Back to Journals » Journal of Asthma and Allergy » Volume 7
Dupilumab: a novel treatment for asthma
Authors Vatrella A ![]() , Fabozzi I, Calabrese C, Maselli R, Pelaia G
, Fabozzi I, Calabrese C, Maselli R, Pelaia G ![]()
Received 1 May 2014
Accepted for publication 30 June 2014
Published 4 September 2014 Volume 2014:7 Pages 123—130
DOI https://doi.org/10.2147/JAA.S52387
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Alessandro Vatrella,1 Immacolata Fabozzi,1 Cecilia Calabrese,2 Rosario Maselli,3 Girolamo Pelaia3
1Department of Medicine and Surgery, University of Salerno, Salerno, 2Department of Cardiothoracic and Respiratory Sciences, Second University of Naples, Naples, 3Department of Medical and Surgical Sciences, University Magna Græcia, Catanzaro, Italy
Abstract: Simultaneously with the steady progress towards a better knowledge of the pathobiology of asthma, the potential usefulness of anticytokine therapies is emerging as one of the key concepts in the newly developing treatments of this widespread airway disease. In particular, given the key role played by interleukin (IL)-4 and IL-13 in the pathophysiology of the most typical aspects of asthma, such as chronic airway inflammation, tissue remodeling, and bronchial hyperresponsiveness, these pleiotropic cytokines are now considered as suitable therapeutic targets. Among the recently developed antiasthma biologic drugs, the monoclonal antibody dupilumab is very promising because of its ability to inhibit the biological effects of both IL-4 and IL-13. Indeed, dupilumab prevents IL-4/13 interactions with the α-subunit of the IL-4 receptor complex. A recent trial showed that in patients with difficult-to-control asthma, dupilumab can markedly decrease asthma exacerbations and improve respiratory symptoms and lung function; these effects were paralleled by significant reductions in T-helper 2-associated inflammatory biomarkers. However, further larger and longer trials are required to extend and validate these preliminary results, and also to carefully study the safety and tolerability profile of dupilumab.
Keywords: Th2-high asthma, interleukin-4, interleukin-13, dupilumab
Introduction
Asthma is a heterogeneous disease, usually characterized by airway inflammation, bronchial hyperresponsiveness, and structural changes in the bronchial walls (airway remodeling).1,2 A complex interplay between genetic determinants and environmental stimuli, mainly including allergens and respiratory viruses, is at the basis of the pathobiology of asthma, which is clinically outlined by recurrent episodes of wheeze, shortness of breath, chest tightness, and cough. This common respiratory disease is traditionally distinguished in two classic subtypes known as extrinsic (allergic) and intrinsic (nonallergic) asthma, respectively.3 The pathophysiology of allergic asthma is mostly sustained by T-helper 2 (Th2) lymphocytes, which orchestrate and coordinate the immune inflammatory response of asthmatic airways. This “Th2-high” signature of bronchial inflammation is the result of multiple interactions between the innate and adaptive branches of the immune system.4,5 Indeed, aeroallergens responsible for allergic asthma derive from both seasonal and perennial triggers, penetrate into the airway epithelium, and stimulate Toll-like receptors, which belong to the so-called “pattern recognition receptors” operating in innate immune responses. Upon stimulation, Toll-like receptors activate a signaling cascade leading to increased airway production of several cytokines, including thymic stromal lymphopoietin, interleukin (IL)-25 and IL-33, which are capable of activating Th2 adaptive responses.
Produced in high amounts by airway epithelial cells and mast cells in subjects with asthma, thymic stromal lymphopoietin acts on dendritic cells, inducing them to synthesize chemokines (CCL17, CCL22) that attract Th2 lymphocytes expressing the CCR4 chemokine receptor.6 Stimulated Th2 cells then migrate from the lymph nodes to the airways, where further antigen penetration and presentation take place. Consequently, Th2 lymphocytes bearing the CCR4 receptor secrete large quantities of Th2-derived cytokines, such as IL-5, IL-4, IL-13, and IL-9. These interleukins promote the development and chemotaxis of inflammatory cells implicated in allergic asthma, including eosinophils and mast cells.7 Namely, maturation of eosinophils is stimulated by IL-5, which cooperates with eosinophil-attracting chemokines such as eotaxin, which is secreted by inflammatory cells and bronchial epithelial cells.8 IL-4 and IL-13 target B-cells and induce these lymphocytes to operate an immunoglobulin (Ig) class switch leading to synthesis of IgE.9 IL-9, released by another subtype of T-cells (Th9) derived from Th2 lymphocytes, recruits mast cells and promotes their growth.10 In addition to thymic stromal lymphopoietin, IL-25 and IL-33 also significantly participate in implementing a Th2-high phenotypic pattern. IL-25 and IL-33 trigger the growth and differentiation of so-called type 2 innate lymphoid cells, thereby stimulating them to produce several Th2 cytokines.11,12
Role of IL-4 and IL-13 in asthma pathobiology
Recent progress in comprehension of the pathophysiology of asthma can have relevant implications in future therapeutic approaches. Within this context, the current more detailed knowledge of the key cellular and molecular mechanisms underlying asthma is unravelling potential targets such as IL-4 and IL-13 for the development and implementation of new biological therapies. IL-4 and IL-13 are mainly secreted by CD4+ Th2 and type 2 innate lymphoid cells, and are also produced in lesser quantities by mast cells, eosinophils, basophils, CD8+ Th cells, and natural killer cells.13,14 These cytokines are noticeably implicated in many aspects of both inflammatory and structural changes characterizing asthmatic airways (Figure 1). Indeed, IL-4 and IL-13 drive Ig class switching from IgM antibodies to IgE at the level of B lymphocytes and plasma cells.9 Furthermore, these cytokines enhance airway smooth muscle contractility, and induce airway recruitment of eosinophils by eliciting eotaxin synthesis and upregulation of endothelial adhesion molecules such as vascular cell adhesion molecule-1. IL-13 also stimulates mucus production and airway epithelial cell expression of inducible nitric oxide synthase. Further, IL-13 promotes significant airway remodeling in asthma by enhancing goblet cell hyperplasia, transformation of bronchial fibroblasts into myofibroblasts, deposition of collagen, and proliferation of airway smooth muscle cells.14 All these proinflammatory and structural changes induced by IL-4 and IL-13 lead to a marked increase in bronchial hyperresponsiveness.
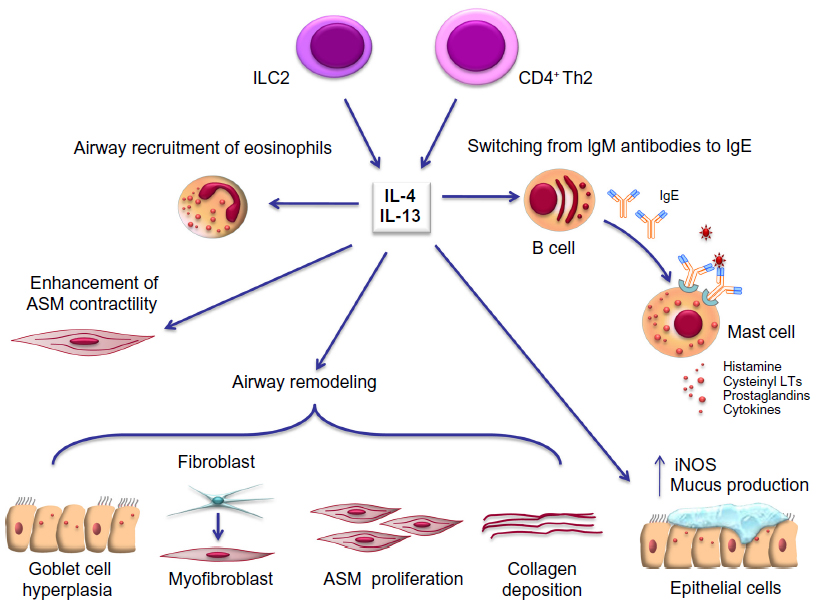 | Figure 1 Pleiotropic effects of IL-4 and IL-13 in asthma pathobiology. See text for details. |
IL-4 and IL-13 play their biological roles by activating a heterodimeric receptor complex consisting of the IL-4 receptor α-subunit (IL-4Rα) and the IL-13 receptor α1-subunit (IL-13Rα1)15 (Figure 2), which are expressed on B lymphocytes, dendritic cells, monocytes/macrophages, eosinophils, basophils, endothelial cells, bronchial epithelial cells, fibroblasts, and airway smooth muscle cells. Overall, the interactions of IL-4 and IL-13 with the IL-4Rα/IL-13Rα1 transmembrane complex stimulate tyrosine kinase proteins Janus kinase 1/2 and tyrosine kinase 2,16,17 that are situated within the cytoplasm and are constitutively associated with IL-4/IL-13 receptor chains (Figure 2). Activation of these two signaling enzymes is responsible for the subsequent phosphorylation-dependent stimulation and nuclear translocation of signal transducer and activator of transcription 616,17 (Figure 2), which is a transcription factor that orchestrates the expression of several target genes. The latter encode many molecules that act as key mediators of the biological effects of IL-4 and IL-13. The mechanism of action of IL-13 can be inhibited by its binding to the IL-13 receptor α2 chain (IL-13Rα2), which does not interact with any other receptor subunit or signaling pathway (Figure 2), thereby mediating an endogenous autoregulatory negative loop that limits IL-13 activity.18 By stimulating the IL-4Rα/IL-13Rα1 heterodimeric receptor expressed by many immune-inflammatory and resident airway cells, IL-4 and IL-13 exert a key pathogenic role in asthma because they elicit several inflammatory and structural changes that are typical of this complex airway disease (Figure 1). In addition to inducing Th2 cell commitment and activation, IgE production, eosinophil recruitment, mast cell growth, and mucous metaplasia, IL-4 also participates in bronchial remodeling via upregulation of collagen and fibronectin synthesis. On the other hand, when compared with nonasthmatic controls, subjects with asthma can show high levels of IL-4 and IL-13 in serum, induced sputum, bronchoalveolar lavage fluid, and airway mucosa.19–21 Indeed, asthmatic patients undergoing segmental allergen challenges experience significant airway increases of IL-4/IL-13 mRNAs and protein levels.19,22 Further, genetic research has also detected relevant linkages of IL-13/IL-13 receptor gene polymorphisms with asthma prevalence and bronchial hyperresponsiveness.23 Thus, genetic control of IL-13 contributes to individual susceptibility to asthma, which is associated with multiple polymorphisms found in the RAD50-IL-13 region of chromosome 5q31.1.24
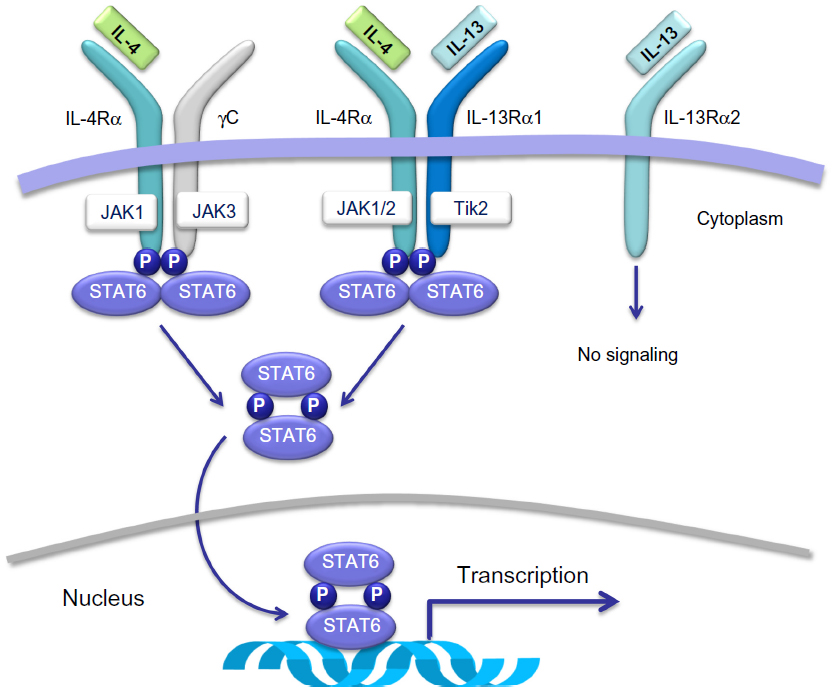 | Figure 2 Membrane receptors and intracellular signaling pathways activated by IL-4 and/or IL-13. IL-4 and IL-13 exert their biological actions by activating a heterodimeric receptor complex consisting of the IL-4 receptor α-subunit (IL-4Rα) and the IL-13 receptor α1-subunit (IL-13Rα1). |
Although the effects of IL-13 largely duplicate and overlap those of IL-4, it can be argued that these apparently very similar cytokines, also sharing common receptor mechanisms and signal transduction pathways, exert distinct biological roles in asthma. In fact, IL-4 primarily and uniquely triggers the initial polarization of naïve CD4+ Th cells towards a Th2 phenotype, whilst IL-13 is probably more relevant than IL-4 in worsening bronchial hyperresponsiveness by enhancing airway inflammation and remodeling via induction of subepithelial fibrosis and airway smooth muscle hyperplasia.25,26 These considerations are corroborated by the detection of higher concentrations of IL-13 with respect to IL-4 in murine lungs sensitized to allergens and in the airways of patients with asthma, where IL-13-synthesizing cells significantly outnumber those producing IL-4.27,28 Anyway, the largely redundant roles of IL-4 and IL-13 in the pathophysiology of asthma make these two cytokines very attractive as suitable targets for specific and effective new treatments.
Therapies targeting IL-4 and IL-13
Many investigations focusing on the potential actions of IL-4-targeted antiasthma therapies have produced discordant findings.15 In mouse models of antigen-induced asthma, experimental evaluations of therapeutic strategies directed against IL-4 or its receptor have detected significant inhibition of bronchial eosinophilic infiltration and IL-5 secretion from T lymphocytes, associated with decreases in airway inflammation as well as in blood concentrations of IgE and bronchial hyperresponsiveness to cholinergic agents.29,30 Nevertheless, although the humanized anti-IL-4 monoclonal antibody pascolizumab is characterized by a good safety and tolerability profile, it is incapable of eliciting relevant antiasthma effects.31 Moreover, in spite of some initial encouraging results with use of the soluble recombinant human IL-4 receptor altrakincept, subsequent studies have not shown any clinical effectiveness.32 Pitrakinra, a mutein obtained by site-directed changes performed on the IL-4 amino acid sequence, that behaves as a dual IL-4/IL-13 antagonist, appears to be characterized by a better therapeutic profile.33 Indeed, when received subcutaneously or via inhalation in asthmatic patients, pitrakinra is well tolerated, attenuates early and late allergic bronchoconstrictive reactions, and also decreases exacerbations of eosinophilic asthma.34,35 Further, in a recent pharmacogenetic, placebo-controlled trial focused on modulation of IL-4/IL-13-mediated mechanisms, several dosages (1 mg, 3 mg, or 10 mg twice a day for 12 weeks) of inhaled pitrakinra were evaluated in asthmatic patients with moderate-to-severe disease.36 This study showed that pitrakinra, although not clinically effective in the entire population of recruited subjects, at the 10 mg dosage significantly reduced asthma exacerbation rates in patients exhibiting specific single nucleotide polymorphisms in the 3′ untranslated region of the IL-4Rα gene (rs8832GG and rs1029489GG genotypes).36
It has been demonstrated in murine models of experimental asthma that monoclonal antibodies directed against intravenously administered IL-13 decrease proliferation of goblet cells, as well as airway inflammation and remodeling caused by allergen challenge.37 For instance, such results have been found when administering tralokinumab, a fully human anti-IL-13 antibody, that markedly inhibits bronchial hyperresponsiveness and airway eosinophilia.38 Later tested at different doses in a double-blind, randomized, placebo-controlled Phase I study, tralokinumab exhibited a linear pharmacokinetic profile and a satisfactory safety pattern when administered intravenously (1.5 or 10 mg/kg) every 28 days.31 In a recent placebo-controlled study performed in 194 asthmatic patients with moderate-to-severe uncontrolled disease, the effects of tralokinumab were investigated administering this drug subcutaneously (150, 300, or 600 mg) at 2-week intervals as add-on therapy to standard controller treatment.39 Comparative evaluation of tralokinumab and placebo did not show any significant difference with regard to symptom score assessment using the Asthma Control Questionnaire. However, tralokinumab significantly decreased the use of as-needed short-acting bronchodilators, and also enhanced percentage forced expiratory volume in one second (FEV1) values with respect to baseline; FEV1 increases were included within an 8.1% (150 mg) to 16.1% (600 mg) range.39 When compared with patients with no sputum IL-13, FEV1 increases were higher in asthmatic subjects exhibiting detectable IL-13 levels in their induced sputum. Tralokinumab displayed a good safety and tolerability profile. Moreover, in a Phase II study performed in subjects with mild allergic asthma, the humanized IL-13-blocking antibody anrukinzumab induced significant inhibition of antigen-elicited late asthmatic reactions which lasted for 14 days, but not for 35 days after subcutaneous administration (2×2 mg/kg, one week apart).40
The most extensively studied anti-IL-13 drug is lebrikizumab, an IgG4 humanized monoclonal antibody that blocks IL-13, thus neutralizing its biological activity.41 Lebrikizumab is able to abrogate IL-13 functions in vitro. In particular, lebrikizumab can inhibit IL-13-dependent signal transducer and activator of transcription 6 phosphorylation and cell proliferation in TF-1 cells (a human erythroleukemic cell line).41 In this regard, it has been shown that lebrikizumab exerts significant therapeutic actions in Th2-mediated asthma, which can be identified by the detection of high serum levels of periostin, a protein component of the extracellular matrix synthesized by airway epithelial cells and lung fibroblasts, expression of which is induced by IL-13.42–44 A recent placebo-controlled trial demonstrated that lebrikizumab is very well tolerated when added to guideline-based inhaled antiasthma treatment.45 This 6-month study, involving 219 adult asthmatic subjects not adequately controlled by inhaled corticosteroids (ICS), evaluated the therapeutic activity of lebrikizumab given by the subcutaneous route every month at a dose of 250 mg. Lebrikizumab elicited its best effects in moderate-to-severe asthmatic patients with high blood concentrations of periostin. Indeed, significant FEV1 improvements, resulting in a 5.5% increase with respect to baseline levels, were found in the whole population of enrolled patients after 12 weeks of treatment with lebrikizumab. In particular, FEV1 increased by 8.2% in the group of patients with high levels of periostin, and by 1.6% (not significant) in the group of patients with low periostin concentrations. These findings suggest that periostin can be reliably utilized in real life as a biomarker of IL-13-induced asthma that is potentially susceptible to anti-IL-13 therapeutic approaches. In the same trial, a post-lebrikizumab FEV1 increase (8.6%) similar to that observed in individuals with high blood levels of periostin was also found in subjects who displayed high pretreatment levels of exhaled nitric oxide fraction (FENO),45 another biomarker whose biosynthesis can be induced by IL-13. Indeed, lebrikizumab caused a 19% mean reduction in FENO after 12 weeks of treatment, which was significantly different (P<0.001) when compared with the 10% increase detected in patients receiving placebo. However, lebrikizumab did not elicit significant changes in secondary end points, including morning peak expiratory flow, Asthma Control Questionnaire score, use of rescue medications, and rate of asthma exacerbations. In another trial carried out in 212 asthmatic patients not receiving ICS, the patients were randomized to undergo therapy with either placebo or three different subcutaneous monthly doses (125, 250, or 500 mg) of lebrikizumab for 3 months. This drug induced a slight increase in mean FEV1, that was not statistically or clinically significant.46 Moreover, with regard to patient subgroups identified on the basis of serum periostin concentrations, no significant differences in FEV1 variations were found between the three lebrikizumab dose groups and the placebo group after 12 weeks of treatment. More recently, GSK679586, another humanized monoclonal antibody that inhibits IL-13 binding to both α1 and α2 subunits of the IL-13 receptor, was evaluated in severe asthmatic subjects who were relatively resistant to high ICS doses.47 When compared with a placebo group, patients receiving a well tolerated 12-week treatment with three once-monthly intravenous infusions of GSK679586 (10 mg/kg) did not experience any significant improvement in several clinical, functional, and laboratory measurements, including Asthma Control Questionnaire symptom score, asthma exacerbation rate, FEV1, serum IgE levels, and blood eosinophil counts.47 The latter findings indicate that biological therapies targeting IL-13 are not always able to exert meaningful therapeutic effects in individuals with asthma that is refractory to maximally indicated ICS doses, equaling or exceeding 1,000 μg/day of fluticasone propionate.
Dupilumab: a new anti-IL-4/IL-13 approach
Although IL-13 undoubtedly represents a valuable therapeutic target for asthma treatment, the recently published studies on anti-IL-13 monoclonal antibodies have also highlighted very relevant limitations. These partial effects are probably due to the overlapping biological actions of IL-4 and IL-13. Hence, a combination approach aimed at blocking the effects of both ILs is likely to be more effective. In this regard, dupilumab, a fully human monoclonal antibody directed against the α-subunit of the IL-4 receptor and capable of blocking the signal transduction pathways activated by IL-4 and IL-13, has been developed and evaluated in asthmatic individuals with persistent, moderate-to-severe disease and airway or peripheral eosinophilia.48 In particular, 104 subjects aged 18–65 years were recruited, who had a mean baseline FEV1 calculated as 72% of predicted, as well as at least 300 eosinophils per μL in peripheral blood or at least 3% eosinophils in induced sputum. They used long-acting β2-adrenergic agonists (LABAs) added to medium/high ICS doses, without achieving satisfactory asthma control. Fifty-two patients were assigned to weekly subcutaneous treatment with dupilumab 300 mg and 52 patients received placebo. Dupilumab was administered for 12 weeks or until patients experienced an asthma exacerbation. Further, the patients were instructed to interrupt LABA administration after 4 weeks and to progressively reduce and then suspend their ICS therapy between weeks 6 and 9. The primary study outcome was the onset of an asthma exacerbation. In this regard, three patients undergoing treatment with dupilumab (6%) had a disease exacerbation, versus 23 subjects in the placebo group (44%), corresponding to an overall 87% reduction in exacerbation rate (P<0.001) in the dupilumab group. In terms of secondary endpoints, dupilumab induced a marked improvement in FEV1, exceeding 200 mL, and also elicited a parallel increase in morning peak expiratory flow, with a smaller increase in evening peak expiratory flow. Moreover, dupilumab progressively improved the Asthma Control Questionnaire score throughout the study, and also decreased asthmatic symptoms occurring during the morning and evening, nocturnal awakenings due to asthma, and use of the short-acting β2-adrenergic agonist salbutamol as rescue medication. Dupilumab was also able to significantly (P<0.001) reduce the levels of Th2-associated inflammatory biomarkers such as FENO concentrations, which were found to be markedly lower after 4 weeks of treatment, and persisted up to week 12 at lower levels than baseline values, in spite of ICS interruption. The FENO decrease was correlated with FEV1 improvement. Similar reductions were also found in serum levels of IgE, eotaxin-3, and TARC (thymus and activation-regulated chemokine).
Conversely, no apparent changes in blood eosinophil counts were detected in patients treated with dupilumab. Taken together, these study findings suggest that targeting Th2-driven uncontrolled asthma with a therapeutic strategy aiming to neutralize the biological effects of both IL-4 and IL-13 at the receptor level can be more effective than blocking the actions of only one of these two cytokines. Because about 80% of the enrolled patients used high ICS doses, it can be argued that dupilumab may be successful in inhibiting residual and relatively ICS/LABA-refractory airway inflammation, mainly sustained by IL-4 and IL-13 in subjects with poorly controlled asthma.
However, despite the evident benefits manifested by asthmatic patients undergoing treatment with dupilumab, there have been some relevant criticisms of this trial.49 Firstly, on the basis of such a study it is not possible to understand whether dupilumab can also be effective in other asthmatic populations, eg, those not exhibiting Th2-mediated airway and/or blood eosinophilia. Secondly, an obvious weakness of this trial is strictly intrinsic to its design, based on an unusual ICS/LABA withdrawal, which is very unlikely to occur in clinical practice. This feature of the experimental protocol precluded the possibility of evaluating the potential additive value of dupilumab with regard to the standard controller treatment of asthma, commonly represented by ICS/LABA associations. On the other hand, in real life, physicians do not withdraw ICS and LABAs in patients with difficult-to-control asthma. For instance, during the first part of the study, when all enrolled subjects were taking ICS and LABAs, no meaningful change in exacerbation rate was found in the dupilumab group when compared with the placebo group. At the same early time points, only very mild improvements in lung function and inflammatory biomarkers were observed in patients receiving dupilumab.
Finally, when considering the safety and tolerability profile with respect to placebo, a higher frequency of injection site reactions, nasopharyngitis, nausea, and headache was reported during treatment with dupilumab. Moreover, progressive cutaneous rash, urticaria, and edema occurred in one patient receiving dupilumab, requiring nonurgent symptomatic therapy (systemic corticosteroids and antihistamine drugs) and immediate interruption of study treatment. Intriguingly and unexpectedly, dupilumab appeared to increase eosinophil levels in four patients. Therefore, given the limited size of the study population (n=52) treated with dupilumab in this trial, the overall range of possible unwanted reactions is not known, and close monitoring for such adverse events is necessary.
Conclusion
Recent advances in our understanding of the pathobiology of asthma, together with an increased awareness of the necessity to pursue better phenotypic stratification of asthmatic patients,1 are opening up novel and exciting therapeutic perspectives. In particular, anticytokine treatments are emerging as potentially effective approaches,50 that are much needed for asthma that is difficult to control with ICS and other standard therapies. Within such a promising new therapeutic scenario, anti-IL-4 and anti-IL-13 treatments could be very important, given the key role exerted by these two cytokines in the pathophysiology of asthma, particularly for phenotypic subgroups identifiable on the basis of a high Th2 profile of asthmatic inflammation. The monoclonal antibody dupilumab, directed against the α-subunit of the IL-4 receptor, utilized by both IL-4 and IL-13 in exerting their biological actions, seems to be very promising with regard to improving the health status of patients suffering from Th2-driven uncontrolled asthma. However, larger trials of longer duration are required to widen and validate these early findings, especially with the aim of evaluating the true therapeutic potential of dupilumab as add on treatment to ICS and LABAs. Dupilumab should thus be tested in well designed studies, without withdrawal or interruption of conventional, inhaled antiasthma control therapies. Moreover, the effects of dupilumab should be comparatively evaluated in asthmatic patients versus other biological treatments targeting Th2-mediated airway inflammation. Finally, close attention should be paid to the safety and tolerability of dupilumab.
Disclosure
The authors report no conflicts of interest in this work.
References
Wenzel SE. Complex phenotypes in asthma: current definitions. Pulm Pharmacol Ther. 2013;26(6):710–715. | |
Murdoch JR, Lloyd CM. Chronic inflammation and asthma. Mutat Res. 2010;690(1–2):24–39. | |
Holt PG, Sly PD. Interaction between adaptive and innate immune pathways in the pathogenesis of atopic asthma: operation of a lung/bone marrow axis. Chest. 2011;139(5):1165–1171. | |
Hammad H, Chieppa M, Perros F, et al. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15(4):410–416. | |
Woodruff PG, Modrek B, Choy DF, et al. T-helper type 2-driven inflammation defines major sub-phenotypes of asthma. Am J Respir Crit Care Med. 2009;180(5):388–395. | |
Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med. 2006;203(2):269–273. | |
Larché M, Robinson DS, Kay AB. The role of T lymphocytes in the pathogenesis of asthma. J Allergy Clin Immunol. 2003;111(3):450–463. | |
Fulkerson PC, Rothenberg ME. Targeting eosinophils in allergy, inflammation and beyond. Nat Rev Drug Discov. 2013;12(2):117–129. | |
Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118(11):3546–3556. | |
Kaiko GE, Foster PS. New insights into the generation of Th2 immunity and potential therapeutic targets for the treatment of asthma. Curr Opin Allergy Clin Immunol. 2011;11(1):39–45. | |
Scanion ST, McKenzie ANJ. Type 2 innate lymphoid cells: new players in asthma and allergy. Curr Opin Immunol. 2012;24(6):707–712. | |
Halim TY, McKenzie AN. New kids on the block – group 2 innate lymphoid cells and type 2 inflammation in the lung. Chest. 2013;144(5):1681–1686. | |
Steinke JW, Borish L. Th2 cytokines and asthma. Interleukin 4: its role in the pathogenesis of asthma, and targeting it for asthma treatment with interleukin-4 receptor antagonists. Respir Res. 2001;2(2):66–70. | |
Corren J. Role of interleukin-13 in asthma. Curr Allergy Asthma Rep. 2013;13(5):415–420. | |
Oh CK, Geba GP, Molfino N. Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur Respir Rev. 2010;19(115):46–54. | |
Andrews R, Rosa L, Daines M, Khurana HG. Reconstitution of a functional human type II IL-4/IL-13 in mouse B cells: demonstration of species specificity. J Immunol. 2001;166(3):1716–1722. | |
Chiba Y, Goto K, Misawa M. Interleukin-13-induced activation of signal transducer and activator of transcription 6 is mediated by an activation of Janus kinase 1 in cultured human bronchial smooth muscle cells. Pharmacol Rep. 2012;64(2):454–458. | |
Zheng T, Liu W, Oh SY, et al. IL-13 receptor α2 selectively inhibits IL-13-induced responses in the murine lung. J Immunol. 2008;180(1):522–529. | |
Maes T, Joos GF, Brusselle GG. Targeting IL-4 in asthma: lost in translation? Am J Respir Cell Mol Biol. 2012;47(3):261–270. | |
Saha SK, Berry MA, Parker D, et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol. 2008;121(3):685–691. | |
Corren J. Anti-interleukin-13 antibody therapy for asthma: one step closer. Eur Respir J. 2013;41(2):255–256. | |
Prieto J, Lensmar C, Roquet AI, et al. Increased interleukin-13 mRNA expression in bronchoalveolar lavage cells of atopic patients with mild asthma after repeated low-dose allergen provocations. Respir Med. 2000;94(8):806–814. | |
Howard TD, Koppelman GH, Xu J, et al. Gene-gene interaction in asthma: IL4RA and IL-13 in a Dutch population with asthma. Am J Hum Genet. 2002;70(1):230–236. | |
Li X, Howard TD, Zheng SL, et al. Genome-wide association study of asthma identifies RAD50-IL13 and HLA-DR/DQ regions. J Allergy Clin Immunol. 2010;125(2):328–335. | |
Grunig G, Warnock M, Wakil AE, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998; 282(5397):2261–2263. | |
Wills-Karp M, Luyimbazi J, Xu X, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282(5397):2258–2261. | |
Munitz A, Brandt EB, Mingler M, et al. Distinct roles for IL-13 and IL-4 via IL-13 receptor α1 and the type II IL-4 receptor in asthma pathogenesis. Proc Natl Acad Sci U S A. 2008;105(20):7240–7245. | |
Kotsimbos TC, Ernst P, Hamid QA, et al. Interleukin-13 and interleukin-4 are coexpressed in atopic asthma. Proc Assoc Am Physicians. 1996;108(5):368–373. | |
Coyle AJ, Le Gros G, Bertrand C, et al. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am J Respir Cell Mol Biol. 1995;13(1):54–59. | |
Swart DA, Anders-Bartholo PM, Tocker JE. Effects of IL-4Rα blockade on lung inflammation and airway hyperresponsiveness using Mu317RAXMu, a murine surrogate for AMG 317, in a treatment model of cockroach allergen-induced asthma in mice. J Allergy Clin Immunol. 2008;121(2):S267. | |
Pelaia G, Vatrella A, Maselli R. The potential of biologics for the treatment of asthma. Nat Rev Drug Discov. 2012;11(12):958–972. | |
Steinke JW. Anti-interleukin-4 therapy. Immunol Allergy Clin North Am. 2004;24(4):599–614. | |
Burmeister Getz E, Fisher DM, Fuller R. Human pharmacokinetics/pharmacodynamics of an interleukin-4 and interleukin-13 dual antagonist in asthma. J Clin Pharmacol. 2009;49(9):1025–1036. | |
Holgate ST. Pathophysiology of asthma: what has our current understanding taught us about new therapeutic approaches? J Allergy Clini Immunol. 2011;128(3):495–505. | |
Wenzel S, Wilbraham D, Fuller R, et al. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370(9596):1422–1431. | |
Slager RE, Otulana BA, Hawkins GA, et al. IL-4 receptor polymorphisms predict reduction in asthma exacerbations during response to an anti-IL-4 receptor α antagonist. J Allergy Clin Immunol. 2012;130(2):516–522. | |
Yang G, Volk A, Petley T, et al. Anti-IL-13 monoclonal antibody inhibits airway hyperresponsiveness, inflammation and airway remodeling. Cytokine. 2004;28(6):224–232. | |
Blanchard C, Mishra A, Saito-Hakei H, et al. Inhibition of human interleukin-13-induced respiratory and oesophageal inflammation by anti-human interleukin-13 antibody (CAT-354). Clin Exp Allergy. 2005;35(8):1096–1103. | |
Piper E, Brightling C, Niven R, et al. A phase II placebo-controlled study of tralokinumab in moderate-to-severe asthma. Eur Respir J. 2013;41(2):330–338. | |
Gauvreau GM, Boulet LP, Cockroft DW, et al. Effects of interleukin-13 blockade on allergen-induced airway responses in mild atopic asthma. Am J Respir Crit Care Med. 2011;183(8):1007–1014. | |
Thomson NC, Patel M, Smith AD. Lebrikizumab in the personalized management of asthma. Biologics. 2012;6:329–335. | |
Yuyama N, Davies DE, Akaiwa M, et al. Analysis of novel disease-related genes in bronchial asthma. Cytokine. 2002;19(6):287–296. | |
Takayama G, Arima K, Kanaji T, et al. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol. 2006;118(1):98–104. | |
Jia G, Erickson RW, Choy DF, et al. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J Allergy Clin Immunol. 2012;130(3):647–654. | |
Corren J, Lemanske RF, Hanania NA, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365(12):1088–1098. | |
Noonan M, Korenblat P, Mosesova S, et al. Dose-ranging study of lebrikizumab in asthmatic patients not receiving inhaled steroids. J Allergy Clin Immunol. 2013;132(3):567–574. | |
De Boever EH, Ashman C, Cahn AP, et al. Efficacy and safety of an anti-IL-13 mAB in patients with severe asthma: a randomized trial. J Allergy Clin Immunol. 2014;133(4):989–996. | |
Wenzel S, Ford L, Pearlman D, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368(26):2455–2466. | |
Wechsler ME. Inhibiting IL-4 and IL-13 in difficult-to-control asthma. N Engl J Med. 2013;368(26):2511–2513. | |
Gallelli L, Busceti MT, Vatrella A, Maselli R, Pelaia G. Update on anti-cytokine treatment for asthma. Biomed Res Int. 2013;2013:104315. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.