Back to Journals » International Journal of Nanomedicine » Volume 14
Drug-loaded exosomal preparations from different cell types exhibit distinctive loading capability, yield, and antitumor efficacies: a comparative analysis
Authors Kanchanapally R, Deshmukh SK, Chavva SR, Tyagi N, Srivastava SK ![]() , Patel GK, Singh AP, Singh S
, Patel GK, Singh AP, Singh S
Received 18 October 2018
Accepted for publication 1 December 2018
Published 11 January 2019 Volume 2019:14 Pages 531—541
DOI https://doi.org/10.2147/IJN.S191313
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Rajashekhar Kanchanapally,1 Sachin Kumar Deshmukh,1 Suhash Reddy Chavva,1 Nikhil Tyagi,1 Sanjeev Kumar Srivastava,1 Girijesh Kumar Patel,1 Ajay Pratap Singh,1,2 Seema Singh1,2
1Department of Oncologic Sciences, Mitchell Cancer Institute, University of South Alabama, Mobile, AL 36604, USA; 2Department of Biochemistry and Molecular Biology, College of Medicine, University of South Alabama, Mobile, AL 36688, USA
Background: Despite tremendous advancement, cancer still remains one of the leading causes of death worldwide. Inefficiency of current drug delivery regimens is one important factor that limits the therapeutic efficacy of existing drugs, thus contributing to cancer mortality. To address this limitation, synthetic nanotechnology-based delivery systems have been developed; however, they raise concern of inducing adverse immunogenic reactions. Exosomes (Exos) are nonimmunogenic nano-sized vesicles that have received significant attention as efficient drug delivery system.
Methods: Drug loading in Exos were achieved by incubating different cell types viz pancreatic cancer cells (PCCs), pancreatic stellate cells (PSCs), and macrophages (MØs) with Doxorubicin (DOX). Differential ultracentrifugation was performed to isolate exosome and their size was determined by dynamic light scattering analysis. The efficacy of drug packaging into Exos was evaluated by HPLC. Flow cytometry was performed to examine the apoptosis. Cell viability was determined using the WST-1 assay.
Results: PCCs shed the most Exos and were the most efficient in drug loading followed by MØs and PSCs as examined by HPLC quantification. However, when compared for antitumor efficacy, MØ-derived Exos loaded with DOX (MØ-Exo-DOX) showed highest activity followed by PSCs and PCCs.
Conclusion: These varying antitumor activities likely resulted from nondrug contents of Exos since we did not observe any significant differences in their uptake by the cancer cells. Altogether, our data suggest that donor cell-specific differences exist in Exos, which could influence their utility as drug carrier for therapeutic purposes.
Keywords: exosome, macrophage, pancreatic cancer, drug delivery, doxorubicin
Introduction
Cancer is the second leading cause of death worldwide, and a major reason for high cancer mortality is the ineffectiveness of existing therapies.1,2 Therapeutic efficacy of current and emerging drugs is limited due to their inability to reach to the target cells in optimally effective doses because of their rapid metabolization in the systemic circulation3 as well as lack of selectivity.4 Moreover, nonspecific delivery of drugs leads to normal tissue toxicity, which further reduces the therapeutic index. To achieve the targeted and protected delivery of drugs, many artificial drug carriers have been developed including dendrimers,5 liposomes,6 plasmonic nanoparticles,7 magnetic nanoparticles,8 and polymer-based nanoparticlces.9 However, being of foreign origin, these artificial nanocarriers are prone to hepatic clearance,10 while also posing concerns for immunotoxicity.11
Exosomes (Exos) are lipid bi-layer membrane vesicular nanostructures of endosomal origin that are released by almost all kinds of cells.12,13 These were initially thought to be garbage bags of unwanted cellular biomaterial; however, they are now established as crucial players in intercellular communication.12–18 Exos efficiently carry all kinds of biological molecules including miRNAs, mRNAs, and proteins protected from enzymatic degradation.15–18 Exosomal delivery of cargo to the recipient cells is highly efficient that occurs either by direct membrane fusion, receptor-mediated endocytosis, macropinocytosis, or phagocytosis.19,20 These properties of Exos have prompted researchers to exploit their utility as drug delivery vehicle for the treatment of a variety of diseases, including cancer.10,19,21–24 Exos derived from the tumor cells have been investigated for tumor-targeted drug delivery since they carry similar surface receptors as tumor cells.25 In other studies, Exos derived from cells in the tumor microenvironment such as fibroblasts and immune cells have been examined for their drug loading and tumor-targeting efficacy.19,22
In this study, we examined the efficacy of different cell types viz pancreatic cancer cells (PCCs), pancreatic stellate cells (PSCs), and macrophages (MØs) for drug (doxorubicin [DOX]) packaging and release into Exos and the subsequent applicability of released Exos as the drug carrier of choice. Our data show that PCCs shed Exos most efficiently followed by MØs and PSCs. Moreover, PCCs exhibited the maximum drug-loading efficiency into the Exos followed by MØs and PSCs. Furthermore, we observed that all types of DOX-treated cells shed slightly more Exos as compared to those treated with vehicle only. In addition, our data show that MØ-derived DOX-loaded Exos had the greatest tumor cell toxicity followed by those derived from PSCs. No changes, however, were reported in the uptake of different exosomal preparations by the cancer cells. Taken together, these findings depict donor cell-specific differences in exosomal yield, drug-loading, and antitumor activities, which should be taken into consideration for their use as effective drug delivery system for cancer therapy.
Materials and methods
Reagents
The following reagents were used in this study: DMEM (Corning Incorporated, Corning, NY, USA); penicillin and streptomycin (10,000 U/mL each; GE Healthcare Life Sciences, Logan, UT, USA); FBS (Atlanta Biologicals, Lawrenceville, GA, USA); DOX hydrochloride (Tocris Bioscience, Bristol, UK); SuperSignal™ West Femto Maximum sensitivity substrate kit (Thermo Fisher Scientific, Waltham, MA, USA), Trypan blue (Corning Incorporated), cell proliferation reagent (WST-1) (Roche Diagnostic Corporation, Indianapolis, IN, USA), PE Annexin V Apoptosis Detection Kit (BD Biosciences, San Diego, CA, USA), Tris-buffered saline (Thermo Fisher Scientific), and Tween 20 (Boston Bioproducts, Ashland, MA, USA).
Cell culture
RAW264.7 (MØs) and MiaPaCa-2 (PCC line) were procured from American Type Culture Collection (Manassas, VA, USA). PSCs were generously gifted by Dr P. K. Singh, Eppley Cancer Institute (Omaha, NE, USA). All the cell lines were maintained as monolayer cultures in DMEM, supplemented with 10% FBS, penicillin (1,000 U/mL), and streptomycin (1,000 U/mL), in a 5% CO2 incubator at 37°C. Intermittent testing was done to authenticate cells and to ensure that they were free of mycoplasma contamination.
Exo extraction, size distribution analysis, and quantitation
For Exos’ extraction, confluent flasks of MØs, PCCs, and PSCs were treated either with vehicle (PBS) or 1 μM DOX for 48 h. After 48 h, conditioned media were collected and the Exos were extracted as reported earlier.26 Briefly, the conditioned media from DOX-treated cells were collected and initially centrifuged at lower speeds (16,500× g for 30 min) to remove cell debris, apoptotic bodies, and large vesicles. The supernatant obtained was further centrifuged at 120,000× g in an ultra-centrifuge, for 2 h, to obtain Exo pellet. A washing step is followed by resuspending the pellet in 5 mL of PBS solution and was centrifuged at 120,000× g for an additional 2 h. The Exos were labeled as PCC-Exo-Veh, PSC-Exo-Veh, and MØ-Exo-Veh (obtained from vehicle-treated PCCs, PSCs, and MØ, respectively) and PCC-Exo-DOX, PSC-Exo-DOX, and MØ-Exo-DOX (obtained from DOX-treated PCCs, PSCs, and MØ, respectively). Exos were stored at 4°C until further analysis. Size distribution of Exos was determined on freshly extracted preps diluted in deionized water (1:1,000 vol ratio) using DelsaMax PRO (Beckman Coulter, Brea, CA, USA) dynamic light scatter analyzer. Exos were quantified indirectly by the surface protein quantitation using the DC™ protein assay kit (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Quantification of DOX loading in exosomal preparations
After washing step, Exo pellets were left to air dry for 2 h. Thereafter, 20 μL of 8 M urea was added and pellets were sonicated in a bath type sonicator for 5–10 min. To the above suspension, 60 μL of 50 mM ammonium bicarbonate/10 mM tris(2-carboxyethyl) phosphine hydrochloride and 1.5 μL of trypsin were added and left overnight for protein digestion. Released DOX was measured using the reverse-phase HPLC, equipped with a UV detector. DOX standards of concentrations ranging from 1 to 500 μM were prepared using a 1:1 mixture of water/acetonitrile. A standard curve was plotted using the area under the curve of the standards. Subsequently, 5.0 μL of the Exo digest was injected onto a C18 guard column using a gradient, starting at 50% Solvent A (96.8% water, 3% acetonitrile, and 0.2% formic acid) and 50% Solvent B (96.8% acetonitrile, 3% water, and 0.2% formic acid) to 70% Solvent B and then a washing step at 90% Solvent B.
Cell viability assay
To examine the effect on cell viability, we used either free DOX or equivalent doses of different Exo-DOX formulations based on their loaded DOX content as determined by HPLC analysis. Cancer cells were seeded in 96-well plates at a density of 5×103 cells/well, and after 24 h of resting period, cells were treated with vehicle or DOX or DOX-loaded exosomal preparations for 24, 48, and 72 h. Subsequently, cell viability was measured using the WST-1 assay following manufacturers’ instructions.
Apoptosis measurement
PCCs were seeded in six-well plates at a density of 2.5×105 cells/well. After a 24 h resting period, cells were treated either with DOX or with Exo-DOX formulations with DOX concentration equivalent of DOX half maximal inhibitory concentration (IC50) at 48 h. Thereafter, cells were stained with 7-amino-actinomycin (7-AAD) and PE Annexin V using the PE Annexin V Apoptosis Detection Kit as per manufacturer’s instructions. The processed cells were analyzed for the percentage of apoptotic cells using flow cytometry.
Uptake studies
For fluorescence imaging, PCCs seeded in a six-well plate at a density of 2.5×105 cells/well. Following a 24 h resting period, cells were treated with Exo-DOX formulations with DOX concentration equivalent to 85.32 nM. After 8 h incubation, the cells were visualized under a microscope for DOX fluorescence (EVOS microscopy). For fluorescence quantification, 5×103 cells/well were seeded in 96-well plate and rested for 24 h. Subsequently, cells were treated with Exo-DOX formulations with DOX concentration equivalent to 85.32 nM. After 8 h incubation, media were removed and cells were washed with PBS. In the end, cells were analyzed for fluorescence intensity using the 96-well plate reader.
Statistical analysis
Data from replicative experiments were analyzed for mean ± SD/error calculation. The unpaired two-tailed Student’s t-test was used to determine P-value, and a P-value of ≤0.05 was considered statistically significant.
Results
Preparation of DOX-loaded exosomal preparations from different cell types and their characterization
To obtain DOX-loaded Exos, we treated near confluent cultures of PCCs, PSCs, and MØs with either vehicle or 1 μM of DOX for 48 h (Figure 1A). Subsequently, culture supernatants were collected and Exos were isolated by ultracentrifugation (Figure 1A). Exos from vehicle- or DOX-treated PCCs, PSCs, and MØs were analyzed for their size distribution using dynamic light scattering (Figure 1B). Data show that the size of the Exos obtained from the vehicle or DOX-treated cells fall under the range of 120–155 nm (Figure 1B). However, the size of Exo-DOX was slightly larger compared to the Exos obtained from cells treated with vehicle only (Figure 1B). Next, we measured the surface protein contents of Exos to assess differences in exosomal shedding by different cell types. Data show that PCCs shed Exos most efficiently followed by MØs, whereas the least quantity of Exos was detected in preparation from PSCs. In addition, we observed that DOX-treated cells shed slightly more Exos than those treated with vehicle only (Figure 1C). Furthermore, immunoblot analyses revealed the presence of CD9 (Exo marker) but the absence of Arf6 (microvesicle marker), indicating the purity of exosomal preparation (Figure S1). UV–Vis spectrophotometry analyses of the Exo-DOX preparations revealed absorption maxima at ~480 nm, which corresponds to DOX absorption, and thus, it is indicative of the DOX loading into these Exos (Figure S2). As expected, no such absorption was observed in Exos obtained from vehicle-treated cells (Exo-Veh). DOX loading in Exos was quantified by HPLC equipped with a UV–Vis detector on a C18 guard column. The HPLC data show that PCC-Exo-DOX contained the maximum amount of DOX (3.00 μg) followed by MØ-Exo-DOX (1.66 μg) and PSC-Exo-DOX (0.86 μg) (Figure 2A). To assess DOX-loading efficiency, we normalized the total DOX content from exosomal preparations to their surface protein content. Resulting data demonstrated maximum loading in PCC-Exo-DOX (14.06 ng/μg), followed by MØ-Exo-DOX (7.27 ng/μg) and PSC-Exo-DOX (3.99 ng/μg) (Figure 2B).
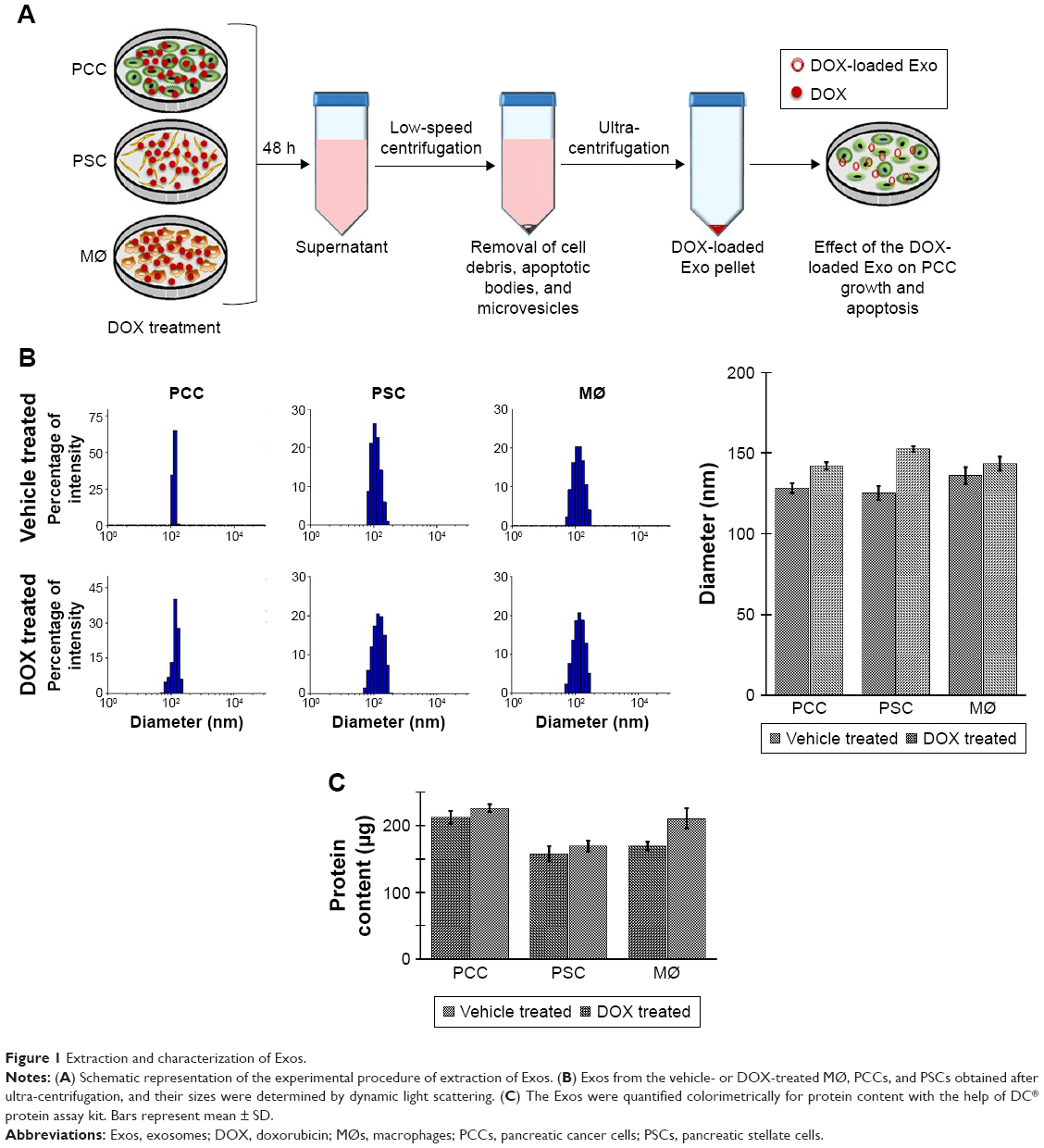 | Figure 1 Extraction and characterization of Exos. |
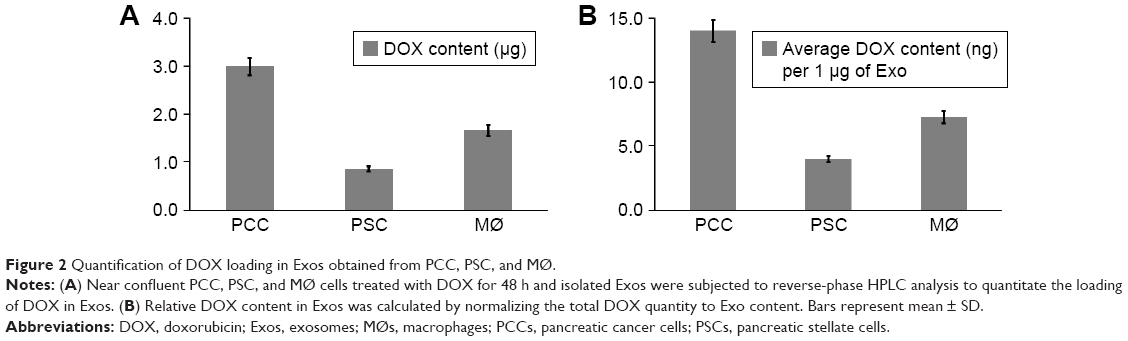 | Figure 2 Quantification of DOX loading in Exos obtained from PCC, PSC, and MØ. |
MØ-derived Exo-DOX preparation shows highest antitumor activity
To assess the efficacy of different DOX-loaded exosomal preparations, we first treated PCCs with free DOX ranging in concentration from 0 to 1,000 nM at three different time points viz 24, 48, and 72 h (Figure 3A). The IC50 was determined to be 121.7, 85.32, and 40.65 nM at 24, 48, and 72 h time points, respectively. Next, we treated PCCs with 85.32 nM (IC50 at 48 h) of free DOX or PCC-Exo-DOX, MØ-Exo-DOX, and PSC-Exo-DOX carrying an equivalent amount of DOX for 24, 48, and 72 h. At the end of the stipulated treatment time, cell viability was determined by the WST-1 assay. At 48 and 72 h time points, Exo-DOX formulations were significantly more toxic to cancer cells than an equivalent amount of free DOX (Figure 3B and C). More importantly, we observed the greatest growth inhibitory potency for MØ-Exo-DOX (Figure 3B and C). Interestingly, we observed that Exos isolated from vehicle-treated PCCs had some growth promoting effect. While this growth stimulation was not statistically significant at 24 h time point when compared with cells treated with vehicle only, we observed a statistically significant difference in growth at 48 h (8.18%) and 72 h (11.92%) time points. Contradictorily, Exos from vehicle-treated MØ caused a decrease in the growth of the PCCs, which was statistically significant at 48 h (5.78%) and 72 h (11.45%) time points. At all the time points (24, 48, and 72 h), PSC-Exo-Veh did not have any significant effect on the growth of PCCs.
 | Figure 3 Comparison of Exo-DOX formulations on cancer cell proliferation and apoptosis. |
MØ-derived Exo-DOX preparation induces greater apoptosis in cancer cells compared to those derived from PCCs or PSCs
Since the induction of apoptotic cell death is a prime mechanism of chemotherapeutic drugs, we were interested to see how different Exo-DOX preparations fared relative to free DOX. PCCs were treated with 85.32 nM DOX or different Exo-DOX preparations containing an equivalent amount of DOX. After 48 h of treatment, the cells were analyzed for the percentage of apoptotic cells by the flow cytometry assessment of Annexin-V-stained cells. We found that all the tested Exo-DOX formulations outperformed the efficacy of free DOX in apoptosis induction (Figure 4). Furthermore, MØ-Exo-DOX preparation was found to be the most toxic to PCCs with an apoptotic index of 80.1% followed by PSC-Exo-DOX and PCC-Exo-DOX with the apoptotic indices of 73.2% and 65.3%, respectively (Figure 4).
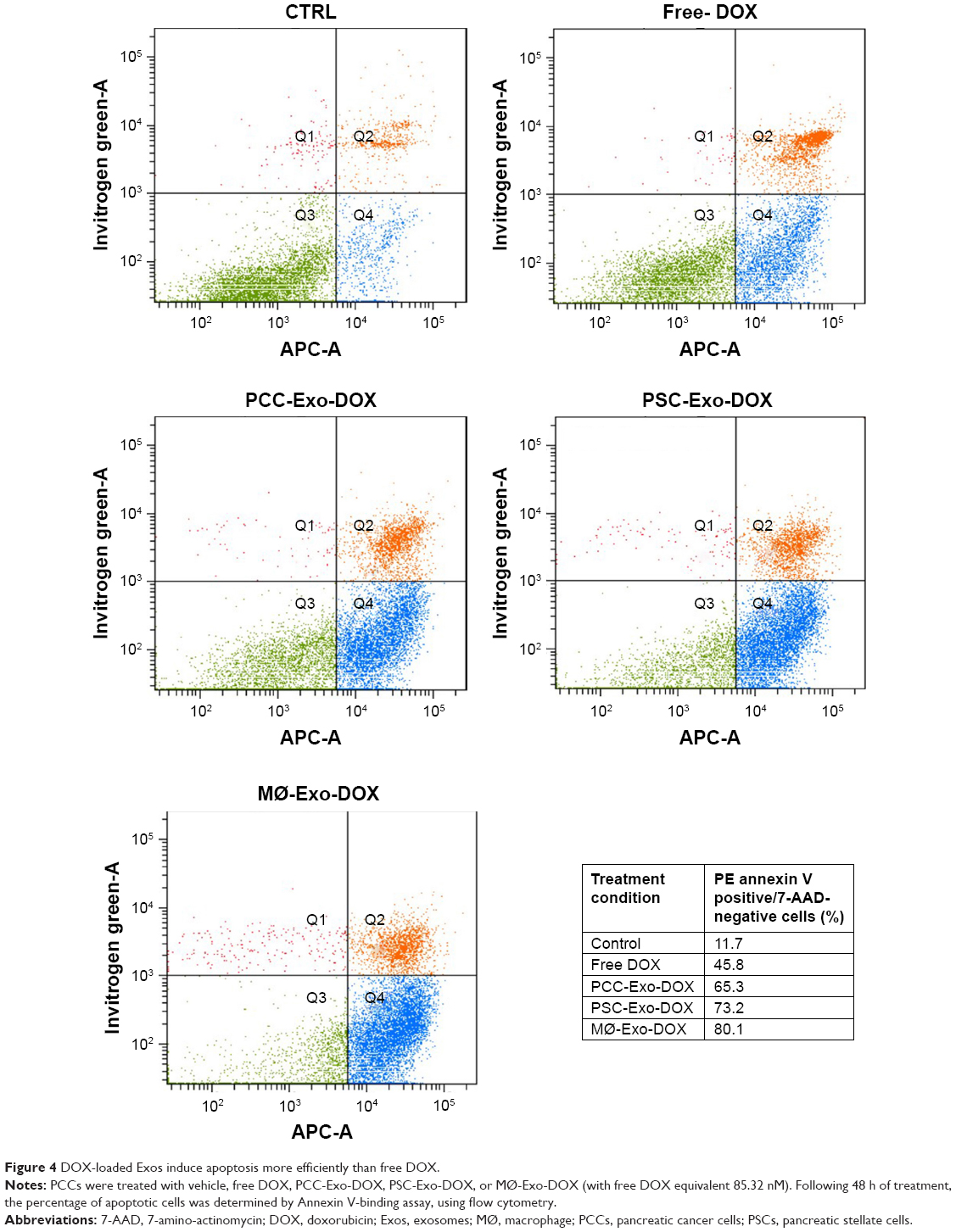 | Figure 4 DOX-loaded Exos induce apoptosis more efficiently than free DOX. |
All Exo-DOX formulations are taken up with equal efficiency by the cancer cells
To determine if the differences in cancer cell toxicity of various Exo-DOX formulations were due to their differential uptake by the cancer cells, we treated PCCs with PCC-Exo-DOX, PSC-Exo-DOX, and MØ-Exo-DOX containing an equal amount of drug (85.32 nM) for 8 h. Thereafter, cellular uptake was monitored under fluorescence microscope as red fluorescent signal of DOX. We observed no apparent difference in the cellular uptake of different Exo-DOX preparations by the PCCs (Figure 5A). Furthermore, to get the quantitative data, we treated PCCs in 96-well opaque bottom plates for 8 h followed by the replacement of culture media. Fluorescence intensity in different wells was recorded at 485 nm excitation and 528 nm emission, which also showed no significant difference in the cellular uptake of Exos (Figure 5B).
 | Figure 5 Comparison of cellular uptake of different Exo-DOX formulations. |
Discussion
Exos are effective carriers of cellular material and being biological membrane nanoparticles that do not induce undesired immunologic response.15–18,21 These properties of Exos have enabled their uses as drug delivery systems.10,19,21,25 Since almost all types of cells shed Exos, various scientific groups have attempted different cell types for drug encapsulation.21,25 In that regard, our study provides useful information on drug loading and release capabilities of Exos from different cell types and the efficacy of drug-loaded Exos in tumor cell killing.
Tumor cells are known to shed greater yield of Exos compared to the normal cells.13,27,28 In line with this, we observed that PCCs were the most efficient in shedding Exos followed by MØs and PSCs. Furthermore, studies from our own laboratory have suggested that tumor cells secrete Exos at a greater frequency when exposed to environmental stress.26,29 Similarly, we observed that drug treatment induced Exo shedding not only in PCCs but also in PSCs and MØs. Though it is known that stress enhances the release of Exos,30,31 the mechanism is not clearly understood. It might be an alternative mechanism to dispose off “waste” from the cell, as degradative pathways are obstructed and/or the lysosomal pathways are overloaded due to stress.32
It is known that DOX accumulates inside the endosomes33,34 and may eventually be exocytosed via Exos. Our findings confirm this nonmechanical way of DOX loading into Exo and demonstrate cell type-specific differences. We chose DOX as our drug of choice due to the ease of analysis by the virtue of its absorption and bright fluorescence properties, and our data support its good antitumor activity in line with other published studies.35–37 We observed that all the tested cell types were capable of shedding of DOX-loaded Exos; however, the highest efficacy was observed for PCCs. This superior efficiency of PCCs can be attributed to the efficiency of cancer cells to pumpout drugs in a more effective way than other cell types.38–40
It is well established that treatment of cancer cells with DOX induces apoptosis.37,41–43 We observed that all the tested Exo-DOX formulations outperformed free DOX in cytotoxicity toward PCCs. This could be due to the fact that drug efflux pumps actively pumpout DOX from the cancer cells.44,45 Moreover, DOX, which has basic pH may likely accumulate in acidic cell organelles such as lysosomes and endosomes and eventually thrown out via exocytosis.33,34 However, when DOX is associated with nanoparticles, it escapes from accumulation in cell organelles and efflux pumps leading to an improved efficacy of DOX.44–47 Though all the tested Exo-DOX formulations have shown improved anticancer efficacy, MØ-Exo-DOX is most toxic toward PCCs, whereas PCC-Exo-DOX is least toxic. From our experimental data, it is evident that there is no significant difference in the uptake efficiency by PCCs of the Exo-DOX formulations from different donor cell types. The reason for the dissimilarity in the efficacy of these Exo-DOX formulations could be that Exos derived from PCCs or PSCs may contain biomolecules that is either tumor supporting or antagonize tumor cell killing by the drug.26,48–53 An alternate explanation could be that immune cell naturally carries antitumor agents, which might have passed on to their corresponding Exos.54
Conclusion
Our data demonstrate differences in yield, drug-loading capability, and anticancer efficacy of Exos derived from different donor cell types. These observations should guide us while considering the use of Exos as effective drug delivery system for cancer therapy.
Acknowledgments
We would like to acknowledge the funding support from the National Institute of Health/National Cancer Institute (CA204801 [to SS] and CA224306 and CA175772 [to APS]) as well as the institutional support from University of South Alabama Mitchell Cancer Institute.
Disclosure
APS and SS are cofounders and serve on the executive management team of Tatva Biosciences LLC, which is involved in the development of tools and models for biological research. The authors report no other conflicts of interest in this work.
References
Roser M, Ritchie H [homepage on the Internet]. Cancer; 2018. Available from: https://ourworldindata.org/cancer. Accessed August 22, 2018. | ||
Chakraborty S, Rahman T. The difficulties in cancer treatment. Ecancermedicalscience. 2012;6:ed16. | ||
Vadlapatla RK, Vadlapudi AD, Pal D, Mitra AK. Mechanisms of drug resistance in cancer chemotherapy: coordinated role and regulation of efflux transporters and metabolizing enzymes. Curr Pharm Des. 2013;19(40):7126–7140. | ||
Heo DN, Yang DH, Moon HJ, et al. Gold nanoparticles surface-functionalized with paclitaxel drug and biotin receptor as theranostic agents for cancer therapy. Biomaterials. 2012;33(3):856–866. | ||
Zhu S, Hong M, Zhang L, Tang G, Jiang Y, Pei Y. PEGylated PAMAM dendrimer-doxorubicin conjugates: in vitro evaluation and in vivo tumor accumulation. Pharm Res. 2010;27(1):161–174. | ||
Gabizon AA. Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest. 2001;19(4):424–436. | ||
Song J, Zhou J, Duan H. Self-assembled plasmonic vesicles of SERS-encoded amphiphilic gold nanoparticles for cancer cell targeting and traceable intracellular drug delivery. J Am Chem Soc. 2012;134(32):13458–13469. | ||
Sun C, Lee JS, Zhang M. Magnetic nanoparticles in MR imaging and drug delivery. Adv Drug Deliv Rev. 2008;60(11):1252–1265. | ||
Masood F. Polymeric nanoparticles for targeted drug delivery system for cancer therapy. Mater Sci Eng C. 2016;60:569–578. | ||
Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles – endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta. 1846;2014(1):75–87. | ||
Lee YK, Choi EJ, Webster TJ, Kim SH, Khang D. Effect of the protein corona on nanoparticles for modulating cytotoxicity and immunotoxicity. Int J Nanomedicine. 2015;10:97–113. | ||
Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. | ||
Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208–1215. | ||
Février B, Raposo G. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol. 2004;16(4):415–421. | ||
Thakur BK, Zhang H, Becker A, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24(6):766–769. | ||
Janas T, Janas MM, Sapoń K, Janas T. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015;589(13):1391–1398. | ||
Bang C, Thum T. Exosomes: new players in cell-cell communication. Int J Biochem Cell Biol. 2012;44(11):2060–2064. | ||
Whiteside TL. Exosomes carrying immunoinhibitory proteins and their role in cancer. Clin Exp Immunol. 2017;189(3):259–267. | ||
Lai RC, Yeo RW, Tan KH, Lim SK. Exosomes for drug delivery – a novel application for the mesenchymal stem cell. Biotechnol Adv. 2013;31(5):543–551. | ||
Mckelvey KJ, Powell KL, Ashton AW, Morris JM, Mccracken SA. Exosomes: mechanisms of uptake. J Circ Biomark. 2015;4:7. | ||
Kim MS, Haney MJ, Zhao Y, et al. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: in vitro and in vivo evaluations. Nanomedicine. 2018;14(1):195–204. | ||
Pascucci L, Coccè V, Bonomi A, et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release. 2014;192:262–270. | ||
Sun D, Zhuang X, Xiang X, et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther. 2010;18(9):1606–1614. | ||
Tian T, Zhang HX, He CP, et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials. 2018;150:137–149. | ||
Saari H, Lázaro-Ibáñez E, Viitala T, Vuorimaa-Laukkanen E, Siljander P, Yliperttula M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J Control Release. 2015;220(Pt B):727–737. | ||
Patel GK, Khan MA, Bhardwaj A, et al. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br J Cancer. 2017;116(5):609–619. | ||
Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-1. Haematologica. 2011;96(9):1302–1309. | ||
Maia J, Caja S, Strano Moraes MC, Couto N, Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Front Cell Dev Biol. 2018;6(18):18. | ||
Patton MC, Zubair H, Patel GK, Singh S, Singh AP. Abstract 2799: exosomes from hypoxic pancreatic cancer cells confer resistance to subsequent hypoxia insult. Cancer Res. 2016;76(14 Suppl):2799. | ||
King HW, Michael MZ, Gleadle JM. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer. 2012;12(1):421. | ||
Xiao X, Yu S, Li S, et al. Exosomes: decreased sensitivity of lung cancer A549 cells to cisplatin. PLoS One. 2014;9(2):e89534. | ||
Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75(2):193–208. | ||
Lee CM, Tannock IF. Inhibition of endosomal sequestration of basic anticancer drugs: influence on cytotoxicity and tissue penetration. Br J Cancer. 2006;94(6):863–869. | ||
Safaei R, Larson BJ, Cheng TC, et al. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol Cancer Ther. 2005;4(10):1595–1604. | ||
Li T, Wang YN, Khokhlova TD, et al. Pulsed high-intensity focused ultrasound enhances delivery of doxorubicin in a preclinical model of pancreatic cancer. Cancer Res. 2015;75(18):3738–3746. | ||
Arachchige MP, Laha SS, Naik AR, Lewis KT, Naik R, Jena BP. Functionalized nanoparticles enable tracking the rapid entry and release of doxorubicin in human pancreatic cancer cells. Micron. 2017;92:25–31. | ||
Xu X, Zhao Y, Zhang M, et al. Inhibition of autophagy by deguelin sensitizes pancreatic cancer cells to doxorubicin. Int J Mol Sci. 2017;18(2):370. | ||
Borst P. Cancer drug pan-resistance: pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biol. 2012;2(5):120066. | ||
Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53(1):615–627. | ||
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. | ||
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170. | ||
Yang XH, Sladek TL, Liu X, Butler BR, Froelich CJ, Thor AD. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Res. 2001;61(1):348–354. | ||
Deshmukh SK, Srivastava SK, Zubair H, et al. Resistin potentiates chemoresistance and stemness of breast cancer cells: implications for racially disparate therapeutic outcomes. Cancer Lett. 2017;396:21–29. | ||
Meng H, Liong M, Xia T, et al. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano. 2010;4(8):4539–4550. | ||
Goren D, Horowitz AT, Tzemach D, Tarshish M, Zalipsky S, Gabizon A. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin Cancer Res. 2000;6(5):1949–1957. | ||
Riva B, Bellini M, Corvi E, et al. Impact of the strategy adopted for drug loading in nonporous silica nanoparticles on the drug release and cytotoxic activity. J Colloid Interface Sci. 2018;519:18–26. | ||
Wang F, Wang YC, Dou S, Xiong MH, Sun TM, Wang J. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano. 2011;5(5):3679–3692. | ||
Tickner JA, Urquhart AJ, Stephenson S-A, Richard DJ, O’Byrne KJ. Functions and therapeutic roles of exosomes in cancer. Front Oncol. 2014;4(10):127. | ||
Chen W, Jiang J, Xia W, Huang J. Tumor-related exosomes contribute to tumor-promoting microenvironment: an immunological perspective. J Immunol Res. 2017;2017(11):1–10. | ||
Hannafon BN, Ding WQ. Intercellular communication by exosome-derived microRNAs in cancer. Int J Mol Sci. 2013;14(7):14240–14269. | ||
Lehmann BD, Paine MS, Brooks AM, et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008;68(19):7864–7871. | ||
Lugea A, Waldron RT. Exosome-mediated intercellular communication between stellate cells and cancer cells in pancreatic ductal adenocarcinoma. Pancreas. 2017;46(1):1–4. | ||
Deshmukh SK, Tyagi N, Khan MA, et al. Gemcitabine treatment promotes immunosuppressive microenvironment in pancreatic tumors by supporting the infiltration, growth, and polarization of macrophages. Sci Rep. 2018;8(1):12000. | ||
Cheng L, Wang Y, Huang L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node. Mol Ther. 2017;25(7):1665–1675. |
Supplementary materials
 | Figure S1 Immunoblot analyses of exosomes shed by PCCs, PSCs, and MØs demonstrate the purity of Exos. |
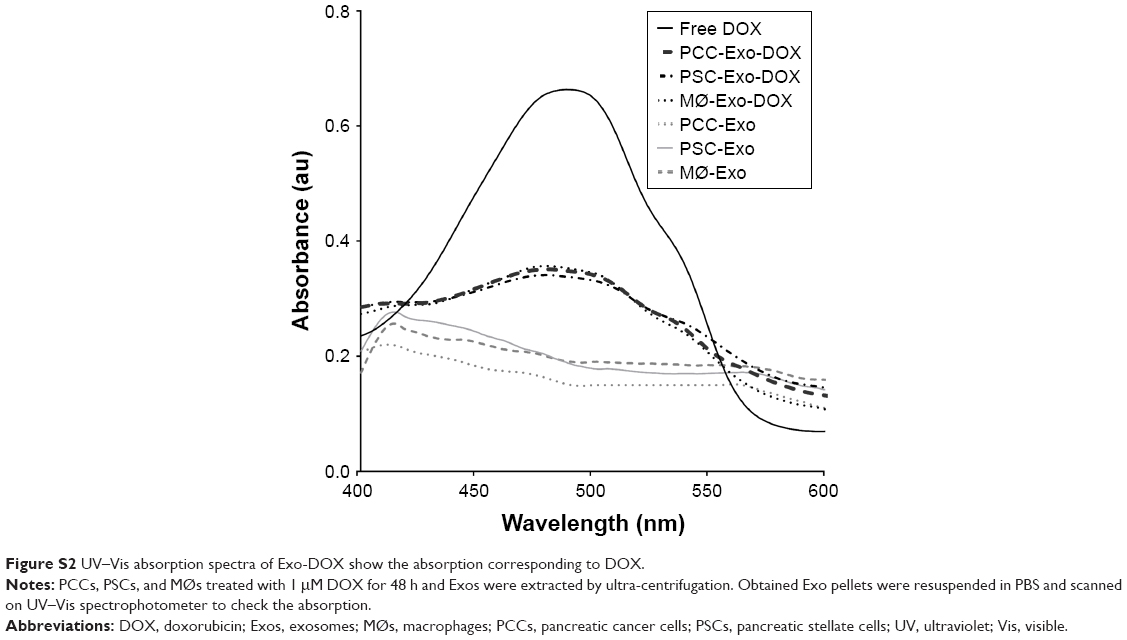 | Figure S2 UV–Vis absorption spectra of Exo-DOX show the absorption corresponding to DOX. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.