Med. 2021;204(11):1340–1343. doi:10.1164/rccm.202103-0736LE
7. Yuan C, Deng D, Yang J, et al. A novel variant and a missense variant identified in the DKC1 gene in three chinese familieswith dyskeratosis congenita. Clin Cosmet Invest Dermatol. 2022;15:1837–1845. doi:10.2147/CCID.S371794
8. Liu H, Rose MJ. Dysplastic megakaryocytes in dyskeratosis congenita with variant in PARN. Blood. 2022;139(26):3779. doi:10.1182/blood.2022016216
9. Choo S, Lorbeer FK, Regalado SG, et al. Editing TINF2 as a potential therapeutic approach to restore telomere length in dyskeratosis congenita. Blood. 2022;140(6):608–618. doi:10.1182/blood.2021013750
10. Gramatges MM, Qi X, Sasa GS, et al. A homozygous telomerase T-motif variant resulting in markedly reduced repeat addition processivity in siblings with Hoyeraal Hreidarsson syndrome. Blood. 2013;121(18):3586–3593. doi:10.1182/blood-2012-08-447755
11. Adams C, Say E, Cheeseman EW. Bilateral exudative retinopathy in a child with revesz syndrome, a severe variant of dyskeratosis congenita. Ophthalmol Retina. 2021;5(11):1106. doi:10.1016/j.oret.2021.08.002
12. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124(18):2775–2783. doi:10.1182/blood-2014-05-526285
13. Zhang X, Dan H, Zhou Y, et al. Extensive and persistent tongue ulceration is an early character of dyskeratosis congenita. Orphanet J Rare Dis. 2025;20(1):192. doi:10.1186/s13023-025-03721-4
14. Niewisch MR, Savage SA. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2019;12(12):1037–1052. doi:10.1080/17474086.2019.1662720
15. Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000;110(4):768–779. doi:10.1046/j.1365-2141.2000.02109.x
16. Kelmenson DA, Hanley M. Dyskeratosis Congenita. N Engl J Med. 2017;376(15):1460. doi:10.1056/NEJMicm1613081
17. Jaju PD, Ransohoff KJ, Tang JY, et al. Familial skin cancer syndromes: increased risk of nonmelanotic skin cancers and extracutaneous tumors. J Am Acad Dermatol. 2016;74(3):437–51; quiz 52–4. doi:10.1016/j.jaad.2015.08.073
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
A Novel Variant and a Missense Variant Identified in the DKC1 Gene in Three Chinese Familieswith Dyskeratosis Congenita
Yuan C, Deng D, Yang J, Liu S, Qian Q, Chen M, Zhou S, Li Y, Li M
Clinical, Cosmetic and Investigational Dermatology 2022, 15:1837-1845
Published Date: 9 September 2022