Back to Journals » International Journal of Nanomedicine » Volume 11
Dual-function synthetic peptide derived from BMP4 for highly efficient tumor targeting and antiangiogenesis
Authors Choi SH, Lee JY, Suh JS, Park YS, Chung CP, Park YJ
Received 15 June 2016
Accepted for publication 5 August 2016
Published 13 September 2016 Volume 2016:11 Pages 4643—4656
DOI https://doi.org/10.2147/IJN.S115044
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Suk Hyun Choi,1,* Jue Yeon Lee,2,* Jin Sook Suh,1 Yoon Shin Park,3 Chong Pyoung Chung,2 Yoon Jeong Park1
1Department of Dental Regenerative Biotechnology, Dental Research Institute, 2Central Research Institute, Nano Intelligent Biomedical Engineering Corporation (NIBEC), School of Dentistry, Seoul National University, Seoul, 3Department in Microbiology, School of Biological Sciences, College of Natural Sciences, Chungbuk National University, Cheongju, South Korea
*These authors contributed equally to this work
Abstract: Angiogenesis plays a critical role in the growth and metastasis of cancer, and growth factors released from cancer promote blood-vessel formation in the tumor microenvironment. The angiogenesis is accelerated via interactions of growth factors with the high-affinity receptors on cancer cells. In particular, heparan sulfate proteoglycans (HSPGs) on the surface of cancer cells have been shown to be important in many aspects of determining a tumor’s phenotype and development. Specifically, the regulation of the interactions between HSPGs and growth factors results in changes in tumor progression. A peptide with heparin-binding (HBP) activity has been developed and synthesized to inhibit tumor growth via the prevention of angiogenesis. We hypothesized that HBP could inhibit the interaction of growth factors and HSPGs on the surface of cancer cells, decrease paracrine signaling in endothelial cells (ECs), and finally decrease angiogenesis in the tumor microenvironment. In this study, we found that HBP had antiangiogenic effects in vitro and in vivo. The conditioned media obtained from a breast cancer cell line treated with HBP were used to culture human umbilical vein ECs (HUVECs) to evaluate the antiangiogenic effect of HBP on ECs. HBP effectively inhibited the migration, invasion, and tube formation of HUVECs in vitro. In addition, the expressions of angiogenesis-mediating factors, including ERK, FAK, and Akt, were considerably decreased. HBP also decreased the levels of invasive factors, including MMP2 and MMP9, secreted by the HUVECs. We demonstrated significant suppression of tumor growth in a breast cancer xenograft model and enhanced distribution of HBP at the site of tumors. Taken together, our results show that HBP has antiangiogenic effects on ECs, and suggest that it may serve as a potential antitumor agent through control of the tumor microenvironment.
Keywords: heparin-binding peptide, HBP, antiangiogenesis, heparan sulfate proteoglycans, HSPGs, endothelial cells, breast cancer xenograft, tumor microenvironment
Introduction
The idea of preventing angiogenesis using bioactive molecules alone or in combination with biomaterials has been a promising strategy for the treatment of cancer.1,2 Tumors require a stable blood supply to grow to a critical volume and metastasize. In fact, increases in tumor mass are limited to 1–2 mm in size within an avascular environment.3 Bioactive agents with antiangiogenic activities, including peptides, small molecules, and antibodies, have emerged as one of the leading tools to treat cancer. Several attempts to target VEGF have been successful in clinical trials, and anti-VEGF therapies have been approved by the US Food and Drug Administration.4,5 However, it has been suggested that targeting VEGF alone may not be sufficient to fully prevent tumor malignancy. Therefore, the need to develop alternative targets related to other factors involved in tumor development is imperative to complement the current VEGF-based therapies.
Heparan sulfate proteoglycans (HSPGs) are present on the surface of every eukaryotic cell, including tumor cells, and are important regulators at the cell surface–extracellular matrix (ECM) interface.6 HSPGs are key components of the ECM, and are involved in tumor progression by regulating growth factor-signaling pathways. Most of the molecular events associated with tumor growth, neovascularization, and metastasis are influenced by interactions between cancer cells and their ECM. The role of HSPGs has not been investigated in depth, but HSPGs do have a role in modulating the activities of heparin-binding angiogenic growth factors, including FGF, PDGF, and VEGF.7–9 HSPGs and heparin are structurally and functionally related, and highly sulfated HSPGs are commonly referred to as heparin.10 HSPGs are the more abundant form, while heparin is restricted to mast cells that line blood vessels and mucosal tissue.11 The heterogeneous structure of HSPGs explains why they are more involved in mediating in vivo signaling than heparin.7 The modulation of neovascularization occurs through the interaction of HSPGs on cancer cells and heparin-binding growth factors involved in angiogenesis. In addition, alterations in the level of HSPG expression can potentially make cancer cells highly versatile in modulating their behavior. For these reasons, HSPGs on the surface of cancer cells can be utilized as a key regulator of the angiogenic system.
In a previous study, we developed a heparin-binding peptide (HBP) derived from the heparin-binding site of BMP4.12 BMP4 has been reported to inhibit angiogenesis in breast cancer cells, in contrast to other angiogenic growth factors.13–15 Herein, we investigated the inhibitory effect of HBP on angiogenesis in tumors via its ability to block the interaction of HSPGs with angiogenic growth factors. The HBP sequence, RKKNPNCRRH, corresponds to residues 15–24 of the heparin-binding site in BMP4, and is similar to the “consensus sequence” of HBP, BBBXTXXBBB, (where X, B, and T indicate a hydropathic residue, a basic residue, or a turn, respectively).12,16 It is known that interactions between heparin and HBP are based on the ionic interactions between positively charged residues (Arg and Lys) of HBP and the negatively charged groups of heparin (an average of 2.7 negative charges per disaccharide).17,18 Therefore, HBP may be a useful antiangiogenic therapeutic agent, because of its ability to block the interactions between HSPGs and angiogenic growth factors.
We evaluated the effect of HBP on angiogenesis in vitro and in vivo. The effects of HBP on cell viability, migration, invasion, and the tubular formation capacity of human umbilical vein endothelial cells (HUVECs) were evaluated. In addition, the therapeutic antiangiogenic activity of HBP in a xenograft tumor model was also examined.
Materials and methods
Materials
Heparin sodium salt from porcine intestinal mucosa and VEGF was purchased from Sigma-Aldrich (St Louis, MO, USA). Rink amide resin (0.75 mM/g) and fluorenylmethoxycarbonyl (Fmoc) amino acids were purchased from BeadTech (Ansan, South Korea). 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate was purchased from AAPPTec (Louisville, KY, USA). Piperidine, diisopropylethylamine, triisopropylsilane, phenol, and thioanisole were obtained from Sigma-Aldrich. Tetrahydrofuran acetonitrile and trifluoroacetic acid were purchased from Honeywell (Morris Plains, NJ, USA). 1,2-Ethanedithiol was purchased from Sigma-Aldrich. Diethyl ether, N, N-dimethylformamide, N-methylpyrrolidone, and other chemicals were of analytical grade. Selective antibodies for FAK (3283), p-FAK (3283S), ERK (9101), p-ERK (9101S), Akt (9271), p-Akt (9271S), MMP2 (4022S), and MMP9 (3862S) were purchased from Cell Signaling Technology (Danvers, MA, USA). All other reagents and products were purchased from Sigma-Aldrich unless noted otherwise.
Peptide synthesis and purification
HBPs and non-HBPs (NHBPs) were prepared in mass quantities using a peptide synthesizer (Apex 396; AAPPTec) based on standard Fmoc chemistry, according to our previous report.12 The synthesized peptides were purified via reverse-phase high-performance liquid chromatography (Waters Corporation, Milford, MA, USA) using a Vydac C18 column and a gradient (0%–100% acetonitrile, 120 minutes) of water/acetonitrile containing 0.1% trifluoroacetic acid. The peptides were characterized in terms of molecular weight via liquid chromatography–mass spectrometry (Waters), and were found to have the expected molecular weights. The purity of the peptides was above 98%.
Binding affinity
The binding affinity of heparin to the peptides was measured via a slot-blot assay. Various concentrations of HBP in phosphate-buffered saline (PBS) were immobilized onto a nitrocellulose membrane in the slot-blot wells, and the wells were blocked with Tris-buffered saline (TBS) with 0.5% skim milk for 1 hour. Then, heparin (20 mg/mL) was incubated in the slot-blot wells for 90 minutes. The wells were then incubated overnight at 4°C with a mouse antiheparin antibody, followed by a reaction with horseradish peroxidase (HRP)-conjugated HBP (Lifespan Technologies, Salt Lake City, UT, USA) in PBS for 1 hour at room temperature. The bound antibodies were detected using chemiluminescence reagents (West-Zol; Intron Biotechnology, Seongnam, South Korea).
Cell lines and cell culture
HUVECs were obtained from Dr Lee Gene of Seoul National University. The procedure for obtaining cells from patients was approved by the Institutional Review Board (IRB) of Seoul National University Dental Hospital (SNUDH76/04-06). Human breast cancer cells (MDA-MB231-Luc-D3H1 cells and MDA-MB231 cells) were purchased from PerkinElmer and the American Type Culture Collection. The cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and a 1% antibiotic–antimycotic solution (Thermo Fisher Scientific, Waltham, MA, USA). HUVECs, MDA-MB231 cells, and MDA-MB231-Luc-D3H1 cells were cultured at 37°C under a humidified 95%:5% (v:v) mixture of air and CO2. Media were changed twice weekly, and cells were subcultured following detachment with a 0.05% trypsin–0.02% ethylenediaminetetraacetic acid solution and seeding into fresh-culture flasks at a ratio of 1:4 upon reaching 80%–90% confluence.
Preparation of tumor cell-conditioned media
Tumor cell-conditioned media (TCM) were generated from MDA-MB231 cells treated with HBP to establish the tumor environment.19 When cultured MDA-MB231 cells reached 90% confluence, they were starved in Dulbecco’s Modified Eagle’s Medium with 0.5% serum for 24 hours. Then, the cells were treated with various concentrations of HBP added to the starvation medium. After 24 hours of incubation, the TCM was centrifuged for 10 minutes at 3,000 rpm at 4°C, and the supernatants were collected separately. TCM samples were stored at −20°C for in vitro tests. NHBP and VEGF were used as treatments instead of HBP as negative and positive controls, respectively.
Cell-viability assay
To determine cell viability, we used the MTT assay. HUVECs or MDA-MB231 (104 cells per well) were seeded in 24-well plates overnight to allow for attachment. Cell viability was determined via MTT assay after 24 hours. After treatment with various concentrations of HBP for 24 hours, the MTT solution was added to the cells to a final concentration of 0.5 mg/mL. The medium was then removed after a 4-hour incubation at 37°C, and 500 μL of dimethyl sulfoxide was added to dissolve the formazan precipitates. A 200 μL aliquot from each well was then transferred to a 96-well plate, and the absorbance was read at 540 nm on a microplate reader (BioTek Instruments, Winooski, VT, USA). The data were analyzed from three independent experiments in each group.
Cell-migration assay
Migration of HUVECs was measured using two assays: a transwell migration assay and a wound-migration assay. The transwell migration assay (transwell plate, 8 μm pore size, 24 wells, Costar; Corning Incorporated, Corning, NY, USA) was performed to assess the invasive capacity of the cells. HUVECs (5×104 cells/well) were seeded into the upper chambers with 1 mL TCM from the MDA-MB231 cells treated with different concentrations of HBP. The bottom chambers were filled with basal EC-culture medium. After 16 hours of incubation, the nonmigrated cells on the upper surface of the membrane were removed with a PBS-soaked cotton swab. Then, the migrated cells were fixed with methanol for 100 minutes. Cells that migrated to the lower side of the membranes were stained with Giemsa. Images were taken using an Olympus DP72 camera (magnification 100×). The number of cells that crossed the membrane in the treated group was compared with that in the control group. For the scratch-migration assay, HUVECs (1×106 cells) were allowed to grow to full confluence in six-well plates. The cells were then rinsed with PBS and starved overnight in 2% fetal bovine-serum media. A single wound was scratched in the center of the cell monolayers with a 200 μL sterile plastic pipette tip. The wounded monolayers were washed twice with PBS to remove nonadherent cells and incubated with HBP for 24 hours. To measure the distance that ECs migrated from the edge of the injured monolayer, images were obtained both immediately after wounding and after a 24-hour incubation period using an Olympus CKX41 inverted microscope (magnification 20×).
Endothelial cell capillary-like tube-formation assay
Matrigel (BD Biosciences, San Jose, CA, USA) was dissolved at 4°C overnight, and prechilled 24-well plates were coated with 150 mL of Matrigel, which was allowed to polymerize at 37°C for 30 minutes. HUVECs (2.2×105 cells/well) were suspended in TCM from MDA-MB231 cells treated with different concentrations of HBP and seeded onto the surface of the Matrigel-coated 24-well culture slides. After a 6-hour incubation period, the morphological changes in the cells and the tubular structures formed were observed and photographed using an Olympus CKX41 inverted microscope (magnification 100×). The total lengths of the tubular structures in three randomly chosen microscopic fields per well were measured.
Western blot analysis
Angiogenesis-mediated protein expression was examined via Western blot assay. Briefly, HUVECs (2×105 cells) were cultured in 100 mm-diameter dishes. When they had reached 90% confluence, cells were starved for 12 hours in a medium containing 2% serum and then treated with TCM from MDA-MB231 cells treated with different concentrations of HBP for 24 hours. At the end of the culture period, cells were lysed for 30 minutes in a cold lysis buffer on ice. Protein concentrations were measured using a Bradford protein assay (Bio-Rad Laboratories Inc, Hercules, CA, USA). Equal aliquots of protein (40 μg) were boiled for 5 minutes in a 5× sample buffer and separated on 10% sodium dodecyl sulfate polyacrylamide-gel electrophoresis. Proteins were transferred to nitrocellulose membranes, washed with TBS + Tween 20 (TBST), and blocked for 60 minutes at room temperature in TBST with 5% skim milk. The membranes were washed and incubated with primary antibodies in TBST containing 5% skim milk for 4 hours at 4°C. After three washes, the membranes were incubated with a secondary antibody (HRP-conjugated goat antirabbit IgG, diluted 1:2,000 in 5% skim milk) for 60 minutes. Protein bands were visualized with chemiluminescence reagents (West-Zol). The relative optical densities of the blotting bands were quantified using ImageJ software (Research Services Branch, National Institute of Mental Health, Bethesda, Maryland, USA).
In vivo tissue distribution of HBP
The in vivo distribution of HBP rhodamine was examined following intraperitoneal injections of each sample (PBS, NHBP rhodamine, and HBP rhodamine). MDA-MB231-Luc-D3H1 cells were inoculated into the right hind flank of nude mice (3×106 cells per mouse in 100 μL Matrigel mixed with PBS). After tumors had grown to 50 mm3, mice received a 100 μL intraperitoneal injection of each sample at a final dose of 1 mg/kg peptide rhodamine. After 24 hours, the mice were sacrificed, and their tissues were collected. The distribution of the peptides was determined via optical imaging using the IVIS® imaging system (PerkinElmer Inc, Waltham, MA, USA), and fluorescence uptake was examined using a confocal microscope to examine frozen heart, kidney, liver, spleen, and tumor sections mounted in optimal cutting-temperature compound (Sakura Finetek, Tokyo, Japan).
Human breast xenograft models
Female BALB/c nude mice (5–6 weeks old; Japan SLC Inc, Hamamatsu, Japan) were used to develop breast cancer xenografts for these studies. Animal selection and management and surgical protocols and preparation followed the guidelines approved by the institutional animal care and use committee of Seoul National University. All studies were performed in accordance with an animal protocol approved by the Seoul National University Institutional Animal Care and Use Committee (protocol number: SNU-12-0161). MDA-MB231-Luc-D3H1 cells were grown to 80%–90% confluence, harvested, prepared as suspensions of 3×106 cells/100 μL in PBS mixed with Matrigel, and inoculated into the right hind flanks of nude mice. Each experimental condition had two animals per group. After tumors had grown to approximately 50 mm3, mice were treated with HBP (1 mg/kg) administered via intraperitoneal injections three times a week for 5 consecutive weeks at the doses indicated. The control mouse group was administered the control solution containing the same amount of NHBP, and untreated mice were injected with PBS. Twice weekly, starting on day 1, mice were weighed and solid-tumor volume determined using Vernier calipers based on the equation A × B2 ×0.52, where A was the longest diameter of the tumor and B the shortest diameter of the tumor.20 In addition, tumor growth was visualized using the IVIS imaging system. Mice were anesthetized using isoflurane and then intraperitoneally injected with D-luciferin at 50 μg/mL 10 minutes before imaging. Images of the bioluminescence were acquired every 3 days. After 5 weeks, mice were sacrificed, and solid tumors were harvested and fixed in formalin for subsequent analysis via immunohistochemistry.
Immunohistochemistry analysis
The vasculature within the tumors was examined via immunohistochemistry techniques specific for angiogenesis-mediated markers (CD31, CD34, CD44, and VEGFR2). Tissues were fixed in 10% formalin prior to paraffin embedding. Sections (4 mm thick) were deparaffinized using xylene and rehydrated in a series of ethanol solutions at concentrations ranging from 95% to 100%. Antigen retrieval was achieved by incubating samples with citrate buffer (pH 6) at room temperature, followed by heating them in microwave for 10 minutes. Endogenous peroxidase activity was blocked using a peroxidase-blocking reagent (Dako Denmark A/S, Glostrup, Denmark) for 5 minutes in a wet chamber. The sections were incubated with CD31 (Abcam ab28364, 1:100), CD34 (Abcam ab81289, 1:100), CD44 (Abcam ab51037, 1:100), and VEGFR2 (Abcam ab39256, 1:100) in EnVision™ Flex antibody diluent (Dako). Detection of antibody binding was performed via incubation with the EnVision Flex/HRP (Dako) for 30 minutes. Slides were reacted with a diaminobenzidine (DAB) solution for 2–3 minutes (EnVision Flex DAB + chromogen). The intensity and distribution of CD31-, CD34-, CD44-, and VEGFR2-specific immunostaining were assessed using the Olympus DP72 camera following hematoxylin counterstaining.
Statistical analysis
All experiments were conducted independently at least three times, unless stated otherwise. All values were expressed as mean ± standard deviation, and the mean values were compared using Student’s t-test.
Results
Ability of HBP to bind to heparin and effect on cell viability
HBP was synthesized based on Fmoc chemistry with a C-terminal amide, and NHBP (GAPPPADSAP) synthesized with a mismatched HBP sequence was used as a control to assess the binding affinity of HBP to heparin and its antiangiogenic activity. The amino acid sequence of HBP corresponded to residues 15–24 of BMP4 (RKKNPNCRRH) (Figure 1A).12 Angiogenic growth factors, including TGFβ1, FGF, and PDGF, have heparin-binding sites composed of positive amino acids (Arg, Lys, and His). It is known that the binding of HBP to HSPGs is based on the ionic interaction between the positively charged residues of HBP and the negatively charged group of HSPGs. Figure 1B shows the coincidence between the heparin-binding consensus sequence (BBBXTXXBBB) and the actual three-dimensional structure of HBP. The proline in HBP acts as a turning site, and might give flexibility to the structure of HBP. Figure 2A shows that HBP had binding activity to heparin at 50, 100, and 200 μM, as determined in a slot-blot assay. The mismatched peptide showed no binding affinity to heparin. The heparin-binding affinity result was consistent with our previous study.12 The current study further confirms the previous results, reaching maximum affinity at a concentration of 100 μM (Figure 2B). Our current data seemed to present low binding affinity starting 50 μM peptide, analyzed by a slot-blot assay. However, HBP possesses higher affinity even at a low concentration (5 nM) with target selectivity, as reported in our previous study.
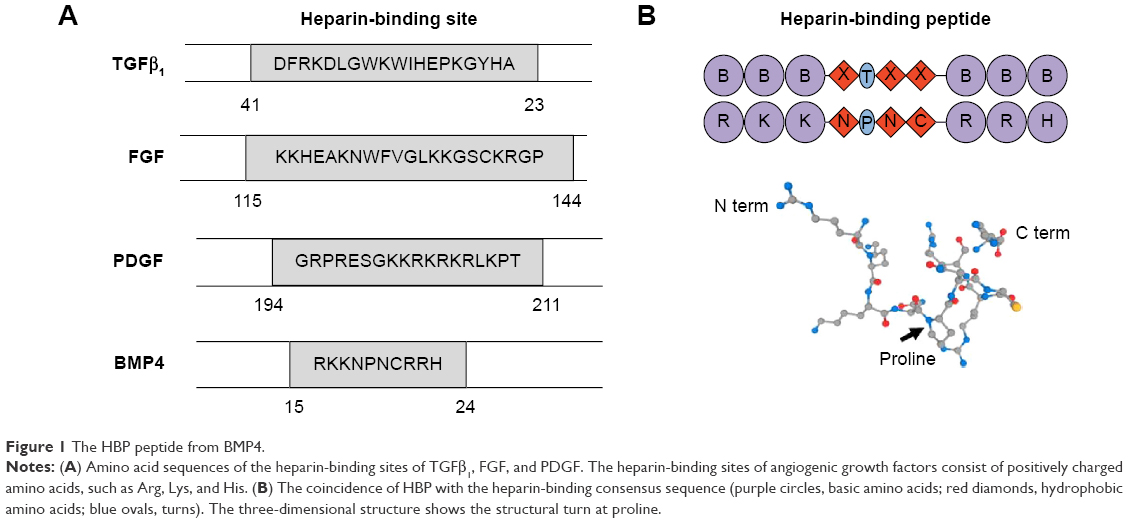 | Figure 1 The HBP peptide from BMP4. |
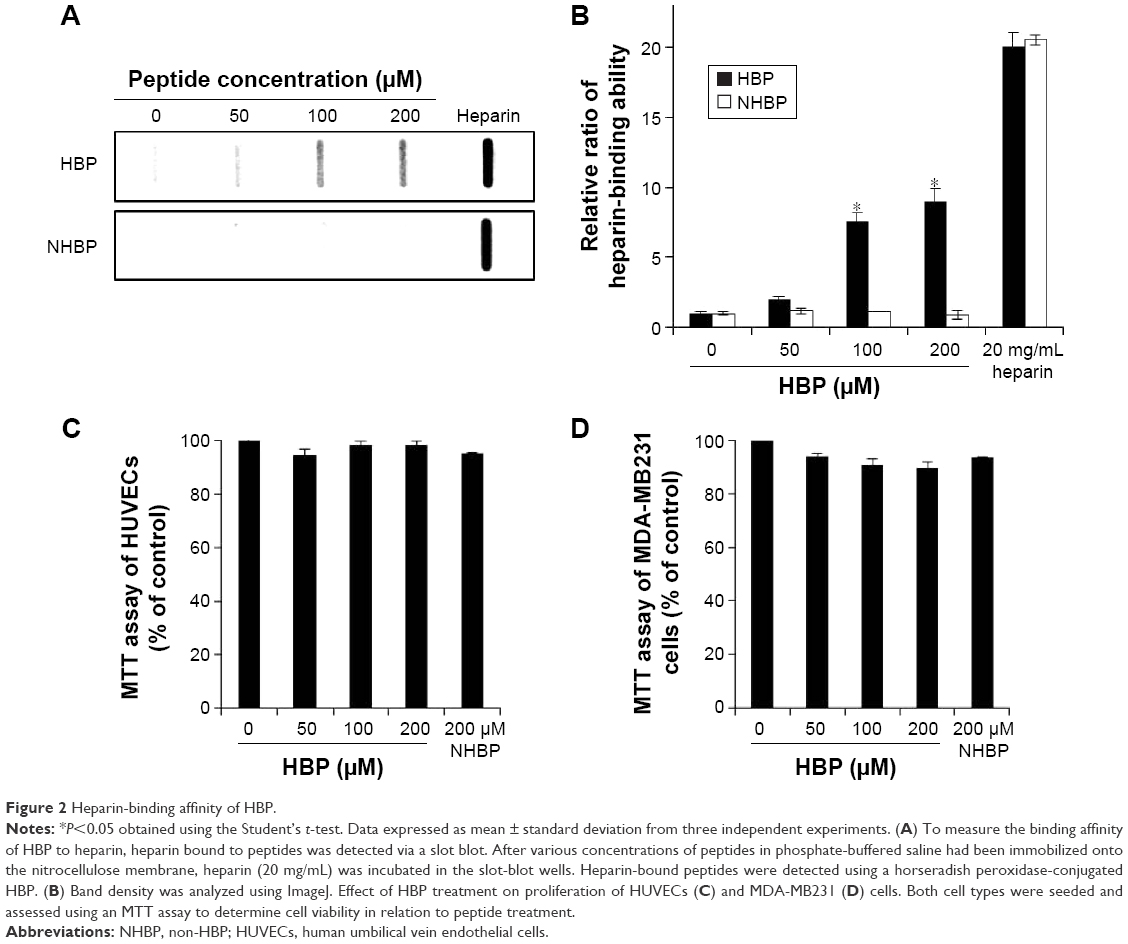 | Figure 2 Heparin-binding affinity of HBP. |
We investigated the cytotoxic effects of HBP on HUVECs and MDA-MB231 cells. The viability of HUVECs and MDA-MB231 cells treated with HBP did not decrease compared with untreated cells or those treated with NHBP (Figure 2C and D). This shows that HBP itself has no dose-dependent cytotoxic effects on HUVECs or MDA-MB231 cells.
Inhibition of endothelial cell migration and the differentiation of capillary-like structures in vitro
To assess the antiangiogenic action of HBP in vitro, we examined its inhibitory function on chemotactic motility via a transwell migration assay and a scratch-migration assay using ECs. Our results showed that the number of invasive ECs in TCM from HBP-treated MDA-MB231 cells were dramatically decreased relative to the number in TCM from NHBP- or VEGF-treated MDA-MB231 cells, and that this decrease occurred in a dose-dependent manner (Figure 3A). NHBP and VEGF showed no inhibitory effect on the migration of ECs. Cell invasion showed a 73% decrease in the group treated with 200 μM HBP relative to the untreated cells (Figure 4A). In addition, the scratch-migration assay confirmed that a greater than 80% reduction was achieved in the group treated with 200 μM HBP, suggesting a potent inhibitory effect of HBP on EC motility (Figures 3B and 4B).
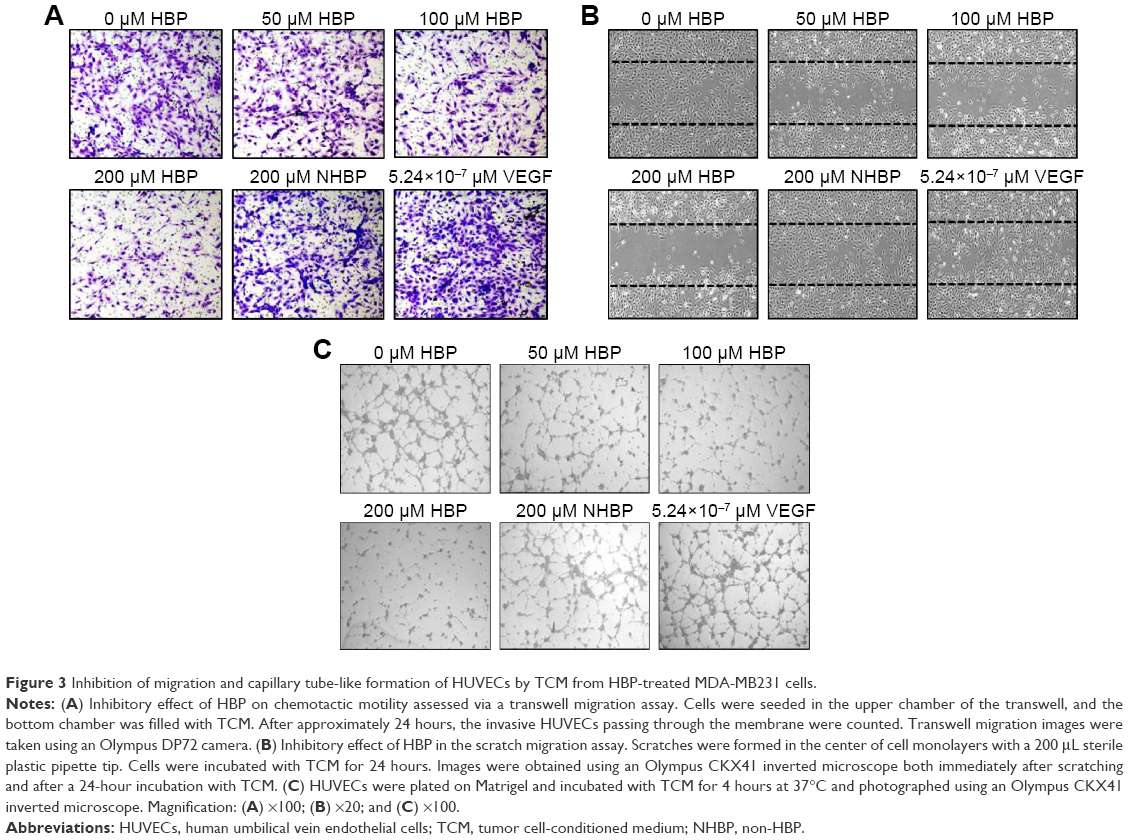 | Figure 3 Inhibition of migration and capillary tube-like formation of HUVECs by TCM from HBP-treated MDA-MB231 cells. |
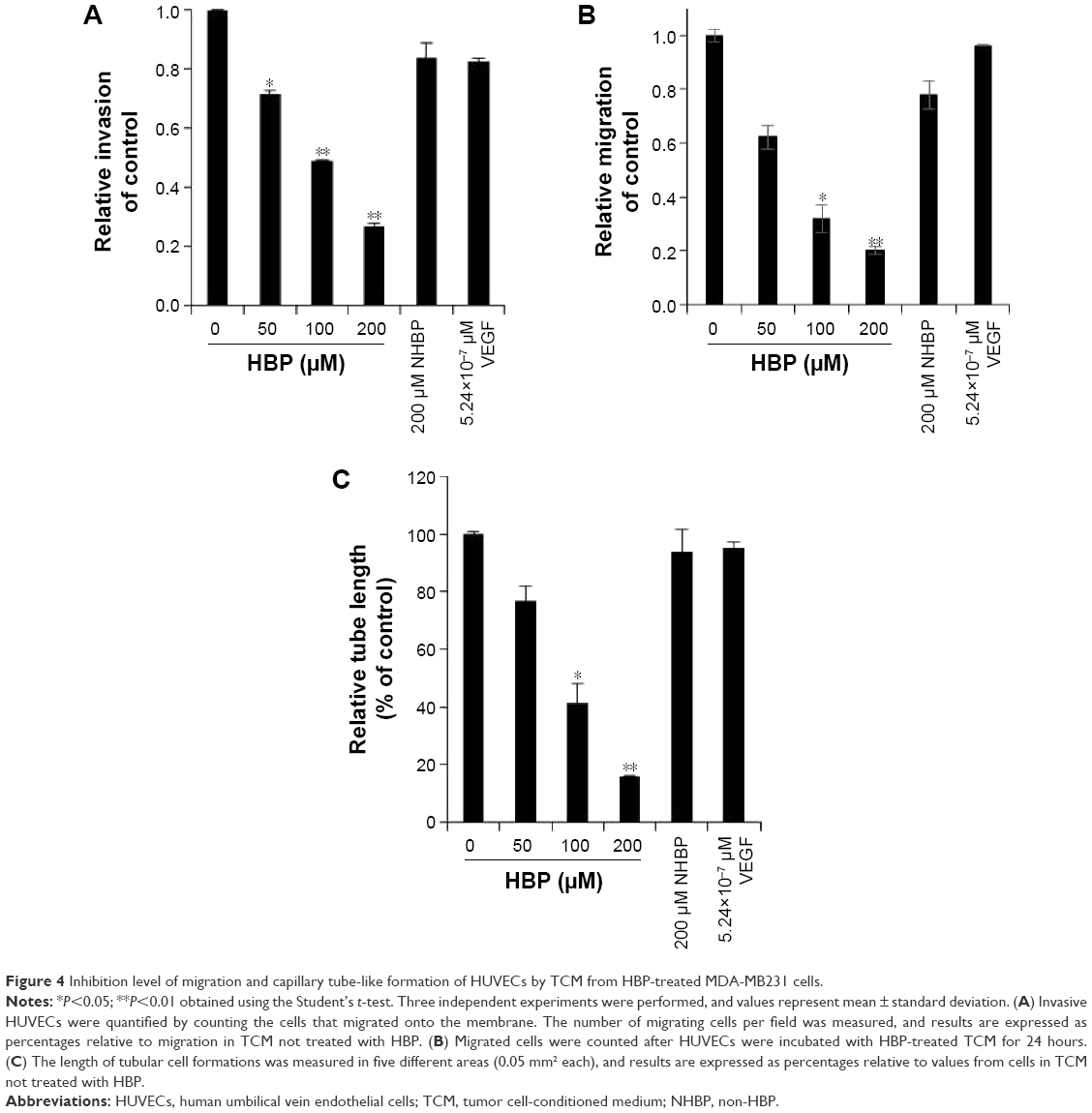 | Figure 4 Inhibition level of migration and capillary tube-like formation of HUVECs by TCM from HBP-treated MDA-MB231 cells. |
To explore the ability of HBP to inhibit angiogenesis, we measured its ability to slow the formation of vascular structures using an in vitro two-dimensional Matrigel assay. In this study, Matrigel was used to replace the basement membrane as a way to observe the effects of TCM from HBP-treated MDA-MB231 cells on the tubular formation of HUVECs. As shown in Figure 3C, when HUVECs were seeded on two-dimensional Matrigel, robust tube-like structures were formed. However, treatment with HBP significantly suppressed the tubular formation by HUVECs in a dose-dependent manner compared with the formation that occurred in the untreated group and the NHBP- and VEGF-treated group. The length of the capillary-like structures was significantly decreased in the group treated with 100 μM HBP, and an 80% decrease in capillary length was observed in the group treated with 200 μM HBP (Figure 4C).
Inhibition of angiogenesis-mediated signaling pathways
Expression levels of the p-FAK, p-ERK, and p-Akt proteins were significantly decreased in a dose-dependent manner in the groups treated with HBP (Figure 5A). More specifically, HBP inhibited the expression of p-Akt and p-FAK by approximately 71% at 200 μM HBP compared with their expression in untreated HUVECs (Figure 5B). The expression of p-ERK was decreased by 60% at 200 μM HBP compared with its expression in untreated HUVECs. The phosphorylation of Akt and FAK was more influenced by HBP than the phosphorylation of ERK. We confirmed that HBP exerted antiangiogenic effects on HUVECs and showed that the suppression of angiogenesis was related to a downstream signaling cascade.
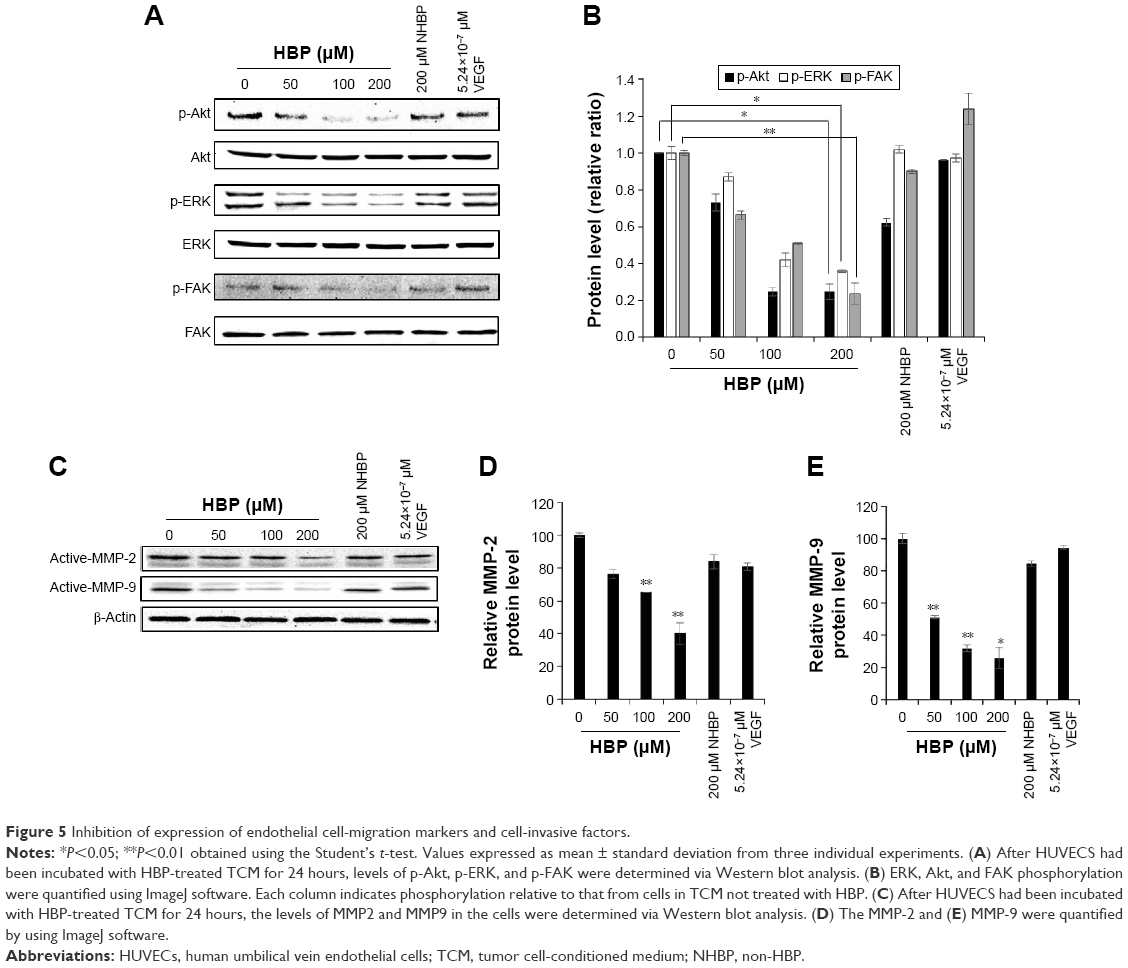 | Figure 5 Inhibition of expression of endothelial cell-migration markers and cell-invasive factors. |
Furthermore, we also examined the expression levels of MMP2 and MMP9 in relation to cell migration in HBP-treated HUVECs. We found that HBP treatment led to the downregulation of these migration-mediated proteins in HUVECs (Figure 5C). The expressions of MMP2 and MMP9 were not affected by VEGF or NHBP. The expression of MMP9 decreased to a greater extent than MMP2 (Figure 5D and E). These data confirm that HBP decreased cell migration in a dose-dependent manner through the downregulation of an MMP2- and MMP9-related pathway.
Targeted distribution of HBP into tumors in vivo
The expression of HSPGs around tumors is dramatically increased in comparison to its expression around other tissues.21 We investigated whether HBP can be targeted to tumors in a breast cancer xenograft mouse model. Rhodamine-labeled peptides were intraperitoneally injected into xenografted mice as a test group. PBS and NHBP were injected into the control groups. The distribution of HBP in these mice was determined 24 hours after the injection using the IVIS imaging system. No fluorescent signal was observed in the PBS-treated mice, and slight fluorescence was observed in the NHBP-treated mice. Rhodamine fluorescence was observed only in tumors and not the heart, liver, spleen, or kidneys in HBP-treated mice. HBP treatment produced appreciable fluorescence in tumors compared with fluorescence in mice given NHBP (Figure 6A). Xenograft tumors were excised and embedded in optimal cutting-temperature compound to evaluate the penetration of HBP into the tumors. Confocal imaging data showed that experimental mice had no significant fluorescence in their spleens, livers, hearts, or kidneys (Figure 6B). A detailed analysis via confocal microscopy revealed that fluorescence staining was predominantly present inside and on the surface of the tumors in the HBP-treated group (Figure 6C). In contrast, only a marginal distribution of fluorescence was observed at the surface of tumors when NHBP was injected. These data demonstrate that tumors were the major site of HBP distribution and that HSPGs can potentially be targeted in breast cancer.
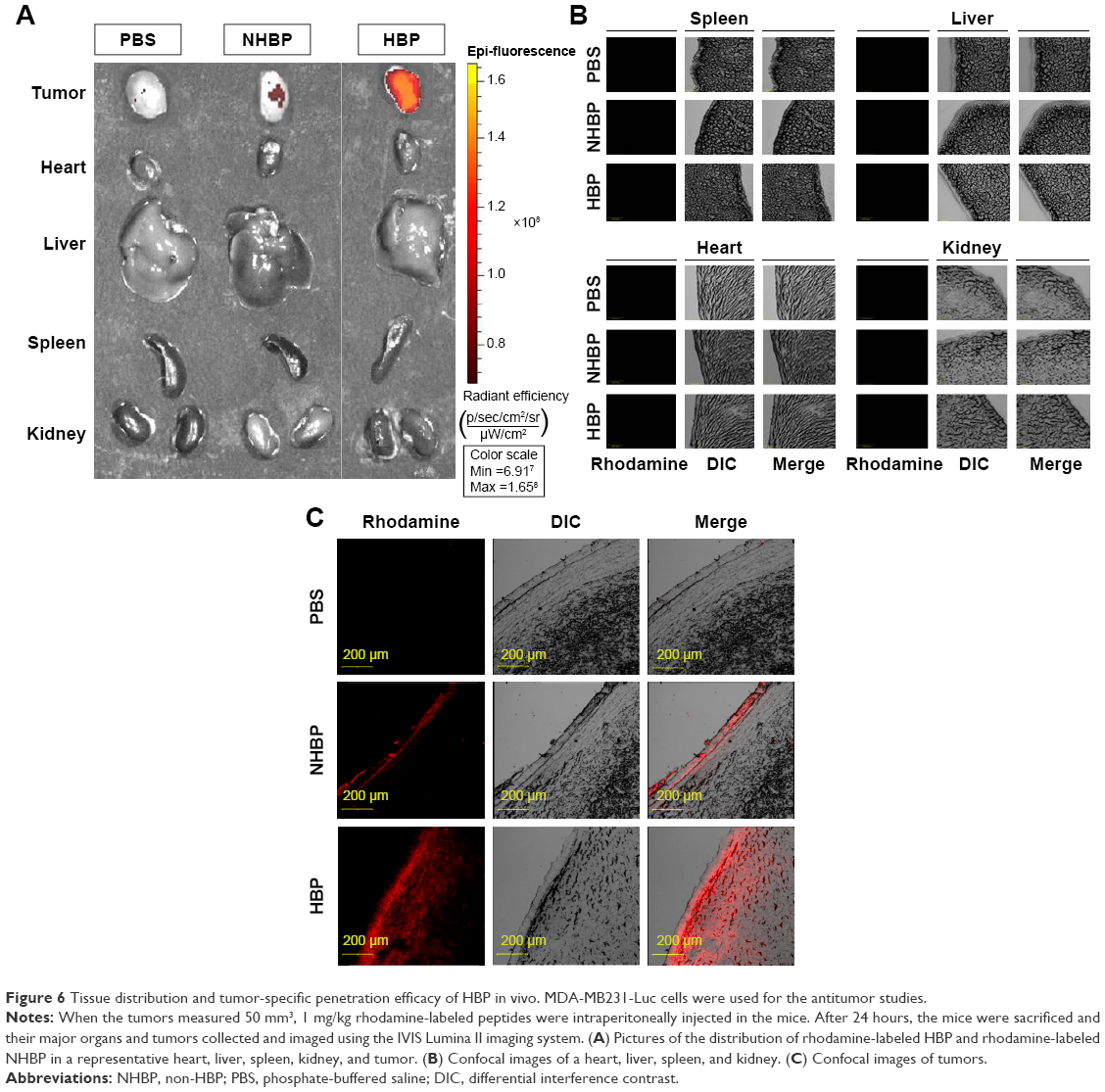 | Figure 6 Tissue distribution and tumor-specific penetration efficacy of HBP in vivo. MDA-MB231-Luc cells were used for the antitumor studies. |
Suppression of tumor growth and angiogenesis in a breast cancer xenograft mouse model
To investigate the anticancer activity of HBP on tumor growth and tumor angiogenesis in vivo, experiments were conducted on a breast cancer xenograft mouse model. We found that the administration of the HBP (1 mg/kg/day, three injections per week) for 5 weeks suppressed tumor volume compared with the effects of PBS and NHBP (Figure 7A). The average tumor volume of the PBS- and NHBP-administered group increased from 50.02±3.21 mm3 to 528.39±80.25 mm3 (10.6-fold increase) and from 50.58±2.01 mm3 to 474.65±62.51 mm3 (9.4-fold increase), respectively, at the end of the experiments, whereas the HBP group showed a smaller increase: from 51.16±3.32 mm3 to 231.75±32.51 mm3 (4.5-fold increase). There was no effect of HBP on the body weight of mice, demonstrating that HBP is a safe indirect agent (Figure 7B). In addition, real-time bioluminescence imaging of tumor grafts expressing luciferase in mice demonstrated a strong correlation between tumor volume and the reduction of luciferase expression as a result of HBP treatment (Figure 7C). Tumor sizes and the luciferase intensity in HBP-treated mice were reduced relative to those in the PBS- and the NHBP-treated mice.
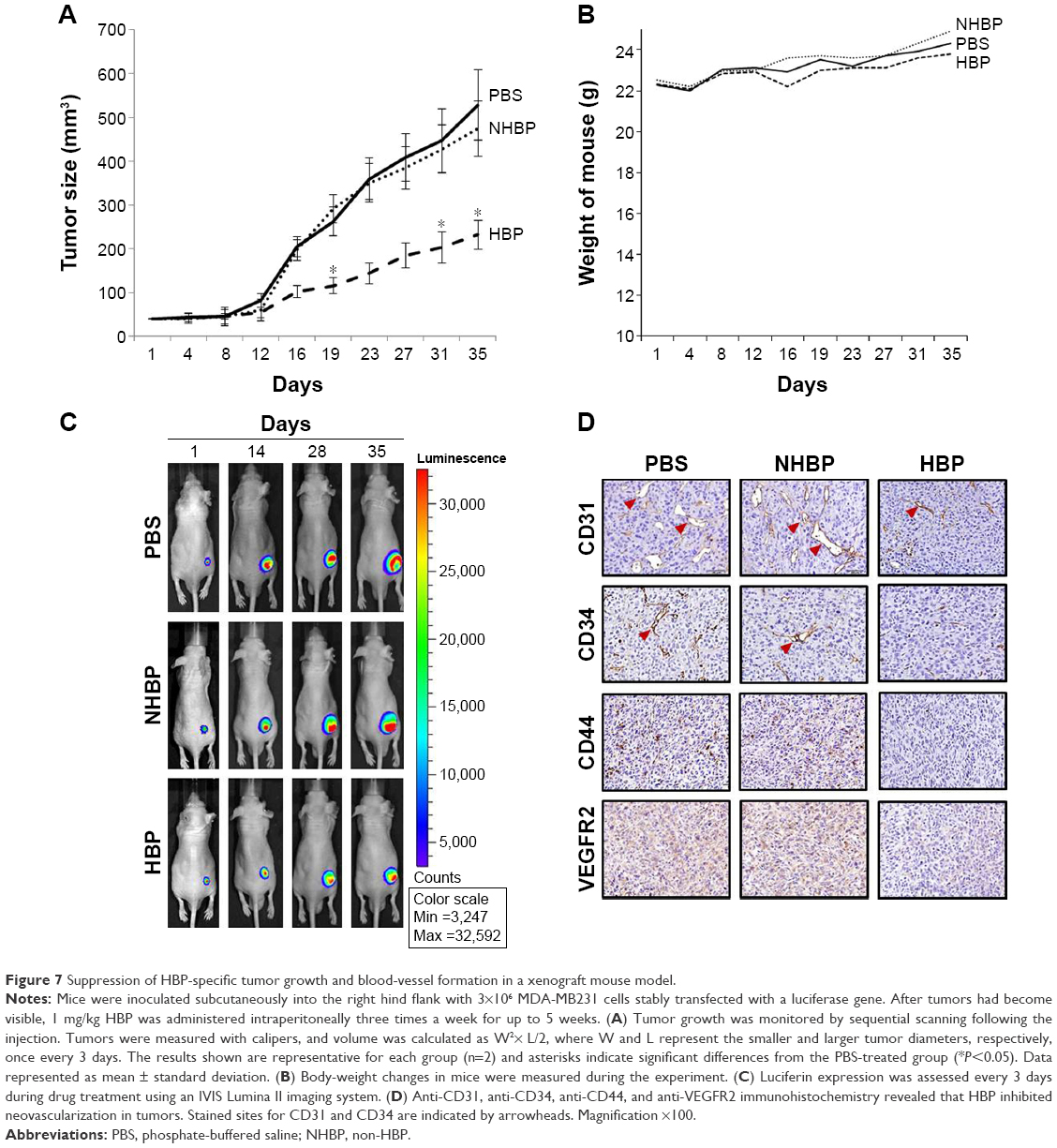 | Figure 7 Suppression of HBP-specific tumor growth and blood-vessel formation in a xenograft mouse model. |
To examine further whether HBP inhibits angiogenesis, we carried out an immunohistochemical analysis on tumor sections using anti-CD31, anti-CD44, anti-VEGFR2, and anti-CD34. Figure 7D shows that four angiogenesis-related markers were decreased and the expressions of CD31 and CD34 remarkably inhibited by HBP. These results demonstrate that the administration of HBP significantly reduces tumor growth by inhibiting neovascularization in a breast cancer xenograft model.
Discussion
Tumors are composed of a complex mixture of many cell types, including ECs, immune cells, and fibroblasts.22 Solid-tumor malignancies, including breast, lung, and prostate carcinomas, are considered to be angiogenesis-dependent. Consequently, angiogenesis inhibitors are receiving increased attention as cancer therapeutics, and several angiogenesis inhibitors are in development.23,24 HSPGs are usually differentially expressed during the development of cancer compared with their expression in healthy tissue and cells. The main importance of HSPGs in tumor growth is their ability to bind key growth factors and stromal signaling molecules that activate tumor cells, influencing signaling, cell–cell interactions, uncontrolled proliferation, microenvironment modulation, migration, and growth-factor storage.25 Due to their multiple roles, HSPGs are considered targets in the treatment of cancer, and active agents that can affect HSPG expression may be applicable for the control of tumor growth.26,27 A previous study reported that heparin-binding sites frequently contain clusters of basic amino acids (XBX, XBBX, XBBBX).16 In addition, HSPGs, including heparin, interact most strongly with peptides containing a complementary binding site with a high positive charge.28 In particular, basic amino acids (arginine and lysine) in heparin-binding sites play a critical role in the interaction of the negatively charged sulfate and carboxylate groups of HSPGs.29 These electrostatic interactions also play a major role in the binding of HSPGs to growth factors and cytokines.30 Many studies have been undertaken to determine whether there is a heparin-binding consensus sequence in HBPs, including FGF4, TGFβ1, HBEGF, and PDGF.31–34 The HBPs are known as angiogenic growth factors, and they promote tumor growth. In contrast, BMP4 has been reported to suppress tumor growth via antiangiogenic effects, even though it has a heparin-binding site; therefore, we developed HBP from BMP4 and evaluated its biological inhibitor role in angiogenesis in vivo and in vitro.
HBP showed a binding affinity to heparin, and was not directly cytotoxic to ECs or cancer cells (Figure 2). Instead, HBP led to an obvious downregulation of angiogenesis in ECs and suppressed cancer cells indirectly by depriving them of nutrients and oxygen. EC migration and tube formation are essential for blood-vessel formation in angiogenesis, and are also critical for tumor growth.35,36 HBP effectively suppressed the migration and matrix gel-induced tube formation of HUVECs in vitro (Figures 3 and 4).
We used a Western blot analysis to examine levels of protein related to the angiogenesis-mediated signaling system in HUVECs treated with TCM from HBP-treated MDA-MB231 cells. To determine whether HBP inhibited downstream signaling molecules, we screened for some key kinases involved in the angiogenic signaling pathway. Treatment with HBP led to an obvious downregulation of the angiogenic signaling system in HUVECs. The phosphorylation of ERK, Akt, and FAK was decreased by HBP treatment (Figure 5A). VEGF induced the activation of ERK1/2, Akt1 (related to survival), and FAK (related to migration).37–39 In addition, the invasive factors MMP2 and MMP9 were considerably decreased by HBP treatment (Figure 5C). Cancers are characterized by their invasiveness into nearby tissues and metastasis to distant locations away from the primary tumor site. In order for these processes to take place, tumor cells break down the surrounding ECM by activating or releasing various proteases, such as MMPs and serine proteases.40–43 These proteases make the ECM more permeable, so that invading cells can pass through established ECM boundaries.44 Recent studies have shown that HSPGs can promote growth-factor signaling and tumor-cell proliferation.8 HBP suppressed intracellular signaling in HUVECs, which might have been a result of blocking of the interaction of growth factors and HSPGs.
These in vitro results provided a rationale for conducting further study related to the control of tumor progression in vivo based on the antiangiogenic activity of HBP. HBP was mainly distributed to tumor sites rather than to organs, and it penetrated tumor tissue (Figure 6). The amino acid sequence of HBP has some similarity with that of cell-penetration peptides. The cell-penetration activity of HBP was evaluated in our preliminary study (data not shown). The capacity for tissue penetration of HBP into tumors was demonstrated in an in vivo model for the first time. It was found that HBP possessed strong antiangiogenic potential that could accelerate tumor-cell death with little effect on other organs in this study. This implies that it can be used as a safe antitumor agent. Despite the reduction capacity of tumor size at 50% by the peptide, it is strongly suggested that the HBP possesses potent antiangiogenic activity with little cytotoxic effects on other organs, because of its target-penetration capacity, based on the current results. This capacity for targeted penetration supports the potential application of HBP as a safe and effective antiangiogenic candidate, anticancer drug-delivery carrier, or a supplement to current anticancer drugs.
Tumor growth in a breast cancer xenograft mouse model was suppressed by 1 mg/kg HBP intraperitoneal injections administered three times per week (Figure 7A). The actual injected dose of HBP was 370–450 μg per mouse for 5 weeks (three times per week) based on the weight of mice (25–30 g). On the other hand, cell migration and tube formation in vitro were significantly decreased at 100 μM (130.8 mg/mL) and mostly inhibited at a peptide concentration of 200 μM (261.6 mg/mL). Therefore, the injected amount of HBP in vivo was not considered a high dose compared with that used in vitro. The 370–450 μg HBP was effective for tumor suppression, and it was indirectly demonstrated that HBP had effective binding affinity to HSPGs. In addition, the antiangiogenesis activity of HBP in vivo was demonstrated by the decreased expression of CD31 and CD34 (Figure 7D). Koskimaki et al reported that their antiangiogenic peptides were effective in a breast cancer xenograft model at a dose of 10 mg/kg and 20 mg/kg when intraperitoneally injected once per day.45 In our study, HBP was effective at 1 mg/kg with three injections per week, which suggests that HBP is more potent and has a longer effective duration time. HBP binds with HSPGs on the surface of cancer cells through ionic interaction, and blocks the interactions between HSPGs and angiogenic growth factors, such as FGF and VEGF. This induces inhibition of the expression of angiogenic protein markers, EC migration, and tube formation, leading to the suppression of tumor growth (Figure 8). The mechanism of action of HBP on the suppression of tumor growth is via control of the tumor microenvironment through inhibiting angiogenesis, rather than via a direct cytotoxic effect on cancer cells. Suppressed angiogenesis determined by wound migration can be explained in two ways. The first is reducing the induction of blood-vessel formation near tumor tissue that will help tumor growth, and the second is reducing the blood vessels inside tumor tissue, thereby resulting in tumor-cell death.
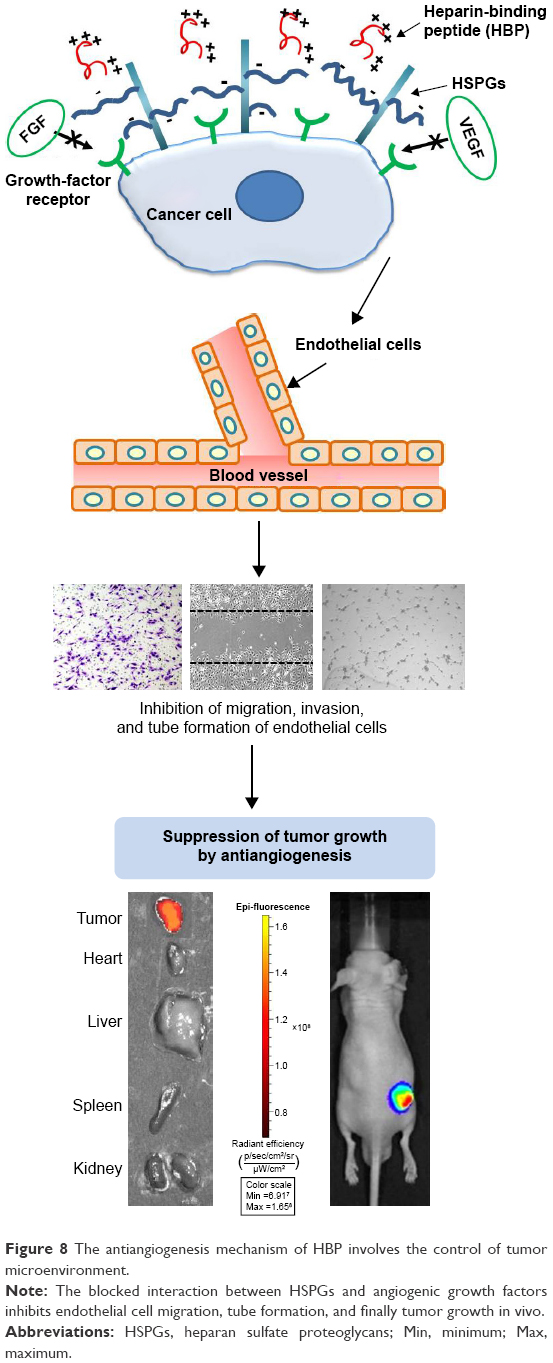 | Figure 8 The antiangiogenesis mechanism of HBP involves the control of tumor microenvironment. |
According to our results, HBP has a role in reducing angiogenesis near tumor tissue and leads to tumor-size suppression. HBP can be utilized as a targeting moiety for diagnostics or drug-delivery purposes, due to the absence of a direct tumor cell-killing effect.
Conclusion
HBP effectively inhibits EC migration in vitro by blocking the interaction of HSPGs with angiogenic growth factors and does not show any cytotoxicity. In addition, HBP suppressed tumor growth in vivo by blocking of angiogenesis and showed targeted distribution into tumor sites. Taken together, these results show that HBP might be useful as a potential antitumor therapeutic agent as an alternative or complementary approach.
Acknowledgments
This study was supported by a National Research Foundation of Korea Grant, through the Oromaxillofacial Dysfunction Research Center for the Elderly (2012000912) at Seoul National University, and in part by the Bio and Medical Technology Development Program of the National Research Foundation (NRF), funded by the Ministry of Science, ICT, and Future Planning (2014M3A9E3064430, 2012M3A9C6049727).
Disclosure
The authors report no conflicts of interest in this work.
References
Risau W. Mechanisms of angiogenesis. Nature. 1997;386(6626):671–674. | ||
Sledge GW Jr, Miller KD. Angiogenesis and antiangiogenic therapy. Curr Probl Cancer. 2002;26(1):1–60. | ||
Muthukkaruppan VR, Kubai L, Auerbach R. Tumor-induced neovascularization in the mouse eye. J Natl Cancer Inst. 1982;69(3): 699–708. | ||
Sachdev JC, Jahanzeb M. Evolution of bevacizumab-based therapy in the management of breast cancer. Clin Breast Cancer. 2008;8(5): 402–410. | ||
Salter JT, Miller KD. Antiangiogenic agents in breast cancer. Cancer Invest. 2007;25(7):518–526. | ||
Liu D, Shriver Z, Venkataraman G, El Shabrawi Y, Sasisekharan R. Tumor cell surface heparan sulfate as cryptic promoters or inhibitors of tumor growth and metastasis. Proc Natl Acad Sci U S A. 2002;99(2):568–573. | ||
Iozzo RV, San Antonio JD. Heparan sulfate proteoglycans: heavy hitters in the angiogenesis arena. J Clin Invest. 2001;108(3):349–355. | ||
Liu D, Shriver Z, Qi Y, Venkataraman G, Sasisekharan R. Dynamic regulation of tumor growth and metastasis by heparan sulfate glycosaminoglycans. Semin Thromb Hemost. 2002;28(1):67–78. | ||
Sasisekharan R, Venkataraman G. Heparin and heparan sulfate: biosynthesis, structure and function. Curr Opin Chem Biol. 2000;4(6):626–631. | ||
Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2(7):521–528. | ||
Niers TM, Klerk CP, DiNisio M, et al. Mechanisms of heparin induced anti-cancer activity in experimental cancer models. Crit Rev Oncol Hematol. 2007;61(3):195–207. | ||
Choi YJ, Lee JY, Park JH, et al. The identification of a heparin binding domain peptide from bone morphogenetic protein-4 and its role on osteogenesis. Biomaterials. 2010;31(28):7226–7238. | ||
Shon SK, Kim A, Kim JY, Kim KI, Yang Y, Lim JS. Bone morphogenetic protein-4 induced by NDRG2 expression inhibits MMP-9 activity in breast cancer cells. Biochem Biophys Res Commun. 2009;385(2):198–203. | ||
Tsuchida R, Osawa T, Wang F, et al. BMP4/thrombospondin-1 loop paracrinically inhibits tumor angiogenesis and suppresses the growth of solid tumors. Oncogene. 2014;33(29):3803–3811. | ||
Xu J, Zhu D, Sonoda S, et al. Over-expression of BMP4 inhibits experimental choroidal neovascularization by modulating VEGF and MMP-9. Angiogenesis. 2012;15(2):213–227. | ||
Cardin AD, Weintraub HJ. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis. 1989;9(1):21–32. | ||
Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC. Heparin structure and interactions with basic fibroblast growth factor. Science. 1996;271(5252):1116–1120. | ||
Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays. 1998;20(2):156–167. | ||
Utoguchi N, Mizuguchi H, Dantakean A, et al. Effect of tumour cell-conditioned medium on endothelial macromolecular permeability and its correlation with collagen. Br Journal Cancer. 1996;73(1):24–28. | ||
Choi YS, Lee JY, Suh JS, et al. The systemic delivery of siRNAs by a cell penetrating peptide, low molecular weight protamine. Biomaterials. 2010;31(6):1429–1443. | ||
Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13(1):9–22. | ||
Ziyad S, Iruela-Arispe ML. Molecular mechanisms of tumor angiogenesis. Genes Cancer. 2011;2(12):1085–1096. | ||
Doggrell SA. Pegaptanib: the first antiangiogenic agent approved for neovascular macular degeneration. Expert Opin Pharmacother. 2005;6(8):1421–1423. | ||
Wedam SB, Low JA, Yang SX, et al. Antiangiogenic and antitumor effects of bevacizumab in patients with inflammatory and locally advanced breast cancer. J Clin Oncol. 2006;24(5):769–777. | ||
Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011;3(7):a004952. | ||
Dedes PG, Gialeli C, Tsonis AI, et al. Expression of matrix macromolecules and functional properties of breast cancer cells are modulated by the bisphosphonate zoledronic acid. Biochim Biophys Acta. 2012;1820(12):1926–1939. | ||
Malavaki CJ, Roussidis AE, Gialeli C, et al. Imatinib as a key inhibitor of the platelet-derived growth factor receptor mediated expression of cell surface heparan sulfate proteoglycans and functional properties of breast cancer cells. FEBS J. 2013;280(10):2477–2489. | ||
Fromm JR, Hileman RE, Caldwell EE, Weiler JM, Linhardt RJ. Pattern and spacing of basic amino acids in heparin binding sites. Arch Biochem Biophys. 1997;343(1):92–100. | ||
Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41(3):391–412. | ||
Fromm JR, Hileman RE, Caldwell EE, Weiler JM, Linhardt RJ. Differences in the interaction of heparin with arginine and lysine and the importance of these basic amino acids in the binding of heparin to acidic fibroblast growth factor. Arch Biochem Biophys. 1995;323(2):279–287. | ||
Bellosta P, Iwahori A, Plotnikov AN, Eliseenkova AV, Basilico C, Mohammadi M. Identification of receptor and heparin binding sites in fibroblast growth factor 4 by structure-based mutagenesis. Mol Cell Biol. 2001;21(17):5946–5957. | ||
Khachigian LM, Chesterman CN. Synthetic peptides representing the alternatively spliced exon of the platelet-derived growth factor A-chain modulate mitogenesis stimulated by normal human serum and several growth factors. J Biol Chem. 1992;267(11):7478–7482. | ||
McCaffrey TA, Falcone DJ, Du B. Transforming growth factor-β1 is a heparin-binding protein: identification of putative heparin-binding regions and isolation of heparins with varying affinity for TGF-β1. J Cell Physiol. 1992;152(2):430–440. | ||
Thompson SA, Higashiyama S, Wood K, et al. Characterization of sequences within heparin-binding EGF-like growth factor that mediate interaction with heparin. J Biol Chem. 1994;269(4):2541–2549. | ||
Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100(6):782–794. | ||
Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. | ||
Abedi H, Zachary I. Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J Biol Chem. 1997;272(24):15442–15451. | ||
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling: in control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359–371. | ||
Takahashi T, Shibuya M. The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-γ pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene. 1997;14(17):2079–2089. | ||
Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277(5323):225–228. | ||
Matrisian LM, Wright J, Newell K, Witty JP. Matrix-degrading metalloproteinases in tumor progression. Princess Takamatsu Symp. 1994;24:152–161. | ||
Murphy AN, Unsworth EJ, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinases-2 inhibits bFGF-induced human microvascular endothelial cell proliferation. J Cell Physiol. 1993;157(2):351–358. | ||
Pratheeshkumar P, Kuttan G. Andrographolide inhibits human umbilical vein endothelial cell invasion and migration by regulating MMP-2 and MMP-9 during angiogenesis. J Environ Pathol Toxicol Oncol. 2011;30(1):33–41. | ||
Liabakk NB, Talbot I, Smith RA, Wilkinson K, Balkwill F. Matrix metalloprotease 2 (MMP-2) and matrix metalloprotease 9 (MMP-9) type IV collagenases in colorectal cancer. Cancer Res. 1996;56(1):190–196. | ||
Koskimaki JE, Karagiannis ED, Rosca EV, et al. Peptides derived from type IV collagen, CXC chemokines, and thrombospondin-1 domain-containing proteins inhibit neovascularization and suppress tumor growth in MDA-MB-231 breast cancer xenografts. Neoplasia. 2009;11(12):1285–1291. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.