Back to Journals » Clinical Ophthalmology » Volume 12
Dry eye syndrome: developments and lifitegrast in perspective
Authors Lollett IV, Galor A ![]()
Received 13 September 2017
Accepted for publication 23 December 2017
Published 15 January 2018 Volume 2018:12 Pages 125—139
DOI https://doi.org/10.2147/OPTH.S126668
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Ivonne V Lollett,1 Anat Galor2,3
1University of Miami, Miller School of Medicine, Miami, FL, 2Ophthalmology Department, Miami Veterans Administration Medical Center, Miami, FL, 3Bascom Palmer Eye Institute, University of Miami, Miller School of Medicine, Miami, FL, USA
Abstract: Dry eye (DE) is a chronic ocular condition with high prevalence and morbidity. It has a complex pathophysiology and is multifactorial in nature. Chronic ocular surface inflammation has emerged as a key component of DE that is capable of perpetuating ocular surface damage and leading to symptoms of ocular pain, discomfort, and visual phenomena. It begins with stress to the ocular surface leading to the production of proinflammatory mediators that induce maturation of resident antigen-presenting cells which then migrate to the lymph nodes to activate CD4 T cells. The specific antigen(s) targeted by these pathogenic CD4+ T cells remains unknown. Two emerging theories include self-antigens by autoreactive CD4 T cells or harmless exogenous antigens in the setting of mucosal immunotolerance loss. These CD4 T cells migrate to the ocular surface causing additional inflammation and damage. Lifitegrast is the second topical anti-inflammatory agent to be approved by the US Food and Drug Administration for the treatment of DE and the first to show improvement in DE symptoms. Lifitegrast works by blocking the interaction between intercellular adhesion molecule-1 and lymphocyte functional associated antigen-1, which has been shown to be critical for the migration of antigen-presenting cells to the lymph nodes as well as CD4+ T cell activation and migration to the ocular surface. In four large multicenter, randomized controlled trials, lifitegrast has proven to be effective in controlling both the signs and symptoms of DE with minimal side effects. Further research should include comparative and combination studies with other anti-inflammatory therapies used for DE.
Keywords: lifitegrast, SAR1118, dry eye syndrome, inflammation, intercellular adhesion molecule 1, lymphocyte function-associated antigen 1
Introduction
Dry eye (DE) is a chronic ocular condition with high prevalence and morbidity.1 It leads to symptoms of ocular pain, discomfort, and visual dysfunction which can greatly impact the quality of life (QOL).2 DE is multifactorial in nature with a complex pathophysiology.3 One important facet is chronic ocular surface inflammation that can perpetuate ocular surface damage.4 Lifitegrast is the second topical anti-inflammatory agent to be approved by the US Food and Drug Administration (FDA) for the treatment of DE and the first to show improvement in DE symptoms.5 This review discusses the contribution of inflammation in DE in both animal models and humans and highlights the place of lifitegrast in the treatment of the disease.
DE disease definition, classification, and epidemiology
As per the Tear Film and Ocular Surface Dry Eye WorkShop II, DE is a multifactorial ocular surface disease causing ocular symptoms.3 It is characterized by a loss of tear film homeostasis as a result of tear film instability and hyperosmolarity, ocular surface inflammation and damage, or neurosensory pathology.3
DE is classified into two major groups based on the effect on tear film stability: aqueous deficient (decreased production of the aqueous component of the tear film) and evaporative (increased evaporation of the tear film).3 Aqueous deficient DE disease (ADDE) is the least common and accounts for 14% of DE.6 ADDE is further classified as Sjögren’s syndrome DE and non-Sjögren’s syndrome DE.3 Evaporative DE (EDE) is far more common and accounts for 50% of DE.6 The most common cause is meibomian gland dysfunction (MGD) leading to meibum deficiency.3 The remaining 36% of DE patients show evidence of both ADDE and EDE.6
DE affects 5%–30% of the world population 50 years and older, with women representing approximately two-thirds of those affected.1,7 Many factors have been linked to DE, including hormonal alterations (eg, menopause),8 medication use (eg, antihistamines),9 and comorbidities (eg, depression).10 DE can also be seen after ophthalmic procedures such as LASIK, where it has been reported in up to 95% of individuals immediately after surgery and 60% of individuals 1 month after surgery.11 DE is also seen to a lesser degree following cataract surgery, with an estimated incidence of 9.8%.12
DE has a significant impact on QOL due to chronic symptoms of ocular pain and discomfort in addition to functional vision impairment. Utility assessment is a tool for quantifying QOL and comparing QOL across different medical conditions. When applied to patients with moderate to severe DE, utility assessment scores showed a decrease in QOL for everyday and leisure activities by 60%, which was comparable to medical conditions such as severe angina, disabling hip fractures, and dialysis.13,14 Furthermore, DE can impact functional vision, interfering with everyday activities such as driving, reading, computer usage, watching television, and work performance.15–17 In addition, DE negatively impacts mental health as DE patients report higher rates of anxiety and depression.18–20 Financially, DE is estimated to cost $3.84 million per year to the health care system with an average annual cost of $783 per patient for treatment alone.21,22
DE symptoms and signs
Currently, there is no “gold standard” test to diagnose DE. Instead, a combination of presenting signs and symptoms is used in clinical judgment. Further complicating the diagnosis of DE is the poor correlation that exists between DE symptoms and signs.23
DE symptoms
The evaluation of DE symptoms can be challenging due to varying presentations and course. For example, some individuals predominantly complain of spontaneous ocular pain/discomfort characterized by different terms (eg, dryness, grittiness, burning, stinging, etc), while other patients may report evoked pain, in particular by wind and light.1 Yet others report visual phenomena in the form of blurry or fluctuating vision.24 Furthermore, some individuals describe transient symptoms, while other have a chronic disease course.1 Several standardized questionnaires have been developed to assess DE symptoms and their effect on QOL. These include the Dry Eye Questionnaire 5,25 the Ocular Surface Disease Index (OSDI),26 the Impact of Dry Eye on Everyday Life,27 and Visual Function Questionnaire-25.28 Most arrive at a DE severity score by lumping together various DE symptoms and there is no consensus on which questionnaire provides the best assessment tool.26,27,29
DE signs
A careful slit-lamp evaluation with a few additional tests can reveal many signs of DE. These signs include decreased tear production measured by Schirmer tear test, tear film instability measured by tear-film breakup time, and corneal or conjunctival epithelial damage measured by the degree of punctate epithelial erosions, best seen after administration of fluorescein, rose bengal, or lissamine green dyes.30 Signs of MGD such as plugging of the meibomian glands, eyelid telangiectasias, and abnormal meibum quality may also be present.30
Point-of-care tests can be used to evaluate subclinical markers of DE, namely, ocular surface inflammation and tear osmolarity. Ocular surface inflammation is assessed by measuring the levels of matrix metallopeptidase (MMP)-9, a protease produced by the epithelial cells in response to inflammatory stress (Inflammadry; Quantel, San Diego, CA, USA).31 Tear film osmolarity is measured with a portable osmometer (TearLAB, San Diego, CA, USA).32,33
Lacrimal functional unit and DE pathophysiology
The lacrimal functional unit (LFU) is composed of the eyelids, lacrimal glands (main and accessory), meibomian glands, cornea, conjunctiva, and their accompanying sensory, autonomic, and motor innervation.34,35 The LFU has three major functions as follows:
- to maintain a pure optical surface for light refraction;36,37
- to maintain ocular surface homeostasis;35 and
- to upregulate and downregulate immune response as needed.38
The LFU accomplishes these goals by regulation of neuronal and hormonal pathways that ultimately control tear flow and quality.35 In a commonly presented schematic, the tear film is statically illustrated as having three layers – an outer lipid layer, a middle aqueous layer, and an inner mucinous layer. The reality, however, is likely more complex as dynamic interactions have been demonstrated between lipids (phospholipids, fatty acids, and cholesterol) in meibum and proteins (lipocalin) in the aqueous layer.39,40
Despite the complexity, it is generally agreed that lipids are produced by meibomian glands located in the tarsal plate.41 They drain a clear oil called meibum that helps reduce the rate of evaporation of the aqueous layer.42
The aqueous layer is the largest component of the tear film and is produced by the lacrimal glands (main and accessory) with smaller contributions from the conjunctival epithelium.43 It is mostly composed of salts that maintain an adequate osmolar gradient and proteins with a wide array of functions. These include growth factors (eg, epidermal growth factor, hepatocyte growth factor, platelet-derived growth factor)44 and defense proteins (eg, lactoferrin, lysozyme, immunoglobulin A)45 that are important for ocular surface repair and immunologic defenses. Anti-inflammatory proteins are also found, including transforming growth factor (TGF)-β, which suppresses antigen-presenting cells (APCs), and tissue inhibitor metalloproteinases (TIMP-1 and TIMP-2), which suppress corneal neovascularization and limit inflammatory cell trafficking into the ocular surface.46–48 Anti-inflammatory omega-3–derived lipids (lipoxins, resolvins, and neuroprotectin) are released into the tear film by resident regulatory polymorphonuclear neutrophils, corneal epithelial cells, and the lacrimal gland.49–54
The mucin layer is produced by conjunctival goblet cells (GCs) which secrete a large gel-forming mucin (MUC5AC) with contributions by the lacrimal glands producing a smaller and soluble mucin (MUC7).55 The mucin layer interacts with the glycocalyx of ocular surface epithelial cells, forming a dense barrier particularly against pathogens, thereby playing an important role in mucosal immunotolerance.55,56 In addition, the mucin layer serves to trap debris and sloughed off epithelial cells as well as facilitates sliding of the eyelid against the corneal and conjunctival epithelium during blinking.57,58
A unifying mechanism in DE is the disruption of one or more components of the LFU, leading to tear film instability and impairment of one or more of its three main functions. The loss of a pure optical surface can lead to aberrant light refraction which can create various visual phenomena.24,37 Tear film instability can give rise to a hyperosmolar tear film either by rapid evaporation of the aqueous component or by normal evaporation of a reduced aqueous component.59 This creates a hostile environment for ocular surface epithelial cells which respond by undergoing apoptosis and releasing proinflammatory mediators as they are injured.60,61 Inflammation is amplified by the loss of anti-inflammatory mechanisms of the LFU.38 Persistent inflammation further damages the ocular surface and can impact sensory nerves, thus dampening reflex tear secretion and causing further tear film instability.1 A feedback cycle can ensue with chronic inflammation leading to more ocular surface damage and, thus, more inflammation.
Inflammation in animal models of DE
Ocular surface inflammation begins with a rapid, but nonspecific innate immune response to stress. In mice, hyperosmolar stress on the ocular surface triggered the release of inflammatory mediators such as interleukins (IL)-1β, tumor necrosis factor (TNF)-α, and MMP-9, which initiate the mitogen-activated protein kinase (MAPK) signaling pathways c-jun n-terminal kinases, extracellular-regulated kinases, and p38.62 In a similar manner, desiccating stress (DS), created by scopolamine plus low humidity, leads to the induction of a wide variety of inflammatory mediators on the ocular surface, including IL-1α, IL-1β, CC chemokine ligand (CCL)2, CCL3, CCL5, C-X-C motif chemokine ligand (CXCL)10, TNF-α, interferon (IFN)-γ, IL-2, IL-6, IL-10, and MMP-9. Again, this inflammatory milieu was found to activate the MAPK signaling pathways.63,64 Interestingly, aging mice were found to spontaneously develop ocular surface inflammation, with elevated IFN-γ and IL-7α on the ocular surface.65
This innate immune response gives way to a slower, but more specific adaptive immune response requiring a complex interaction between APCs, namely, macrophages and dendritic cells (DCs), and CD4+ T cells. In a study by Schaumburg et al,66 acute cytokine production from the initial innate immune response phase of DS-induced DE mice was associated with an increased number of CD11c+ DCs and increased expression of DC maturation markers (major histocompatibility complex II, CD83, CD86, C-C motif receptor 7) which preceded activation of CD4+ T cells.
Once activated by APCs, CD4+ T cells infiltrate and damage the ocular surface with corneal irregularity, corneal barrier disruption, and decreased conjunctival GCs noted.67 The specific antigen(s) targeted by these pathogenic CD4+ T cells remains unknown.
One possibility is that T cell response is aimed at self-antigens of the cornea and conjunctiva. In fact, adoptive transfer of CD4+ T cells from DS-induced DE mice to nude mice was enough to reproduce disease with CD4+ T cells localizing to the lacrimal gland, cornea, and conjunctiva in the nude mice.67 Adoptive transfer of CD4+ T cells from aging mice with signs of DE was similarly effective in replicating disease.65 The important interplay between the APCs and T cells became evident as APC depletion prior to DS induction mitigated the ability of T cells to recreate the disease in nude mice recipients.66
T regulatory cells (Tregs) also play an important role as inflammatory modulators in DE mice. Adoptive transfer of pathogenic CD4+ T cells from DS-induced DE mice to nude mice is effectively attenuated by reconstitution of Treg cells in nude recipient mice.67–69 In DE mice, Treg dysfunction contributes to pathogenesis as Tregs from DS-induced DE mice were less capable of suppressing proliferation of T helper (Th)17 cells, leading to higher expression of IL-17 and higher number of Th17 cells in regional lymph nodes.70
Another possibility is that T cell response is aimed at harmless exogenous antigens as part of a derailed immunologic response in the setting of dysfunctional mucosal immunotolerance.56 Guzman et al studied the role of mucosal tolerance in DS-induced DE mice.71 Ocular exposure to ovalbumin (OVA) antigen in wild-type (WT) mice led to immunotolerance demonstrated by reduced in vitro antigen-specific T cell proliferation and in vivo delayed-type hypersensitivity (DTH) response to OVA immunization. Immunotolerance was also retained early on in DS-induced DE mice when they were exposed to ocular OVA on day 1 of DS. However, when OVA instillation was applied on day 4 of DS, DS-induced DE mice exhibited loss of immunotolerance with elevated in vitro antigen-specific T cell proliferation and in vivo DTH response to OVA immunization, suggesting time-dependent deterioration of ocular mucosal tolerance. Inhibition of NF-kB, a key regulator of mucosal tolerance, restored mucosal tolerance and decreased corneal staining and inflammatory markers (IL-1B and IL-6) in DS-induced DE mice. Similarly, time-dependent mucosal tolerance loss and mitigation of corneal damage were demonstrated in a DE mice model created by resection of extraorbital lacrimal glands.72
Loss of GCs is often noted in DE models and may contribute to mucosal immunotolerance loss, given their critical role in modulating antigen distribution and exposure to adjacent APCs.56,67,73,74 Barbosa et al73 studied the role of GCs in immune tolerance using SAM pointed domain ETS transcription factor knockout (Spdef−/−) mice, a DE mice model that lacks GCs and exhibits signs of DE. In WT mice, topically applied antigen OVA was effectively delivered to the stroma through GC-associated passages for uptake by adjacent APCs (CD11b+ F4/80+ macrophages), while Spdef−/− mice retained OVA within the epithelium. APCs isolated from conjunctival draining cervical lymph nodes of Spdef−/− mice showed stronger induction of antigen-specific lymphoproliferation, greater IFN-γ production, and lesser Treg proliferation, compared to WT. These findings were consistent with the loss of immune tolerance observed in Spdef−/− mice compared to WT, as assessed by cutaneous DTH to OVA following immunization with complete Freund’s adjuvant mixed with OVA.
In addition to regulating antigen exposure to DCs, GCs play a role in modulation of DCs phenotype. Studies by Contreras-Ruiz and Masli revealed that GCs TSP-1–dependent expression of TGF-β, particularly the TGF-β2 isoform, plays a role in modulation of DC phenotype.75 When DCs were co-cultured with WT globet cells from mice, they were found to have reduced major histocompatibility complex II and costimulatory molecules (CD80 and CD86) expression, compared to cultures of DCs alone. This effect was dependent on TSP-1 expression by GCs.
Irrespective of whether T cell activation is triggered by self-antigens or exogenous antigens, T cell migration to the ocular surface is a key event in DE and is driven by a variety of inflammatory cytokines and chemokines as well as other proteins. MRL/lpr mice are homozygous for the recessive lpr (lymphoproliferative) gene and used as Sjögren’s syndrome DE models.76 These mice have high expression of intercellular adhesion molecule (ICAM)-1, an adhesion molecule important for homing and activation of infiltrating lymphocytes, in the conjunctival epithelium and vascular endothelium along with infiltrating lymphocytes within the lacrimal gland tissue.77 In fact, ICAM-1 expression is positively correlated with disease progression and severity.77
The importance of these inflammatory mediators is further highlighted by mice studies showing improvement of DE with the use of anti-inflammatory therapy targeting IL-1,78 IL-17,70,79 and C-C motif receptor 2.80 Furthermore, T cell infiltration decreased with the use of monoclonal antibodies against ICAM-1 and its receptor lymphocyte functional associated antigen-1 (LFA-1).77
Inflammation in humans with DE
Hyperosmolarity is a known trigger of inflammation. Cell culture experiments demonstrated increased proinflammatory mediators (eg, IL-1, IL-6, TNF-α, and MMP-9) after human corneal epithelium was subjected to hyperosmolar stress.81,82 Similar results were noted in limbal epithelial cells with elevated levels of IL-1β, IL-8, and TNF-α found through the c-jun n-terminal kinase and extracellular-regulated kinase MAPK signaling pathway.61
Similar to mice, patients with DE (defined in a number of different ways) have increased tear levels of proinflammatory cytokines, chemokines, and chemokine receptors including IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12, IL-13, IL-17, IL-21, IFN-γ, TNF-α, CXCL9, CXCL10, CXCL11, CXCR3, IL-1Ra, IP-10/CXCL10, fractalkine/CX3CL1, CCL-5, and vascular endothelial growth factor.83–90 In fact, levels of IL-6, IL-17, IL-21, TNF-α, CXCL-9, CXCL-10, and CXCL-11 were found to be positively correlated with DE signs and symptoms (eg, Schirmer test, tear-film breakup time, punctate epithelial erosions, GC density, OSDI).88,90–92
Patients with DE have other features in common with mouse models, including T cell infiltration of the ocular surface and elevated ICAM expression. Stern et al93 studied patients with moderate to severe ADDE and found CD4+ T cell infiltration in the conjunctiva, along with increased markers of immune activation (HLA-DR and HLA-DQ). ADDE patients were also found to have increased ICAM-1 expression in the conjunctival epithelium and lacrimal gland venule endothelium, near CD4+ T cells.77,93–95
Inflammation in DE can also affect the corneal nerves, disrupting normal tearing reflex and blink rate.96 Many studies using in vivo confocal microscope, a minimally invasive tool for imaging ocular surface, have shown decreased sub-basal nerve density of the cornea97–103 and morphologic changes of the sub-basal nerves (increased tortuosity, nerve sprouting, increased bead-like formation, and decreased reflectivity) in DE patients.97–99,101,104,105 Additionally, DE patients generally show higher DC density and morphologic changes including larger DC size and additional dendrites at the sub-basal nerve plexus.98,100,104,106,107 A few studies have attempted to correlate in vivo confocal microscope parameters to DE signs and symptoms. Decreased sub-basal nerve density is correlated to decreased corneal sensitivity,99,102 increased DC density at the sub-basal nerve plexus,98 and increased DE symptoms and signs.97,98,108 On the other hand, studies correlating DC density to DE signs and symptoms have yielded conflicting results.100,107,109
Treatment modalities: targeting inflammation
Corticosteroids
Mechanism of action
Topical corticosteroids decrease inflammation primarily by binding to glucocorticoid receptors and regulating the expression of anti-inflammatory and proinflammatory genes.110 Specifically, corticosteroids suppress NF-kB, a key transcription factor in inflammation, leading to suppression of proinflammatory mediators and promoting lymphocyte apoptosis. There is a wide range of proinflammatory mediators that are suppressed by corticosteroids, including ICAM-1, MMPs, prostaglandins, cytokines, chemokines, and phospholipase A2. Non-genomic effects of corticosteroids also aid in suppressing leukocyte infiltration into areas of inflammation.111
Clinical data in DE
Topical corticosteroids have proven effective in the treatment of DE in several studies.112–116 One of these studies included a multicenter, randomized, double-masked, placebo-controlled clinical trial with 64 moderate to severe ADDE patients receiving either 0.5% loteprednol or placebo four times a day for 4 weeks.115 This study evaluated symptoms on a visual analog scale and signs by corneal staining, conjunctival injection, and lid margin injection. At 2 weeks, treatment with 0.5% loteprednol improved the signs of DE, including lid margin injection and conjunctival injection. Furthermore, in a subset analysis of patients with moderate clinical inflammatory component, treatment with 0.5% loteprednol improved corneal staining, conjunctival injection, and DE symptoms (eye redness).
Side effects
The use of corticosteroids is limited due to a wide range of side effects including glaucoma, ocular infection, corneal thinning, and formation of cataracts.117 Given the chronic nature of DE, topical corticosteroid use tends to be limited to short-term treatment of DE exacerbations.
Cyclosporine
Mechanism of action
Cyclosporine is FDA approved for the treatment of DE signs. It suppresses inflammation by binding to cyclophilins and inhibiting calcineurin, a calcium-dependent phosphatase, thereby preventing nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) dephosphorylation which reduces IL-2 levels suppressing T cell activation.118
Formulation
Cyclosporine requires suspension and emulsion forms due to its poor solubility in water. Restasis is formulated with 0.05% cyclosporine in an emulsion of glycerin (2.2%), castor oil (1.25%), polysorbate 80 (1.00%), carbomer copolymer type A (0.05%), purified water (to 100%), and sodium hydroxide for pH adjustment.119
Clinical data in DE
Studies with topical cyclosporine treatment in patients with moderate to severe DE generally showed significant improvement of DE signs and variable improvement of DE symptoms.120 Large multicenter, randomized, double-blind, controlled studies with moderate to severe DE patients have shown improvement in DE signs (corneal staining, tear production) over emulsion placebo.121–123 Symptomatic improvement in these studies was more variable, with the most consistent improvement found in reports of “dryness” symptoms.121–124 Similarly, smaller prospective randomized studies of moderate to severe DE patients treated with cyclosporine or combinations of cyclosporine (cyclosporine plus artificial tears [AT] or cyclosporine plus vitamin A) consistently showed improvement in DE signs (corneal staining, conjunctival staining, GC density, tear production, tear film stability)125–131 and, in some cases, DE symptoms (OSDI, blurry vision, burning, and photophobia).125,127–129
Side effects
Cyclosporine use is limited by its side effects of mild to moderate burning and irritation of the eye.123 Tolerance is improved with the use of concomitant topical corticosteroids.132,133
Lifitegrast
Mechanism of action
Lifitegrast (formerly SAR1118) is the only FDA-approved treatment for both signs and symptoms of DE. Lifitegrast suppresses inflammation by mimicking ICAM-1, thus blocking the interaction between ICAM-1 and LFA-1, a cell surface protein belonging to the β2 family of integrins and found on leukocytes (Figure 1).134,135 LFA-1/ICAM-1 interaction has been implicated in various aspects of lymphocyte activation and migration.136 It promotes migration of DC to lymph nodes where they can activate naïve T cells.137,138 It also plays a direct role in T cell activation by maintaining initial contact between naïve T cells and APCs in lymph nodes139 and enhancing T cell sensitivity to antigens by stabilization of the immunologic synapse between naïve T cells and APCs in the lymph nodes.140–142 Beyond T cell activation, LFA-1/ICAM-1 interaction also plays a role in T cell migration by allowing firm adhesion to the endothelium with subsequent transmigration into inflamed tissue.143,144 By blocking the interaction between LFA-1 and ICAM-1, lifitegrast is able to disrupt various key steps in T cell–mediated inflammation.
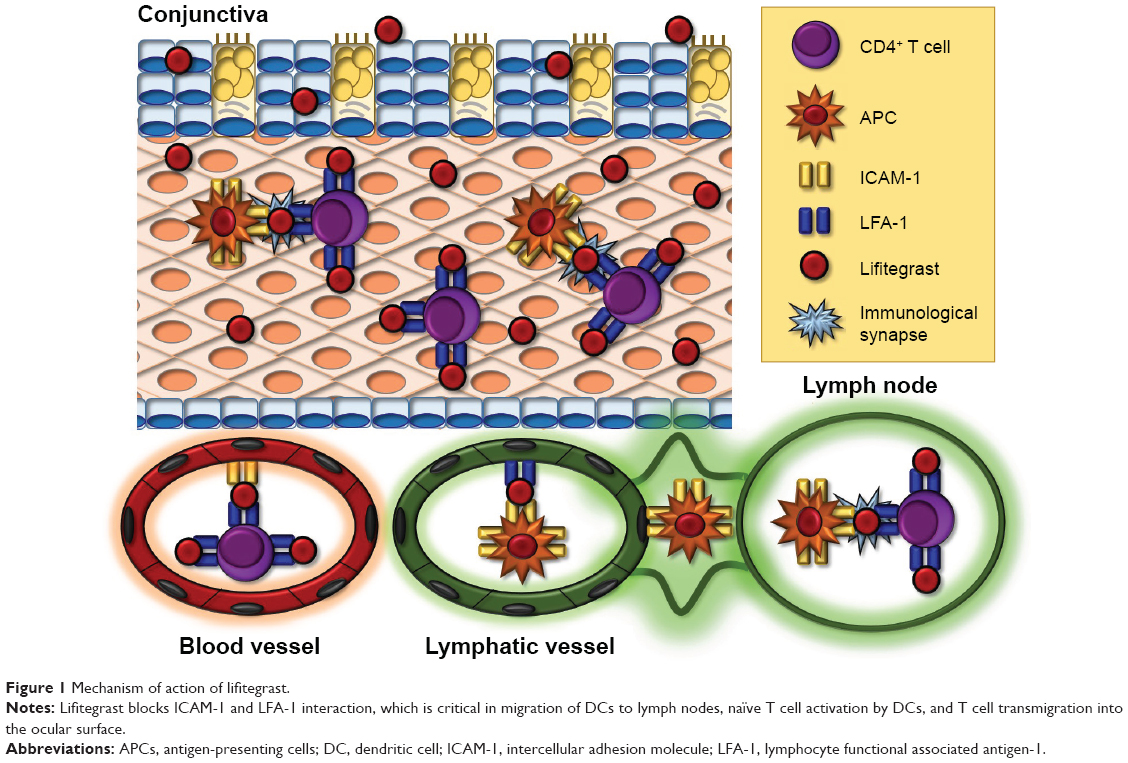 | Figure 1 Mechanism of action of lifitegrast. |
Formulation
Lifitegrast is prepared in PBS with a pH, tonicity, and osmolarity range consistent with other currently approved topical ophthalmic solutions.145 It is preservative-free and comes in single-unit dose ampules.145
Animal studies with lifitegrast in non-DE conditions
A few animal studies have shown the anti-inflammatory effect of lifitegrast in various inflammatory-based ocular diseases. A study by Sun et al tested the efficacy of lifitegrast in inhibiting inflammation in mice with corneal inflammation induced by antibiotic-killed Pseudomonas aeruginosa and Staphylococcus aureus in the presence of contact lenses. Lifitegrast reduced neutrophilic infiltration into the cornea, reduced clinical signs of corneal inflammation, and prevented P. aeruginosa- and S. aureus- induced inflammation compared to controls.146 In rat streptozotocin model of diabetic retinopathy, lifitegrast was able to significantly reduce the number of adherent leukocytes and level of myeloperoxidase, a leukocyte-derived proinflammatory protein, to levels comparable to those of normal retina.147 Lifitegrast also significantly reduced blood–retinal barrier breakdown, compared to vehicle-control.147
Animal studies with lifitegrast in DE
A study of 10 idiopathic keratoconjunctivitis sicca dogs receiving 1% lifitegrast three times a day for 12 weeks showed a significant increase in tear production from baseline for 11 of 18 study eyes, which corresponded with a decreased inflammatory cell infiltrate of the conjunctiva from baseline.
Human studies with lifitegrast
In vitro studies demonstrate lifitegrast’s ability to inhibit human T cell binding to ICAM-1 and inhibit the release of proinflammatory cytokines including IFN-γ, TNF-α, macrophage inflammatory protein-1α, IL-1α, IL-1β, IL-2, IL-4, and IL-6 from activated lymphocytes.148 Additionally, imaging studies reveal inhibition of immunologic synapse formation in the presence of lifitegrast.149
The clinical efficacy of lifitegrast in the treatment of signs and symptoms of DE has been demonstrated across four prospective, multicenter, randomized, double-masked, vehicle-controlled trials (Table 1). The first of these trials was a phase II clinical trial of 230 patients with moderate to severe DE receiving either 0.1%, 1%, or 5% lifitegrast vs vehicle twice a day for 84 days.150 Key eligibility criteria included adult DE patients with corneal staining score ≥2, un-anesthetized Schirmer >1 and <10, no active lid margin disease, and worsening of signs (inferior corneal staining score [ICSS] increase >1 point) and symptoms (ocular discomfort score [ODS] increase >1 point) in response to controlled adverse environment (CAE), an environment with specified humidity, temperature, airflow, and lighting designed to worsen DE. The use of topical cyclosporine and AT was prohibited 6 weeks and 3 days from visit 1, respectively. Three visits occurred during the treatment period at days 14, 42, and 84 of treatment during which patients were exposed to additional 90-minute sessions of CAE. Lifitegrast (1% and 5%) showed significant improvement in ICSS (primary sign endpoint) at 84 days from baseline, compared to vehicle. This study also showed improvement in tear production with 5% lifitegrast at 14 days from baseline, compared to vehicle. In terms of symptoms, all lifitegrast groups showed significant improvement in the OSDI at 14 days and 1% and 5% lifitegrast showed significant improvement in the vision-related subscale score of OSDI at 14 and 84 days from baseline, compared to vehicle. Baseline ODS showed trends toward improvement, but was not significantly different from vehicle.
 | Table 1 Summary of studies using LIF for the treatment of dry eye |
This was followed by the first Phase III trial, OPUS-1, consisting of 588 patients with moderate to severe DE receiving 5% lifitegrast vs vehicle twice a day for 84 days.151 Unlike the previously described Phase II study, CAE was only used during screening visits and not during the in-treatment visits. Once again, lifitegrast significantly improved ICSS (primary end point) at 84 days from baseline, compared to vehicle. Additional improvements in DE signs over vehicle included superior corneal staining, total corneal staining, nasal conjunctival staining, and conjunctival staining, with some of these changes starting as early as at 14 days of treatment. Lifitegrast significantly improved DE symptoms over vehicle, including decreased ODS and visual analog eye dryness score (VAS-EDS); however, this study failed to show significant improvement in vision-related subscale score of OSDI (primary symptom end point) over vehicle.
The next Phase III trial was OPUS-2 consisting of 718 patients with moderate to severe DE receiving 5% lifitegrast vs vehicle twice a day for 84 days.152 Study methods were similar to the previous two studies; however, no CAE was used in this study and AT use within 30 days and VAS-EDS ≥40 were added to the inclusion criteria based on post hoc analysis from OPUS-1, suggesting that the drug effect is increased in patients with recent AT use and VAS-EDS ≥40. In this study, primary symptom end point was met as lifitegrast showed significant improvement from baseline in VAS-EDS as early as 14 days and continuing to 84 days, over vehicle. Additionally, lifitegrast was superior in decreasing ODS at 84 days from baseline, compared to vehicle. Primary sign end point of ICSS showed improvement from baseline, but did not reach significant difference compared to vehicle. The authors hypothesize that the lack of ICSS improvement was related to patients’ recent AT use which hindered detection of drug effect, or the enrollment of more severe DE patients with more advanced ocular surface damage.
The final phase III trial was OPUS-3 consisting of 711 patients with moderate to severe DE receiving 5% lifitegrast vs vehicle twice a day for 84 days.153 Study methods were similar to those of OPUS-2. This study also met its primary symptom end point of VAS-EDS improvement from baseline compared to vehicle starting at 14 days and continuing to 84 days. Other symptoms significantly improved with the use of lifitegrast over vehicle, including visual analog scale for itching, foreign body sensation, and eye discomfort at 42 days of treatment.
Safety studies with lifitegrast
The safety profile of lifitegrast has been explored in several dose-escalation tolerance studies (Table 2). Dose-escalation tolerance studies using healthy dogs with a maximum dosage of 10% lifitegrast administered three times per day for 1 month did not demonstrate any adverse effect on the ocular surface.148 Similarly, dose-escalation tolerance assessment with a maximum dosage of 3% lifitegrast administered three times per day for 13 weeks did not show any adverse effect on the ocular surface, but was associated with a transient period of blinking and squinting upon drop administration, which appeared to improve after the first few days of instillation.148
 | Table 2 Summary of safety studies using LIF for the treatment of dry eye |
Short-term dose-escalation tolerance studies of healthy individuals and patients undergoing pars plana vitrectomies have tested lifitegrast in dosages ranging from 0.1% to 5% at frequencies as high as three times per day.154,155 In these studies, adverse events from lifitegrast were mild to moderate in severity and consisted of transient ocular irritation and ocular hyperemia. Non-ocular adverse events included dysgeusia, headaches, erythema, and musculoskeletal pain. No effects were seen in intraocular pressure, visual acuity, ocular surface exams, healing time, and hematologic and chemistry panels. SONATA was a multicenter, randomized, double-blind, vehicle-controlled trial that was conducted to assess the long-term safety profile of lifitegrast 5% vs vehicle twice daily for 1 year in 331 DE patients.156 Eligibility criteria included Schirmer tear test result ≥1 and ≤10 mm in 5 minutes, no active lid margin disease, corneal staining score ≥2.0, EDS ≥40, and use and/or desire to use AT in the past 6 months. Patients receiving lifitegrast had a higher percentage of treatment-emergent adverse effects (TEAEs; 53.6% lifitegrast vs 34.2% vehicle), most of which were mostly transient and mild to moderate in severity. TEAEs included instillation site irritation (15.0% lifitegrast vs 4.5% vehicle), instillation site reaction (13.2% lifitegrast vs 1.8% vehicle), reduction in visual acuity (11.4% lifitegrast vs 6.3% vehicle), DE (1.8% lifitegrast vs 5.4% vehicle), and dysgeusia (16.4% lifitegrast vs 1.8% vehicle). Drop comfort improved at each visit. Dysgeusia was likely the result of tear drainage into the oropharynx, while decreased visual acuity may have been due to transient alterations in the tear film leading to altered refractive properties. Despite the decreased visual acuity, changes in best-corrected visual acuity from baseline to 1 year were minimal in both groups. No serious ocular adverse events occurred in this study. All non-ocular TEAEs, except for dysgeusia, were considered to be unrelated to lifitegrast. There was no evidence of immunosuppression as per CD3, CD4 and CD8 serum levels and no alterations in hematologic, renal and liver panels. Concomitant AT use appeared to increase the rates of ocular TEAEs in both groups, but was associated with lower discontinuation rates due to TEAEs in both groups.
Conclusion and remaining questions
Evidence suggests that ocular surface inflammation is an important component of DE. Stress to the ocular surface stimulates production of proinflammatory mediators inducing maturation of resident APCs that migrate to the lymph nodes to activate autoreactive CD4+ T cells which migrate to the ocular surface causing more inflammation and damage. Lifitegrast blocks the interaction between ICAM-1 and LFA-1, which has been shown to be critical for APC migration to the lymph nodes as well as CD4+ T cell activation and migration to the ocular surface. In four large multicenter, randomized controlled trials, lifitegrast has proven to be effective at controlling both the signs and symptoms of DE with minimal side effects.
Despite its success, many questions remain. It is known that not all individuals with DE symptoms have detectable levels of inflammation as measured by ocular surface MMP-9 levels.157 DE patients with ADDE are more likely to have inflammation, especially in the setting of systemic inflammation such as Sjögren’s syndrome and graft vs host disease.157 Could these patients also be more likely to respond to anti-inflammatory therapy such as lifitegrast?
Another question is whether anti-inflammatory therapies for DE could work better in combination. While studies of topical cyclosporine and methylprednisolone combinations showed faster symptomatic relief, at this time, no combination studies have been done with lifitegrast. Additionally, no comparative studies have been done with lifitegrast to determine if one anti-inflammatory agent could be better than the others. These are all important avenues of future investigation.
Of note, we focused our review on the inflammatory component of DE, given our focus on lifitegrast as a new therapeutic modality in DE. However, it is known that not all patients with DE symptoms have ocular surface inflammation (as measured by Inflammadry).157 Other contributors to DE include MGD and resultant EDE.3 The role of inflammation in the initiation and propagation of MGD is less well clarified.158 Furthermore, many other exposures contribute to DE, which do not clearly fit into the autoreactive and/or loss of mucosal tolerance story. These include environmental exposures such as air pollution and low humidity,159 dietary patterns such as a high consumption of free fatty acids,160 and psychosocial considerations such as depression and chronic widespread pain, to name a few.161 The role of inflammation, along with the other contributors, needs to be considered when evaluating a patient with DE.
Acknowledgments
This study was supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Clinical Sciences Research EPID-006-15S (Dr Galor), R01EY026174 (Dr Galor), NIH Center Core Grant P30EY014801, and Research to Prevent Blindness Unrestricted Grant.
Disclosure
The authors report no conflicts of interest in this work.
References
The epidemiology of dry eye disease: report of the Epidemiology Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007;5(2):93–107. | ||
Pouyeh B, Viteri E, Feuer W, et al. Impact of ocular surface symptoms on quality of life in a United States veterans affairs population. Am J Ophthalmol. 2012;153(6):1061.e3–1066.e3. | ||
Craig JP, Nichols KK, Akpek EK, et al. TFOS DEWS II definition and classification report. Ocul Surf. 2017;15(3):276–283. | ||
Stern ME, Schaumburg CS, Pflugfelder SC. Dry eye as a mucosal autoimmune disease. Int Rev Immunol. 2013;32(1):19–41. | ||
Godin MR, Gupta PK. Lifitegrast ophthalmic solution in the treatment of signs and symptoms of dry eye disease: design, development, and place in therapy. Clin Ophthalmol. 2017;11:951–957. | ||
Lemp MA, Crews LA, Bron AJ, Foulks GN, Sullivan BD. Distribution of aqueous-deficient and evaporative dry eye in a clinic-based patient cohort: a retrospective study. Cornea. 2012;31(5):472–478. | ||
Schaumberg DA, Sullivan DA, Buring JE, Dana MR. Prevalence of dry eye syndrome among US women. Am J Ophthalmol. 2003;136(2):318–326. | ||
Sullivan DA. Tearful relationships? Sex, hormones, the lacrimal gland, and aqueous-deficient dry eye. Ocul Surf. 2004;2(2):92–123. | ||
Moss SE, Klein R, Klein BE. Prevalence of and risk factors for dry eye syndrome. Arch Ophthalmol. 2000;118(9):1264–1268. | ||
Galor A, Feuer W, Lee DJ, et al. Depression, post-traumatic stress disorder, and dry eye syndrome: a study utilizing the national United States Veterans Affairs administrative database. Am J Ophthalmol. 2012;154(2):340.e2–346.e2. | ||
Yu EY, Leung A, Rao S, Lam DS. Effect of laser in situ keratomileusis on tear stability. Ophthalmology. 2000;107(12):2131–2135. | ||
Kasetsuwan N, Satitpitakul V, Changul T, Jariyakosol S. Incidence and pattern of dry eye after cataract surgery. PLoS One. 2013;8(11):e78657. | ||
Buchholz P, Steeds CS, Stern LS, et al. Utility assessment to measure the impact of dry eye disease. Ocul Surf. 2006;4(3):155–161. | ||
Schiffman RM, Walt JG, Jacobsen G, Doyle JJ, Lebovics G, Sumner W. Utility assessment among patients with dry eye disease. Ophthalmology. 2003;110(7):1412–1419. | ||
Deschamps N, Ricaud X, Rabut G, Labbe A, Baudouin C, Denoyer A. The impact of dry eye disease on visual performance while driving. Am J Ophthalmol. 2013;156(1):184.e3–189.e3. | ||
Ridder WH 3rd, Zhang Y, Huang JF. Evaluation of reading speed and contrast sensitivity in dry eye disease. Optom Vis Sci. 2013;90(1):37–44. | ||
Miljanović B, Dana R, Sullivan DA, Schaumberg DA. Impact of dry eye syndrome on vision-related quality of life. Am J Ophthalmol. 2007;143(3):409.e2–415.e2. | ||
Li M, Gong L, Sun X, Chapin WJ. Anxiety and depression in patients with dry eye syndrome. Curr Eye Res. 2011;36(1):1–7. | ||
Kim KW, Han SB, Han ER, et al. Association between depression and dry eye disease in an elderly population. Invest Ophthalmol Vis Sci. 2011;52(11):7954–7958. | ||
Le Q, Zhou X, Ge L, Wu L, Hong J, Xu J. Impact of dry eye syndrome on vision-related quality of life in a non-clinic-based general population. BMC Ophthalmol. 2012;12:22. | ||
McDonald M, Patel DA, Keith MS, Snedecor SJ. Economic and humanistic burden of dry eye disease in Europe, North America, and Asia: a systematic literature review. Ocul Surf. 2016;14(2):144–167. | ||
Yu J, Asche CV, Fairchild CJ. The economic burden of dry eye disease in the United States: a decision tree analysis. Cornea. 2011;30(4):379–387. | ||
Nichols KK, Nichols JJ, Mitchell GL. The lack of association between signs and symptoms in patients with dry eye disease. Cornea. 2004;23(8):762–770. | ||
Goto E, Yagi Y, Matsumoto Y, Tsubota K. Impaired functional visual acuity of dry eye patients. Am J Ophthalmol. 2002;133(2):181–186. | ||
Chalmers RL, Begley CG, Caffery B. Validation of the 5-Item Dry Eye Questionnaire (DEQ-5): Discrimination across self-assessed severity and aqueous tear deficient dry eye diagnoses. Cont Lens Anterior Eye. 2010;33(2):55–60. | ||
Schiffman RM, Christianson MD, Jacobsen G, Hirsch JD, Reis BL. Reliability and validity of the ocular surface disease index. Arch Ophthalmol. 2000;118(5):615–621. | ||
Abetz L, Rajagopalan K, Mertzanis P, Begley C, Barnes R, Chalmers R. Development and validation of the impact of dry eye on everyday life (IDEEL) questionnaire, a patient-reported outcomes (PRO) measure for the assessment of the burden of dry eye on patients. Health Qual Life Outcomes. 2011;9:111. | ||
Mangione CM, Lee PP, Gutierrez PR, et al. Development of the 25-item National Eye Institute Visual Function Questionnaire. Arch Ophthalmol. 2001;119(7):1050–1058. | ||
Nichols KK, Mitchell GL, Zadnik K. Performance and repeatability of the NEI-VFQ-25 in patients with dry eye. Cornea. 2002;21(6):578–583. | ||
Messmer EM. The pathophysiology, diagnosis, and treatment of dry eye disease. Dtsch Arztebl Int. 2015;112(5):71–81; quiz 2. | ||
Lanza NL, Valenzuela F, Perez VL, Galor A. The matrix metalloproteinase 9 point-of-care test in dry eye. Ocul Surf. 2016;14(2):189–195. | ||
Messmer EM, Bulgen M, Kampik A. Hyperosmolarity of the tear film in dry eye syndrome. Dev Ophthalmol. 2010;45:129–138. | ||
Lemp MA, Bron AJ, Baudouin C, et al. Tear osmolarity in the diagnosis and management of dry eye disease. Am J Ophthalmol. 2011;151(5):792.e1–798.e1. | ||
Stern ME, Beuerman RW, Fox RI, Gao J, Mircheff AK, Pflugfelder SC. The pathology of dry eye: the interaction between the ocular surface and lacrimal glands. Cornea. 1998;17(6):584–589. | ||
Stern ME, Gao J, Siemasko KF, Beuerman RW, Pflugfelder SC. The role of the lacrimal functional unit in the pathophysiology of dry eye. Exp Eye Res. 2004;78(3):409–416. | ||
Dursun D, Monroy D, Knighton R, et al. The effects of experimental tear film removal on corneal surface regularity and barrier function. Ophthalmology. 2000;107(9):1754–1760. | ||
Liu Z, Pflugfelder SC. Corneal surface regularity and the effect of artificial tears in aqueous tear deficiency. Ophthalmology. 1999;106(5):939–943. | ||
Barabino S, Chen Y, Chauhan S, Dana R. Ocular surface immunity: homeostatic mechanisms and their disruption in dry eye disease. Prog Retin Eye Res. 2012;31(3):271–285. | ||
Dean AW, Glasgow BJ. Mass spectrometric identification of phospholipids in human tears and tear lipocalin. Invest Ophthalmol Vis Sci. 2012;53(4):1773–1782. | ||
Yeh PT, Casey R, Glasgow BJ. A novel fluorescent lipid probe for dry eye: retrieval by tear lipocalin in humans. Invest Ophthalmol Vis Sci. 2013;54(2):1398–1410. | ||
McCulley JP, Shine WE. The lipid layer of tears: dependent on meibomian gland function. Exp Eye Res. 2004;78(3):361–365. | ||
Bron AJ, Tiffany JM, Gouveia SM, Yokoi N, Voon LW. Functional aspects of the tear film lipid layer. Exp Eye Res. 2004;78(3):347–360. | ||
Holly FJ, Lemp MA. Tear physiology and dry eyes. Surv Ophthalmol. 1977;22(2):69–87. | ||
Klenkler B, Sheardown H, Jones L. Growth factors in the tear film: role in tissue maintenance, wound healing, and ocular pathology. Ocul Surf. 2007;5(3):228–239. | ||
Garreis F, Gottschalt M, Paulsen FP. Antimicrobial peptides as a major part of the innate immune defense at the ocular surface. Dev Ophthalmol. 2010;45:16–22. | ||
Sack RA, Conradi L, Krumholz D, Beaton A, Sathe S, Morris C. Membrane array characterization of 80 chemokines, cytokines, and growth factors in open- and closed-eye tears: angiogenin and other defense system constituents. Invest Ophthalmol Vis Sci. 2005;46(4):1228–1238. | ||
Walter SD, Gronert K, McClellan AL, Levitt RC, Sarantopoulos KD, Galor A. ω-3 tear film lipids correlate with clinical measures of dry eye. Invest Ophthalmol Vis Sci. 2016;57(6):2472–2478. | ||
Gupta A, Monroy D, Ji Z, Yoshino K, Huang A, Pflugfelder SC. Transforming growth factor beta-1 and beta-2 in human tear fluid. Curr Eye Res. 1996;15(6):605–614. | ||
Biteman B, Hassan IR, Walker E, et al. Interdependence of lipoxin A4 and heme-oxygenase in counter-regulating inflammation during corneal wound healing. FASEB J. 2007;21(9):2257–2266. | ||
Gao Y, Min KJ, Zhang YB, Su J, Greenwood M, Gronert K. Female-specific downregulation of tissue polymorphonuclear neutrophils drives impaired regulatory T cell and amplified effector T cell responses in autoimmune dry eye disease. J Immunol. 2015;195(7):3086–3099. | ||
Leedom AJ, Sullivan AB, Dong B, Lau D, Gronert K. Endogenous LXA4 circuits are determinants of pathological angiogenesis in response to chronic injury. Am J Pathol. 2010;176(1):74–84. | ||
Wang SB, Hu KM, Seamon KJ, Mani V, Chen Y, Gronert K. Estrogen negatively regulates epithelial wound healing and protective lipid mediator circuits in the cornea. FASEB J. 2012;26(4):1506–1516. | ||
Cortina MS, He J, Li N, Bazan NG, Bazan HE. Neuroprotectin D1 synthesis and corneal nerve regeneration after experimental surgery and treatment with PEDF plus DHA. Invest Ophthalmol Vis Sci. 2010;51(2):804–810. | ||
Kenchegowda S, Bazan HE. Significance of lipid mediators in corneal injury and repair. J Lipid Res. 2010;51(5):879–891. | ||
Gipson IK, Argueso P. Role of mucins in the function of the corneal and conjunctival epithelia. Int Rev Cytol. 2003;231:1–49. | ||
Galletti JG, Guzman M, Giordano MN. Mucosal immune tolerance at the ocular surface in health and disease. Immunology. 2017;150(4):397–407. | ||
Norn MS. Dead, degenerate, and living cells in conjunctival fluid and mucous thread. Acta ophthalmologica. 1969;47(5):1102–1115. | ||
Hodges RR, Dartt DA. Tear film mucins: front line defenders of the ocular surface; comparison with airway and gastrointestinal tract mucins. Exp Eye Res. 2013;117:62–78. | ||
Baudouin C, Aragona P, Messmer EM, et al. Role of hyperosmolarity in the pathogenesis and management of dry eye disease: proceedings of the OCEAN group meeting. Ocul Surf. 2013;11(4):246–258. | ||
Luo L, Li DQ, Pflugfelder SC. Hyperosmolarity-induced apoptosis in human corneal epithelial cells is mediated by cytochrome c and MAPK pathways. Cornea. 2007;26(4):452–460. | ||
Li DQ, Luo L, Chen Z, Kim HS, Song XJ, Pflugfelder SC. JNK and ERK MAP kinases mediate induction of IL-1beta, TNF-alpha and IL-8 following hyperosmolar stress in human limbal epithelial cells. Exp Eye Res. 2006;82(4):588–596. | ||
Luo L, Li DQ, Corrales RM, Pflugfelder SC. Hyperosmolar saline is a proinflammatory stress on the mouse ocular surface. Eye Contact Lens. 2005;31(5):186–193. | ||
Rashid S, Jin Y, Ecoiffier T, Barabino S, Schaumberg DA, Dana MR. Topical omega-3 and omega-6 fatty acids for treatment of dry eye. Arch Ophthalmol. 2008;126(2):219–225. | ||
Luo L, Li DQ, Doshi A, Farley W, Corrales RM, Pflugfelder SC. Experimental dry eye stimulates production of inflammatory cytokines and MMP-9 and activates MAPK signaling pathways on the ocular surface. Invest Ophthalmol Vis Sci. 2004;45(12):4293–4301. | ||
McClellan AJ, Volpe EA, Zhang X, et al. Ocular surface disease and dacryoadenitis in aging C57BL/6 mice. Am J Pathol. 2014;184(3):631–643. | ||
Schaumburg CS, Siemasko KF, De Paiva CS, et al. Ocular surface APCs are necessary for autoreactive T cell-mediated experimental autoimmune lacrimal keratoconjunctivitis. J Immunol. 2011;187(7):3653–3662. | ||
Niederkorn JY, Stern ME, Pflugfelder SC, et al. Desiccating stress induces T cell-mediated Sjogren’s Syndrome-like lacrimal keratoconjunctivitis. J Immunol. 2006;176(7):3950–3957. | ||
Siemasko KF, Gao J, Calder VL, et al. In vitro expanded CD4+ CD25+ Foxp3+ regulatory T cells maintain a normal phenotype and suppress immune-mediated ocular surface inflammation. Invest Ophthalmol Vis Sci. 2008;49(12):5434–5440. | ||
Ratay ML, Glowacki AJ, Balmert SC, et al. Treg-recruiting microspheres prevent inflammation in a murine model of dry eye disease. J Control Release. 2017;258:208–217. | ||
Chauhan SK, El Annan J, Ecoiffier T, et al. Autoimmunity in dry eye is due to resistance of Th17 to Treg suppression. J Immunol. 2009;182(3):1247–1252. | ||
Guzman M, Keitelman I, Sabbione F, Trevani AS, Giordano MN, Galletti JG. Desiccating stress-induced disruption of ocular surface immune tolerance drives dry eye disease. Clin Exp Immunol. 2016;184(2):248–256. | ||
Guzman M, Keitelman I, Sabbione F, Trevani AS, Giordano MN, Galletti JG. Mucosal tolerance disruption favors disease progression in an extraorbital lacrimal gland excision model of murine dry eye. Exp Eye Res. 2016;151:19–22. | ||
Barbosa FL, Xiao Y, Bian F, et al. Goblet cells contribute to ocular surface immune tolerance-implications for dry eye disease. Int J Mol Sci. 2017;18(5):1–13. | ||
McDole JR, Wheeler LW, McDonald KG, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature. 2012;483(7389):345–349. | ||
Contreras-Ruiz L, Masli S. Immunomodulatory cross-talk between conjunctival goblet cells and dendritic cells. PLoS One. 2015;10(3):e0120284. | ||
Hoffman RW, Alspaugh MA, Waggie KS, Durham JB, Walker SE. Sjogren’s syndrome in MRL/l and MRL/n mice. Arthritis Rheum. 1984;27(2):157–165. | ||
Gao J, Morgan G, Tieu D, et al. ICAM-1 expression predisposes ocular tissues to immune-based inflammation in dry eye patients and Sjogrens syndrome-like MRL/lpr mice. Exp Eye Res. 2004;78(4):823–835. | ||
Okanobo A, Chauhan SK, Dastjerdi MH, Kodati S, Dana R. Efficacy of topical blockade of interleukin-1 in experimental dry eye disease. Am J Ophthalmol. 2012;154(1):63–71. | ||
De Paiva CS, Chotikavanich S, Pangelinan SB, et al. IL-17 disrupts corneal barrier following desiccating stress. Mucosal Immunol. 2009;2(3):243–253. | ||
Goyal S, Chauhan SK, Zhang Q, Dana R. Amelioration of murine dry eye disease by topical antagonist to chemokine receptor 2. Arch Ophthalmol. 2009;127(7):882–887. | ||
Wang C, Shi X, Chen X, et al. 17-beta-estradiol inhibits hyperosmolarity-induced proinflammatory cytokine elevation via the p38 MAPK pathway in human corneal epithelial cells. Mol Vis. 2012;18:1115–1122. | ||
Li DQ, Chen Z, Song XJ, Luo L, Pflugfelder SC. Stimulation of matrix metalloproteinases by hyperosmolarity via a JNK pathway in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004;45(12):4302–4311. | ||
Lee SY, Han SJ, Nam SM, et al. Analysis of tear cytokines and clinical correlations in Sjogren syndrome dry eye patients and non-Sjogren syndrome dry eye patients. Am J Ophthalmol. 2013;156(2):247.e1–253.e1. | ||
Meadows JF, Dionne K, Nichols KK. Differential profiling of T-cell cytokines as measured by protein microarray across dry eye subgroups. Cornea. 2016;35(3):329–335. | ||
Lam H, Bleiden L, de Paiva CS, Farley W, Stern ME, Pflugfelder SC. Tear cytokine profiles in dysfunctional tear syndrome. Am J Ophthalmol. 2009;147(2):198.e1–205.e1. | ||
Enriquez-de-Salamanca A, Castellanos E, Stern ME, et al. Tear cytokine and chemokine analysis and clinical correlations in evaporative-type dry eye disease. Mol Vis. 2010;16:862–873. | ||
He C, Lai P, Weng J, et al. Toll-like receptor 2-mediated NF-kappaB inflammatory responses in dry eye associated with cGVHD. Mol Vis. 2011;17:2605–2611. | ||
Yoon KC, Park CS, You IC, et al. Expression of CXCL9, -10, -11, and CXCR3 in the tear film and ocular surface of patients with dry eye syndrome. Invest Ophthalmol Vis Sci. 2010;51(2):643–650. | ||
Massingale ML, Li X, Vallabhajosyula M, Chen D, Wei Y, Asbell PA. Analysis of inflammatory cytokines in the tears of dry eye patients. Cornea. 2009;28(9):1023–1027. | ||
Yoon KC, Jeong IY, Park YG, Yang SY. Interleukin-6 and tumor necrosis factor-alpha levels in tears of patients with dry eye syndrome. Cornea. 2007;26(4):431–437. | ||
Mrugacz M, Ostrowska L, Bryl A, Szulc A, Zelazowska-Rutkowska B, Mrugacz G. Pro-inflammatory cytokines associated with clinical severity of dry eye disease of patients with depression. Adv Med Sci. 2017;62(2):338–344. | ||
Lim SA, Nam DH, Lee JH, Kwok SK, Park SH, Chung SH. Association of IL-21 cytokine with severity of primary Sjogren syndrome dry eye. Cornea. 2015;34(3):248–252. | ||
Stern ME, Gao J, Schwalb TA, et al. Conjunctival T-cell subpopulations in Sjogren’s and non-Sjogren’s patients with dry eye. Invest Ophthalmol Vis Sci. 2002;43(8):2609–2614. | ||
Saito I, Terauchi K, Shimuta M, et al. Expression of cell adhesion molecules in the salivary and lacrimal glands of Sjogren’s syndrome. J Clin Lab Anal. 1993;7(3):180–187. | ||
Jones DT, Monroy D, Ji Z, Atherton SS, Pflugfelder SC. Sjogren’s syndrome: cytokine and Epstein-Barr viral gene expression within the conjunctival epithelium. Invest Ophthalmol Vis Sci. 1994;35(9):3493–3504. | ||
Dartt DA. Dysfunctional neural regulation of lacrimal gland secretion and its role in the pathogenesis of dry eye syndromes. Ocul Surf. 2004;2(2):76–91. | ||
Villani E, Galimberti D, Viola F, Mapelli C, Ratiglia R. The cornea in Sjogren’s syndrome: an in vivo confocal study. Invest Ophthalmol Vis Sci. 2007;48(5):2017–2022. | ||
Tepelus TC, Chiu GB, Huang J, et al. Correlation between corneal innervation and inflammation evaluated with confocal microscopy and symptomatology in patients with dry eye syndromes: a preliminary study. Graefes Arch Clin Exp Ophthalmol. Epub 2017 May 20. | ||
Labbe A, Liang Q, Wang Z, et al. Corneal nerve structure and function in patients with non-sjogren dry eye: clinical correlations. Invest Ophthalmol Vis Sci. 2013;54(8):5144–5150. | ||
Shetty R, Sethu S, Deshmukh R, et al. Corneal dendritic cell density is associated with subbasal nerve plexus features, ocular surface disease index, and serum vitamin D in Evaporative dry eye disease. BioMed Res Int. 2016;2016:4369750. | ||
Erdelyi B, Kraak R, Zhivov A, Guthoff R, Nemeth J. In vivo confocal laser scanning microscopy of the cornea in dry eye. Graefes Arch Clin Exp Ophthalmol. 2007;245(1):39–44. | ||
Benitez-Del-Castillo JM, Acosta MC, Wassfi MA, et al. Relation between corneal innervation with confocal microscopy and corneal sensitivity with noncontact esthesiometry in patients with dry eye. Invest Ophthalmol Vis Sci. 2007;48(1):173–181. | ||
Benitez del Castillo JM, Wasfy MA, Fernandez C, Garcia-Sanchez J. An in vivo confocal masked study on corneal epithelium and subbasal nerves in patients with dry eye. Invest Ophthalmol Vis Sci. 2004;45(9):3030–3035. | ||
Tuisku IS, Konttinen YT, Konttinen LM, Tervo TM. Alterations in corneal sensitivity and nerve morphology in patients with primary Sjogren’s syndrome. Exp Eye Res. 2008;86(6):879–885. | ||
Tuominen IS, Konttinen YT, Vesaluoma MH, Moilanen JA, Helinto M, Tervo TM. Corneal innervation and morphology in primary Sjogren’s syndrome. Invest Ophthalmol Vis Sci. 2003;44(6):2545–2549. | ||
Kheirkhah A, Rahimi Darabad R, Cruzat A, et al. Corneal epithelial immune dendritic cell alterations in subtypes of dry eye disease: a pilot in vivo confocal microscopic study. Invest Ophthalmol Vis Sci. 2015;56(12):7179–7185. | ||
Machetta F, Fea AM, Actis AG, de Sanctis U, Dalmasso P, Grignolo FM. In vivo confocal microscopic evaluation of corneal langerhans cells in dry eye patients. Open Ophthalmol J. 2014;8:51–59. | ||
Zhang X, Chen Q, Chen W, Cui L, Ma H, Lu F. Tear dynamics and corneal confocal microscopy of subjects with mild self-reported office dry eye. Ophthalmology. 2011;118(5):902–907. | ||
Villani E, Garoli E, Termine V, Pichi F, Ratiglia R, Nucci P. Corneal Confocal Microscopy in Dry Eye Treated with Corticosteroids. Optom Vis Sci. 2015;92(9):e290–e295. | ||
Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol. 2008;4(10):525–533. | ||
Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711–1723. | ||
Marsh P, Pflugfelder SC. Topical nonpreserved methylprednisolone therapy for keratoconjunctivitis sicca in Sjogren syndrome. Ophthalmology. 1999;106(4):811–816. | ||
Avunduk AM, Avunduk MC, Varnell ED, Kaufman HE. The comparison of efficacies of topical corticosteroids and nonsteroidal anti-inflammatory drops on dry eye patients: a clinical and immunocytochemical study. Am J Ophthalmol. 2003;136(4):593–602. | ||
Jung HH, Ji YS, Sung MS, Kim KK, Yoon KC. Long-term outcome of treatment with topical corticosteroids for severe dry eye associated with Sjogren’s Syndrome. Chonnam Med J. 2015;51(1):26–32. | ||
Pflugfelder SC, Maskin SL, Anderson B, et al. A randomized, double-masked, placebo-controlled, multicenter comparison of loteprednol etabonate ophthalmic suspension, 0.5%, and placebo for treatment of keratoconjunctivitis sicca in patients with delayed tear clearance. Am J Ophthalmol. 2004;138(3):444–457. | ||
Pinto-Fraga J, Lopez-Miguel A, Gonzalez-Garcia MJ, et al. Topical fluorometholone protects the ocular surface of dry eye patients from desiccating stress: a randomized controlled clinical trial. Ophthalmology. 2016;123(1):141–153. | ||
McGhee CN, Dean S, Danesh-Meyer H. Locally administered ocular corticosteroids: benefits and risks. Drug Saf. 2002;25(1):33–55. | ||
Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology. 2000;47(2–3):119–125. | ||
Allergan. Prescribing information for RESTASIS®. Available from: http://www.allergan.com/assets/pdf/restasis_pi.pdf. Accessed June 25, 2017. | ||
Ames P, Galor A. Cyclosporine ophthalmic emulsions for the treatment of dry eye: a review of the clinical evidence. Clin Investig (Lond). 2015;5(3):267–285. | ||
Stevenson D, Tauber J, Reis BL. Efficacy and safety of cyclosporin A ophthalmic emulsion in the treatment of moderate-to-severe dry eye disease: a dose-ranging, randomized trial. The Cyclosporin A Phase 2 Study Group. Ophthalmology. 2000;107(5):967–974. | ||
Chen M, Gong L, Sun X, et al. A comparison of cyclosporine 0.05% ophthalmic emulsion vs vehicle in Chinese patients with moderate to severe dry eye disease: an eight-week, multicenter, randomized, double-blind, parallel-group trial. J Ocul Pharmacol Ther. 2010;26(4):361–366. | ||
Sall K, Stevenson OD, Mundorf TK, Reis BL. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase 3 Study Group. Ophthalmology. 2000;107(4):631–639. | ||
Baiza-Duran L, Medrano-Palafox J, Hernandez-Quintela E, Lozano-Alcazar J, Alaniz-de la OJ. A comparative clinical trial of the efficacy of two different aqueous solutions of cyclosporine for the treatment of moderate-to-severe dry eye syndrome. Br J Ophthalmol. 2010;94(10):1312–1315. | ||
Rao SN. Topical cyclosporine 0.05% for the prevention of dry eye disease progression. J Ocul Pharmacol Ther. 2010;26(2):157–164. | ||
Kunert KS, Tisdale AS, Gipson IK. Goblet cell numbers and epithelial proliferation in the conjunctiva of patients with dry eye syndrome treated with cyclosporine. Arch Ophthalmol. 2002;120(3):330–337. | ||
Laibovitz RA, Solch S, Andriano K, O’Connell M, Silverman MH. Pilot trial of cyclosporine 1% ophthalmic ointment in the treatment of keratoconjunctivitis sicca. Cornea. 1993;12(4):315–323. | ||
Deveci H, Kobak S. The efficacy of topical 0.05% cyclosporine A in patients with dry eye disease associated with Sjogren’s syndrome. Int Ophthalmol. 2014;34(5):1043–1048. | ||
Kim EC, Choi JS, Joo CK. A comparison of vitamin a and cyclosporine a 0.05% eye drops for treatment of dry eye syndrome. Am J Ophthalmol. 2009;147(2):206.e3–213.e3. | ||
Prabhasawat P, Tesavibul N, Mahawong W. A randomized double-masked study of 0.05% cyclosporine ophthalmic emulsion in the treatment of meibomian gland dysfunction. Cornea. 2012;31(12):1386–1393. | ||
Demiryay E, Yaylali V, Cetin EN, Yildirim C. Effects of topical cyclosporine a plus artificial tears vs artificial tears treatment on conjunctival goblet cell density in dysfunctional tear syndrome. Eye Contact Lens. 2011;37(5):312–315. | ||
Sheppard JD, Scoper SV, Samudre S. Topical loteprednol pretreatment reduces cyclosporine stinging in chronic dry eye disease. J Ocul Pharmacol Ther. 2011;27(1):23–27. | ||
Byun YJ, Kim TI, Kwon SM, et al. Efficacy of combined 0.05% cyclosporine and 1% methylprednisolone treatment for chronic dry eye. Cornea. 2012;31(5):509–513. | ||
Gadek TR, Burdick DJ, McDowell RS, et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science. 2002;295(5557):1086–1089. | ||
Zhong M, Gadek TR, Bui M, et al. Discovery and development of potent LFA-1/ICAM-1 antagonist SAR 1118 as an ophthalmic solution for treating dry eye. ACS Med Chem Lett. 2012;3(3):203–206. | ||
Pflugfelder SC, Stern M, Zhang S, Shojaei A. LFA-1/ICAM-1 interaction as a therapeutic target in dry eye disease. J Ocul Pharmacol Ther. 2017;33(1):5–12. | ||
Teijeira A, Garasa S, Pelaez R, et al. Lymphatic endothelium forms integrin-engaging 3D structures during DC transit across inflamed lymphatic vessels. J Invest Dermatol. 2013;133(9):2276–2285. | ||
Rouzaut A, Garasa S, Teijeira A, et al. Dendritic cells adhere to and transmigrate across lymphatic endothelium in response to IFN-alpha. Eur J Immunol. 2010;40(11):3054–3063. | ||
Varga G, Nippe N, Balkow S, et al. LFA-1 contributes to signal I of T-cell activation and to the production of T(h)1 cytokines. J Invest Dermatol. 2010;130(4):1005–1012. | ||
Hartman NC, Nye JA, Groves JT. Cluster size regulates protein sorting in the immunological synapse. Proc Natl Acad Sci U S A. 2009;106(31):12729–12734. | ||
Fooksman DR, Vardhana S, Vasiliver-Shamis G, et al. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol. 2010;28:79–105. | ||
de la Fuente H, Mittelbrunn M, Sanchez-Martin L, et al. Synaptic clusters of MHC class II molecules induced on DCs by adhesion molecule-mediated initial T-cell scanning. Mol Biol Cell. 2005;16(7):3314–3322. | ||
Porter JC, Bracke M, Smith A, Davies D, Hogg N. Signaling through integrin LFA-1 leads to filamentous actin polymerization and remodeling, resulting in enhanced T cell adhesion. J Immunol. 2002;168(12):6330–6335. | ||
Smith A, Stanley P, Jones K, Svensson L, McDowall A, Hogg N. The role of the integrin LFA-1 in T-lymphocyte migration. Immunol Rev. 2007;218:135–146. | ||
Semba CP, Gadek TR. Development of lifitegrast: a novel T-cell inhibitor for the treatment of dry eye disease. Clin Ophthalmol. 2016;10:1083–1094. | ||
Sun Y, Zhang R, Gadek TR, O’Neill CA, Pearlman E. Corneal inflammation is inhibited by the LFA-1 antagonist, lifitegrast (SAR 1118). J Ocul Pharmacol Ther. 2013;29(4):395–402. | ||
Rao VR, Prescott E, Shelke NB, et al. Delivery of SAR 1118 to the retina via ophthalmic drops and its effectiveness in a rat streptozotocin (STZ) model of diabetic retinopathy (DR). Invest Ophthalmol Vis Sci. 2010;51(10):5198–5204. | ||
Murphy CJ, Bentley E, Miller PE, et al. The pharmacologic assessment of a novel lymphocyte function-associated antigen-1 antagonist (SAR 1118) for the treatment of keratoconjunctivitis sicca in dogs. Invest Ophthalmol Vis Sci. 2011;52(6):3174–3180. | ||
Dustin ML, Groves JT. Receptor signaling clusters in the immune synapse. Annu Rev Biophys. 2012;41:543–556. | ||
Semba CP, Torkildsen GL, Lonsdale JD, et al. A phase 2 randomized, double-masked, placebo-controlled study of a novel integrin antagonist (SAR 1118) for the treatment of dry eye. Am J Ophthalmol. 2012;153(6):1050.e1–1060.e1. | ||
Sheppard JD, Torkildsen GL, Lonsdale JD, et al. Lifitegrast ophthalmic solution 5.0% for treatment of dry eye disease: results of the OPUS-1 phase 3 study. Ophthalmology. 2014;121(2):475–483. | ||
Tauber J, Karpecki P, Latkany R, et al. Lifitegrast ophthalmic solution 5.0% vs placebo for treatment of dry eye disease: results of the randomized Phase III OPUS-2 Study. Ophthalmology. 2015;122(12):2423–2431. | ||
Holland EJ, Luchs J, Karpecki PM, et al. Lifitegrast for the treatment of dry eye disease: results of a Phase III, Randomized, Double-Masked, Placebo-Controlled Trial (OPUS-3). Ophthalmology. 2017;124(1):53–60. | ||
Semba CP, Swearingen D, Smith VL, et al. Safety and pharmacokinetics of a novel lymphocyte function-associated antigen-1 antagonist ophthalmic solution (SAR 1118) in healthy adults. J Ocul Pharmacol Ther. 2011;27(1):99–104. | ||
Paskowitz DM, Nguyen QD, Gehlbach P, et al. Safety, tolerability, and bioavailability of topical SAR 1118, a novel antagonist of lymphocyte function-associated antigen-1: a phase 1b study. Eye (Lond). 2012;26(7):944–949. | ||
Donnenfeld ED, Karpecki PM, Majmudar PA, et al. Safety of lifitegrast ophthalmic solution 5.0% in patients with dry eye disease: a 1-year, multicenter, randomized, placebo-controlled study. Cornea. 2016;35(6):741–748. | ||
Lanza NL, McClellan AL, Batawi H, et al. Dry eye profiles in patients with a positive elevated surface matrix metalloproteinase 9 point-of-care test versus negative patients. Ocul Surf. 2016;14(2):216–223. | ||
Baudouin C, Messmer EM, Aragona P, et al. Revisiting the vicious circle of dry eye disease: a focus on the pathophysiology of meibomian gland dysfunction. Br J Ophthalmol. 2016;100(3):300–306. | ||
Galor A, Kumar N, Feuer W, Lee DJ. Environmental factors affect the risk of dry eye syndrome in a United States veteran population. Ophthalmology. 2014;121(4):972–973. | ||
Miljanovic B, Trivedi KA, Dana MR, Gilbard JP, Buring JE, Schaumberg DA. Relation between dietary n-3 and n-6 fatty acids and clinically diagnosed dry eye syndrome in women. Am J Clin Nutr. 2005;82(4):887–893. | ||
Galor A, Felix ER, Feuer W, et al. Dry eye symptoms align more closely to non-ocular conditions than to tear film parameters. Br J Ophthalmol. 2015;99(8):1126–1129. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.