Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Drug-induced impairment of renal function
Authors Pazhayattil GS, Shirali A
Received 17 February 2014
Accepted for publication 23 April 2014
Published 12 December 2014 Volume 2014:7 Pages 457—468
DOI https://doi.org/10.2147/IJNRD.S39747
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
George Sunny Pazhayattil, Anushree C Shirali
Section of Nephrology, Yale University School of Medicine, New Haven, CT, USA
Abstract: Pharmaceutical agents provide diagnostic and therapeutic utility that are central to patient care. However, all agents also carry adverse drug effect profiles. While most of these are clinically insignificant, some drugs may cause unacceptable toxicity that impacts negatively on patient morbidity and mortality. Recognizing adverse effects is important for administering appropriate drug doses, instituting preventive strategies, and withdrawing the offending agent due to toxicity. In the present article, we will review those drugs that are associated with impaired renal function. By focusing on pharmaceutical agents that are currently in clinical practice, we will provide an overview of nephrotoxic drugs that a treating physician is most likely to encounter. In doing so, we will summarize risk factors for nephrotoxicity, describe clinical manifestations, and address preventive and treatment strategies.
Keywords: acute kidney injury, chronic kidney disease, drug nephrotoxicity, chemotherapy, NSAIDs
Introduction
In the era of modern medicine, patients are exposed to an expanding variety of drugs for diagnostic and therapeutic purposes. Unfortunately, some of these agents cause adverse drug effects linked with systemic toxicity, including impairment of renal function. Nephrotoxicity results in serious clinical syndromes, including acute kidney injury (AKI). Nephrotoxic agents have been implicated as etiologic factors in 17%–26% of in-hospital AKI.1,2 Given the hospitalization rates, morbidity, and mortality associated with AKI, knowledge of the typical agents associated with nephrotoxicity is essential in improving AKI rates and outcomes.3 Drug-induced renal impairment involves many classes of drugs and includes prescription agents as well as commonly encountered over-the-counter drugs. There are drug-specific and patient-specific risk factors that influence the development of drug-related nephropathy. In the present article, we will review these factors and discuss the various classes of agents associated with nephrotoxicity.
Risk factors for nephrotoxicity
Drug- and kidney-specific factors
Toxicity of therapeutic and diagnostic agents may be inherent to the pharmacological compound itself and the potential for toxicity may be heightened in the kidney microenvironment. For example, the aim of chemotherapy is to kill malignant cells via various mechanisms aimed at arresting cellular division. Since the cell cycle operates normally in nonmalignant cells, healthy tissues, including renal parenchymal cells, are also affected. However, in particular ways, the kidney is especially poised as a target for toxicity. Since it receives a significant percentage of cardiac output, robust blood flow through the kidney regularly exposes it to drugs and drug metabolites. Some of these agents may have the requisite charge and size for filtration at the glomerulus and subsequently gain entry into renal tubular epithelial cells via pinocytosis or endocytosis.4 Other drugs are transported via peritubular capillaries and gain access to renal tubular epithelial cells at the basolateral surface, where they are taken up by organic anion and organic cation transporters (OATs and OCTs, respectively) and eventually effluxed into tubular lumens.5 Genetic polymorphisms that affect the function of these transporters may explain differences in susceptibility to nephrotoxicity of drugs such as cisplatin.6 As tubular fluid flows down the loop of Henle, water is reabsorbed, thus increasing the tubular concentration of drug to potentially injurious levels. Tubular cells in the collecting duct and loop of Henle are at greater risk for nephrotoxicity because they are highly metabolically active and, as a result, reside in a relatively hypoxic microenvironment. In addition, the kidney oxidizes drugs via cytochrome p450 and other enzyme systems into smaller metabolites, suggesting that intrarenal drug toxicity may be mechanistically linked to reactive oxygen species as well as direct effects of drug metabolites.4,7 Lastly, certain therapeutic agents may gain toxic potential within the kidney microenvironment. For example, methotrexate (MTX) nephrotoxicity depends upon crystallization of the parent compound and its metabolites. This crystallization is highly favored with the acidic urine pH that exists in the normal host with average protein intake. Thus, both inherent properties of a particular drug and the kidney-specific environment contribute to clinically significant nephrotoxicity. While these are general mechanisms for kidney injury, specific drugs may exploit unique pathways to induce nephrotoxicity. We will examine these in greater depth in later drug-specific discussions.
Patient-specific risk factors
Certain patient characteristics predispose to drug-induced nephrotoxicity. Among these, older age and female sex are associated with reduced muscle mass and lower total body water. This can impact on drug dosing in two ways. First, reduced muscle mass is reflected in lower serum creatinine. Depending on the type of estimating equation used to approximate glomerular filtration rate (GFR), this may be falsely interpreted as high GFR, leading to inappropriately high drug dosing. Second, decreased total body water increases the concentration of drug in serum. Both factors work in concert to raise serum drug concentration to potentially toxic levels. In addition to these factors, hypoalbuminemia also carries the risk of inducing toxic drug levels by increasing the unbound drug fraction in the serum.
Risk of drug nephrotoxicity is increased in the patient with AKI or chronic kidney disease (CKD). Thus, the patient who is on diuretic therapy or has vomiting or diarrhea that results in true volume depletion is vulnerable to toxic drug effects on the kidney. Similarly, the patient with congestive heart failure or hepatic failure with ascites who has effective volume depletion may experience prerenal AKI and become more susceptible to the nephrotoxic effects of certain agents. Hepatic failure is a particular risk factor for drug-induced renal impairment because cirrhotic patients tend to have reduced muscle mass and hypoalbuminemia, which, as discussed earlier, increases the chance of inadvertent drug overdosing. Additionally, the presence of hyperbilirubinemia is the highest predictive factor for nephrotoxicity among patients with liver failure, putatively because of tubular damage from bile salts.8
Like patients with AKI, those with CKD are also vulnerable to drug nephrotoxicity via several shared mechanisms, including excessive drug dosing relative to kidney function and decreased renal reserve. Other populations at particular risk for drug-induced renal impairment include the elderly and neonates. For elderly patients, reduced muscle mass and total body water may be factors, as previously discussed in this article. Additional variables in older patients include comorbid conditions that predispose to AKI as well as an increased likelihood of polypharmacy with nephrotoxic drugs. In neonates, particularly those with premature delivery, drug nephrotoxicity bears a significant burden for AKI compared to adult patients, with some data suggesting that drug-induced renal impairment leads to 16% of AKI cases in newborns.9 Several factors may explain this, including increased susceptibility of the neonatal immature kidney to nephrotoxic insults as well as the use of multiple nephrotoxic agents in critically ill newborns.10,11
Some drugs induce renal injury in the context of a systemic syndrome. For example, a particular drug may possess inherent nephrotoxic potential, which is increased in the setting of sepsis and decreased effective circulating volume. Certain agents contribute to renal failure within a rare but life-threatening hypersensitivity syndrome known as drug reaction and eosinophilia with systemic symptoms (DRESS). While DRESS has features in common with Stevens–Johnson syndrome as well as other cutaneous drug eruption diseases, it is distinguished by eosinophilia, atypical lymphocytosis, and visceral involvement.12,13 An ever-expanding list of agents is associated with development of DRESS.13 While the liver is the most commonly implicated organ, kidney involvement has been reported in the form of acute tubulointerstitial nephritis with sulfasalazine and leflunomide.14,15 Though the specific mechanistic link to renal failure is not clear, a recent study examining peripheral T-cell responses in 40 patients with DRESS found that a majority of cases involved human herpesvirus 6, human herpesvirus 7, and Epstein–Barr virus reactivation.16
Overview of drug-induced renal impairment
In discussing drug-induced renal impairment, it is helpful to recall that renal function is a composite of several steps which includes renal arterial blood flow, glomerular filtration, tubular fluid formation, and exit of urine into the urogenital system. Drugs can impair renal function by interfering with any of these steps. Thus, one method of classification divides drug-induced nephrotoxicity into prerenal, intrarenal, or postrenal mechanisms. However, while many drugs have a single mechanism of injury, some classes of drugs possess multiple ways of inducing renal dysfunction. The following subsections will expand upon the different categories of drugs that have nephrotoxic potential, with examples of the most commonly encountered drugs in each category.
Nonsteroidal anti-inflammatory drugs
Classes of nephrotoxic drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used to relieve pain and signs of inflammation. Their effectiveness and relative safety make them among the most frequently sold over-the-counter medications worldwide. The worldwide incidence of renal side effects is reported to be around 1%–5%.17 However, widespread use makes NSAIDs among the most common causes of drug-induced renal injury. NSAIDs can result in a variety of renal complications (Table 1).
 | Table 1 Renal complications of nonsteroidal anti-inflammatory drug use |
Prostaglandin (PG) inhibition mediated by NSAIDs explains many of its renal complications. PGs play a significant role in maintaining normal renal physiology. PG-induced renal vasodilation is critical for maintaining adequate renal perfusion. NSAIDs impair this renal vasodilation and alter renal hemodynamics. This effect is magnified in patients who are hypovolemic or are concomitantly using angiotensin converting enzyme (ACE) inhibitors. It is important to note that AKI can occur with either nonselective NSAIDs or selective (COX-2-specific) NSAIDs.18
PGs have also been shown to play a role in stimulating renin and angiotensin-mediated aldosterone release.19 Thus, NSAID-mediated PG inhibition can result in hyperkalemia and metabolic acidosis (hyporeninemic hypoaldosteronism). Hyponatremia induced by NSAIDs is possibly related to release of inhibitory effect of PGs on antidiuretic hormone (ADH)-facilitated water absorption at the distal collecting tubules.20,21 NSAID-mediated PG inhibition is also responsible for sodium retention, which can lead to hypertension and edema.
Angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers
These groups of drugs are widely used in the treatment of hypertension and congestive heart failure and for delaying the progression of diabetic nephropathy. Angiotensin II constricts both the afferent and efferent arterioles, but the effect is more pronounced on the efferent arteriole.22 The net effect of angiotensin II is an increased intraglomerular pressure. This mechanism is critical in renal autoregulation, ie, maintaining a stable GFR across a wide range of renal perfusion pressures. ACE inhibitors and angiotensin II receptor blockers (ARBs) antagonize the activity of angiotensin II, thereby interfering with the renal autoregulation of GFR. Though this effect is well tolerated in most patients, in certain situations the loss of autoregulation could precipitate or potentiate AKI. These situations include: 1) bilateral renal artery stenosis or renal artery stenosis in solitary kidney; 2) volume depletion; and 3) concomitant use of NSAIDs, cyclosporine, and tacrolimus.23 ACE inhibitors or ARBs should be withheld if the patient has or is at risk for volume depletion. Furthermore, coadministration of NSAIDs along with ACE inhibitors or ARBs should be avoided, especially in the setting of preexisting CKD or volume depletion. Combination therapy involving both ACE inhibitors and ARBs is associated with more adverse events, including a higher risk for AKI and hyperkalemia.24
Antimicrobial agents
Aminoglycosides
Aminoglycosides (AGs) are well known to cause nephrotoxicity and ototoxicity. In spite of these risks, they are still widely used in the treatment of Gram-negative bacterial infections. Even at therapeutic doses, the incidence of nephrotoxicity is reported to be as high as 10%–25%.25,26 Risk for nephrotoxicity is higher with prolonged duration of therapy, hypovolemic states, preexisting CKD, and older age. Clinically, AKI manifests after 5–7 days of therapy. Renal complications are usually dose-dependent, but subtherapeutic doses of AGs have been implicated in causing chronic tubulointerstitial nephritis.27 AGs are non-protein bound and freely filtered at the glomeruli. Due to their cationic structure, AGs can undergo proximal tubule reabsorption by megalin-mediated endocytosis. This leads to a preferential accumulation of the drug in the cortical tubular cells, which results in tubular cytotoxicity. The risk for nephrotoxicity is directly proportional to the cationic charge. Neomycin is the most toxic drug in this group, followed by gentamicin, tobramycin, amikacin, and streptomycin (least toxic). Intracellular accumulation results either in tubular cell death or in functional alteration of cell membrane transporters. The latter explains the electrolyte abnormalities (hypokalemia, hypomagnesemia, and hypocalcemia) commonly associated with AG use. Rarely, AGs have been reported to cause Fanconi syndrome where impaired proximal tubule reabsorption results in renal wasting of sodium, potassium phosphate, amino acids, glucose, bicarbonate, and low-molecular-weight proteins.28 AGs also accumulate in the distal tubules and collecting ducts.29 This could result in distal tubular dysfunction leading to impaired concentration capability and polyuria. To prevent nephrotoxicity, loading and maintenance doses should be calculated based on estimated creatinine clearance.30 Monitoring peak and trough serum AG levels, expanding volume, and limiting dosing to once daily versus multiple times daily are other strategies by which to minimize the risk of AG nephrotoxicity.30,31
Sulfamethoxazole–trimethoprim and sulfa-based antibiotics
Sulfamethoxazole (SMX) is probably the most widely used sulfa-based antibiotic. It is generally prescribed along with synergistically acting trimethoprim (TMP) as a combination antimicrobial agent. Low cost and excellent spectrum of antimicrobial activity make SMX-TMP a popular choice among clinicians. The recent rise in incidence of methicillin-resistant Staphylococcus aureus-related skin infections has further increased use of this drug. SMX–TMP has several adverse renal effects. First, it is important to note that TMP inhibits proximal tubular secretion of creatinine and can result in elevation of measured serum creatinine.32,33 This effect is not accompanied by a decrease in actual GFR and hence should not be considered as AKI. TMP can also result in hyperkalemia by inhibiting the epithelial sodium channel at the distal convoluted tubule, which provides the driving force for potassium excretion.34,35 While overall incidence of SMX–TMP-associated renal disease was reported as low in a previous retrospective cohort, newer data suggest the overall incidence may be as high as 11.2%.36,37
AKI from sulfa-based antimicrobial agents is most commonly secondary to acute interstitial nephritis (AIN). Use of high-dose sulfadiazine can, on rare occasions, also cause crystal nephropathy.38,39 Urine microscopy in this case may reveal the characteristic birefringent, needle-shaped crystals arranged in a “shocks of wheat” pattern. Crystal precipitation typically occurs in acidic urine and in low-flow states. Thus, alkalinization of the urine and high fluid intake could potentially prevent development of crystalluria.38
Vancomycin
Vancomycin (VCM), a glycopeptide antibiotic, is commonly used in the critical care setting since it is a first-line agent in the treatment of severe methicillin-resistant Staphylococcus aureus infections. VCM-related nephrotoxicity is generally due to acute tubular necrosis (ATN) or AIN.40 High trough levels (>15 mg/L), long duration of therapy, and concomitant administration of other nephrotoxins (eg, AGs) have been described as significant risk factors for development of nephrotoxicity.41,42 The exact nephrotoxic mechanism of VCM-related ATN is not yet fully established. It has been suggested that VCM-induced oxidative stress results in the tubular damage.43 Given its widespread use, clinicians should be aware of this risk and should monitor patients for development of nephrotoxicity. Overall risk for nephrotoxicity is difficult to pinpoint as there is wide variability within the reported numbers from different studies. A recent meta-analysis found incidence of nephrotoxicity ranged between 5% and 43% across 15 different prospective and retrospective cohorts.41 In patients with CKD, the dosing should be strictly based on estimated creatinine clearance.
Ciprofloxacin
Ciprofloxacin, a commonly prescribed fluoroquinolone antibiotic, has been reported to cause AIN and crystalluria.44 Ciprofloxacin crystallizes in alkaline urine.45 Crystals can sometimes be visualized by polarized microscopy as birefringent needles, stars, or sheaves.44,46 Crystallization could be avoided by making sure the patients are volume replete and by dosing the drug appropriately for the level of renal function.
Other antibiotics
Penicillins and cephalosporins are commonly used antimicrobial agents and have been found, although rarely, to cause AIN and ATN.47,48 Polymyxins (colistin and polymyxin B) cause AKI by toxic tubular injury.49 While the polymyxins have been largely replaced by newer antimicrobial agents, clinicians should be aware that they are being reintroduced for the treatment of certain multidrug-resistant infections.
Antiviral agents
Acyclovir
High-dose intravenous use of acyclovir can induce AKI secondary to crystal precipitation in the renal tubules.50,51 Acyclovir crystals can sometimes be detected by urine sediment microscopy. Typically, they are birefringent, needle-shaped crystals. Nephrotoxicity can be prevented by adequate volume expansion, low-dose infusion, and slower rate of infusion. Treatment of acyclovir nephrotoxicity is limited to saline diuresis to prevent further crystallization by inducing high urine flow rates.
Foscarnet
Foscarnet is nephrotoxic by inciting ATN,52 although a case report also suggested the possibilty of a crystal-related injury.53 In addition to AKI, foscarnet can also cause significant electrolyte abnormalities. For example, foscarnet can result in symptomatic hypocalcemia by chelating free (ionized) calcium.54 Other electrolyte disturbances include hypomagnesemia, hypokalemia, and hypo- or hyperphosphatemia.55
Antiretroviral drugs
Since the introduction of highly active antiretroviral therapy, there has been a dramatic improvement in mortality and morbidity associated with HIV disease. Since antiretroviral therapy is generally lifelong, it is important for clinicians to be able to recognize the common nephrotoxic manifestations of these drugs.
The most prominent of the nephrotoxic antiretroviral agents is tenofovir, a nucleoside reverse transcriptase inhibitor that can cause AKI with or without proximal tubulopathy.56 AKI results from direct toxicity to tubular cells, mediated by mitochondrial injury, resulting in ATN.57 Proximal tubular dysfunction presents as electrolyte derangements, including an overt Fanconi syndrome.58 Discontinuation of tenofovir generally reverses these pathological changes; however, severe cases of AKI may result in residual CKD.
Protease inhibitors, another class of antiretroviral drugs, can also be nephrotoxic.59,60 Indinavir, a once-prominent protease inhibitor, can crystallize in renal tubules, resulting in crystal-related kidney injury and nephrolithiasis. Fortunately, the use of indinavir is on the decline as it is being replaced by newer and safer agents. Atazanavir is a newer protease inhibitor that is also associated with nephrolithiasis, but to a much lesser extent compared to indinavir. Cases of AIN have also been reported with use of protease inhibitors, including indinavir, abacavir, ritonavir, and atazanavir.60
Antifungal agents
Amphotericin B
Amphotericin B (AmB) is frequently used in the treatment of serious, life-threatening fungal infections. Therapeutic use is often limited by dose-dependent nephrotoxicity. AmB is available for use in two forms: a conventional form and the more recent liposomal form. Liposomal AmB has an improved renal safety profile compared to conventional AmB.61 Two pathogenic mechanisms have been suggested to explain AKI with AmB use.62 The antifungal effect of AmB is related to its ability to alter membrane permeability of fungal cells, which leads to cell death. AmB can bind to cholesterol molecules in cellular membranes, thereby altering membrane permeability. This effect can be toxic to renal tubular cells and result in ATN and tubular dysfunction. Tubular dysfunction manifests as renal tubular acidosis (RTA), urinary concentration defects, and electrolyte disturbances.63 The second mechanism for AKI is renal vasoconstriction. Volume depletion can worsen the nephrotoxic effects, so volume expansion is standard of care to prevent nephrotoxicity with AmB infusions.
Chemotherapeutic agents
Chemotherapeutic drugs play a central role in the treatment of various neoplasms. Unfortunately, they can result in serious multisystem complications. Nephrotoxicity is common with many chemotherapeutic agents and can result in a wide spectrum of renal complications. An overview of chemotherapy-related nephrotoxicity is listed in Table 2. While a detailed explanation of all mechanisms underlying chemotherapy nephrotoxicity is beyond the scope of this article, we will highlight some of the commonly used chemotherapeutic agents.
 | Table 2 Overview of nephrotoxicity from oncologic drugs |
Cisplatin
Due to its efficacious tumoricidal activity, cisplatin is a standard component in the treatment regimens for various solid organ tumors. Cisplatin use can result in a variety of clinical renal syndromes (Table 3). The main mechanism for causing AKI is thought to be a direct cellular toxic injury primarily to the proximal tubule. The selective injury to proximal tubules is due to preferential accumulation of the drug in proximal tubular cells. This cellular uptake happens via the copper transporter-1 and OCT2.64–66 Other platinum-based chemotherapeutic agents such as carboplatin and oxaliplatin are not as nephrotoxic as cisplatin because of reduced uptake via OCT2. In addition to measuring kidney function, serum electrolytes should be monitored during cisplatin therapy to detect metabolic abnormalities. Some strategies to prevent cisplatin nephrotoxicity include intravenous volume expansion, avoidance of other potential nephrotoxins, and limiting cisplatin use in patients with preexisting CKD.67 Various strategies, such as coadministration of hypertonic saline and sodium thiosulfate, have been tried to prevent cisplatin nephrotoxicity, but thus far none of them have demonstrated definite positive clinical results.67
 | Table 3 Renal complications from cisplatin use |
Ifosfamide
Ifosfamide, a structural isomer of cyclophosphamide, is an alkylating agent used in the treatment of solid organ tumors, including those affecting children. Like cisplatin, ifosfamide also undergoes cellular uptake at the proximal tubule via OCT2.68 Once inside the cell, the drug is then metabolized into chloroacetaldehyde, which is chiefly responsible for cellular toxicity.69 Proximal tubular dysfunction resulting in Fanconi-like syndrome is a common manifestation of ifosfamide nephrotoxicity. Tubular cell death could also result in AKI. It is interesting to note that cyclophosphamide, despite its structural similarity, is not transported via OCT2 and hence is not associated with similar nephrotoxic events.68 Prevention of ifosfamide nephrotoxicity is limited to volume expansion with saline.
MTX and pemetrexed
MTX is an anti-folate agent widely used as chemotherapy against several malignancies. MTX-induced AKI occurs with high-dose (1,000–33,000 mg/m2) intravenous use and results from crystallization in the renal tubules as well as direct tubular toxicity resulting in ATN.70 The crystallization is enhanced by high urinary MTX concentration, low urine volume, and acidic urine pH. Since 90% of MTX is cleared by kidneys, the urine concentration of this drug closely follows its serum concentration. Preventive strategies include maintaining high urine output with intravenous volume expansion and urinary alkalinization. Leucovorin rescue is frequently provided to prevent systemic toxicity of MTX by restoring the reduced folate pool. Once AKI is established, management is usually supportive. High-flux hemodialysis efficiently clears MTX but is limited by postdialysis rebound of MTX levels.71,72 Newer potential therapies include carboxypeptidase G2 ([CPDG2] glucarpidase), a recombinant bacterial enzyme that rapidly hydrolyzes MTX to inactive metabolites. CPDG2 has been shown to efficiently reduce toxic MTX levels.73 At this time, the use of CPDG2 is restricted by high cost and limited availability, but, despite these limitations, CPDG2 is promising therapy as it results in sustained reduction in MTX levels.
Pemetrexed, a structural analog of MTX, is another antifolate agent used in cancer treatment. Nephrotoxicity of pemetrexed is mostly due to direct tubular injury resulting in ATN.74,75 However, nephrogenic diabetes insipidus (NDI) and distal RTA has also been reported following pemetrexed use.75
Targeted cancer therapies
Recent advancements in tumor research have led to the development of targeted molecular therapies that allow effective cancer therapy with fewer systemic side effects compared to conventional chemotherapeutic agents. Anti-angiogenic drugs, which include monoclonal antibodies against vascular endothelial growth factor (VEGF) and inhibitors of VEGF receptor (VEGF-R), act by disrupting tumor vascular development. Unfortunately, these agents interfere with VEGF signaling in normal tissues as well, resulting in adverse renal effects. In podocytes, VEGF plays an important role in the maintenance of a fenestrated glomerular endothelium. Disruption of this pathway by VEGF inhibition may result in hypertension, proteinuria, and thrombotic microangiopathy.76
Cetuximab, a monoclonal antibody against epidermal growth factor receptor (EGFR), is another drug used for targeted tumor therapy. EGFRs are also present on basolateral membrane of distal convoluted of the distal convoluted tubule. Activation of these receptors stimulates magnesium reabsorption via TRPM6 channels (transient receptor potential cation channel, subfamily M, member 6). Thus, inhibition of EGFR in renal tissue by cetuximab can result in renal magnesium wasting and hypomagnesemia.77
Bisphosphonates
Certain malignancies commonly metastasize to bone and stimulate osteoclast-mediated bone resorption. Bisphosphonates can inhibit bone resorption, thereby preventing or delaying skeletal complications. They are also first-line agents in treating tumor-associated hypercalcemia. While oral bisphosphonates have no adverse renal effects, nephrotoxicity is an important complication with high-dose intravenous administration. Intravenous pamidronate use has resulted in collapsing focal segmental glomerulosclerosis.78 On the other hand, intravenous zoledronate is associated with ATN.78 The risk of nephrotoxicity correlates with a short infusion time and high dosage. Hence, the risk can be attenuated by decreasing the infusion rate, adjusting the dose for level of renal function, and avoiding the drug in patients with CKD.79 Ibandronate is another intravenous bisphosphonate, which has a better renal side effect profile and should be considered as an alternative in patients with renal dysfunction.
Radiocontrast agents
Intravenous iodinated radiocontrast agents are necessary for several diagnostic and interventional radiology procedures. Contrast-induced nephropathy (CIN), a well-known complication associated with iodinated radiocontrast media, is one of the most common causes of in-hospital AKI. CIN classically presents with a rise in serum creatinine within the first 24–48 hours of exposure. In most cases, the AKI is reversible and the serum creatinine returns to baseline in 7–10 days.80 There are multiple mechanisms involved in causing AKI. Soon after administration, the initial insult is triggered by an intense renal afferent vasoconstriction. This effect can result in renal medullary hypoxia and subsequent ATN. Another mechanism for causing AKI is by osmotic nephrosis (see “Osmotic agents”). Several factors contribute to the risk and severity of CIN (Table 4).81
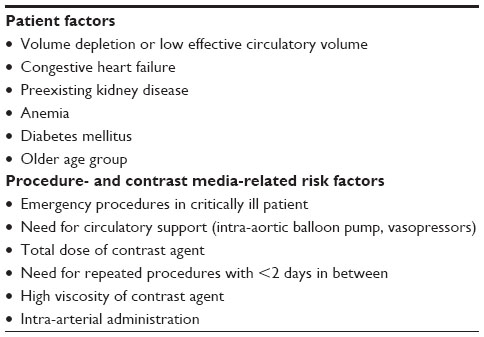 | Table 4 Important risk factors for contrast-induced nephropathy |
Risk of CIN can be attenuated with certain preventive strategies. Intravenous hydration before and after radiocontrast agent exposure is the cornerstone of preventing the incidence of CIN. There is some controversy surrounding the appropriate choice of intravenous fluid. Several randomized trials, as demonstrated in a recent systematic review, have compared the benefit of isotonic sodium bicarbonate infusion versus isotonic saline infusion. The results are variable and there is no definite evidence favoring sodium bicarbonate infusion.82 Another frequently used prophylactic agent is the antioxidant drug N-acetylcysteine (NAC). Results from a recent clinical trial concluded that NAC has no benefit in preventing CIN.83 Meanwhile, other data seems to indicate potential benefit.84,85 Hence, the available evidence is conflicting, and benefits of NAC are yet to be conclusively proven at this time, although future trials may provide definite answers.86 Despite the lack of evidence, NAC is still widely used, mainly because of its favorable drug safety profile, low cost, and ease of administration. Other preventive strategies that are obvious and should be followed whenever possible include avoiding concomitant use of nephrotoxic drugs, limiting contrast volume, avoiding repeated contrast exposure, and achieving hemodynamic stability before the test or procedure.
Immunosuppressive agents
Calcineurin inhibitors
Tacrolimus and cyclosporine are widely used calcineurin inhibitors (CNIs). They are currently considered to be the cornerstone of immunosuppressive therapy after solid organ transplantation. CNIs possess a very narrow therapeutic window and are susceptible to multiple drug–drug interactions, which often lead to toxic serum drug levels. CNI-related nephrotoxicity occurs in multiple ways.87 Acute toxicity is usually related to altered renal hemodynamics secondary to afferent arteriolar vasoconstriction. Rarely, AKI could also result from thrombotic microangiopathy.88 Long-term CNI exposure can cause interstitial fibrosis and tubular atrophy, leading to CKD.89,90
Miscellaneous
Lithium
Lithium has been the mainstay of treatment for patients with bipolar disorder for many years. Chronic lithium use can result in various forms of nephrotoxicity. The most common renal disorder associated with chronic lithium use is NDI. Clinically significant NDI can occur in up to 20% of patients on lithium. The mechanism for lithium-induced NDI is possibly by downregulation of acquaporin-2 (AQP2) channels in the collecting duct.91 Long-term lithium therapy can also lead to CKD secondary to chronic tubulointerstitial nephritis. Rarely, nephrotic syndrome secondary to minimal change disease or focal segmental glomerulosclerosis have been shown to occur after lithium therapy.92,93 Lithium use has also resulted in hypercalcemia secondary to hyperparathyroidism.94 The exact mechanism leading to hyperparathyroidism is not clear at this time.
Proton-pump inhibitors
Proton-pump inhibitors (PPIs) are currently the mainstay of treatment for gastroesophageal reflux disease. They are generally well tolerated but can, rarely, lead to AKI from AIN.95 Although many drugs can result in AIN, PPI-induced AIN requires special mention because it is one of the most common causes of AIN in some regions.96 Since PPIs are widely prescribed drugs, the incidence of PPI-induced AIN is on the rise. As with most cases of drug-induced AIN, withdrawal of the drug usually results in recovery.
Acetaminophen
Acetaminophen is probably the most common over-the-counter analgesic and antipyretic medication. Acetaminophen overdose is well described to cause acute fulminant liver failure. Rarely, acetaminophen toxicity can lead to AKI. The incidence of AKI in acetaminophen overdose is around 2%.97 The incidence rises with severity of the overdose. Rate of AKI is higher in patients with concomitant acute liver failure, which may be related to hemodynamic instability or development of hepatorenal syndrome. AKI can also occur without acute liver failure where the mechanism of nephrotoxicity is acute tubular injury leading to ATN.97 Treatment is usually supportive in nature. It is interesting to note that NAC, which is widely used in the treatment of acetaminophen toxicity, prevents liver failure but has no effect on incidence or progression of AKI.97,98
HMG-CoA reductase inhibitors
HMG-CoA reductase inhibitors (statins) are generally considered as the first-line agents in treating hypercholesterolemia. A common side effect of statin therapy is statin-induced myopathy, which is reported in up to 7% of patients on chronic statin therapy.99 Statin-induced myopathy could range from nonspecific myalgia without elevation of creatine kinase to life-threatening rhabdomyolysis.99 The occurrence of rhabdomyolysis is rare, and one study revealed an average incidence of 0.44 per 10,000 person-years.100 The incidence is higher in patients on combination therapy with statins and fibrates. AKI from statin use is usually in the setting of rhabdomyolysis; however, a direct tubular toxicity independent of rhabdomyolysis has also been reported.101
Osmotic agents
AKI from osmotic agents, frequently referred to as osmotic nephrosis or osmotic nephropathy, occurs when renal tubules are exposed to hyperosmolar substances. Kidney biopsy is diagnostic and reveals severe swelling of tubular cells and apparent obliteration of tubular lumens. Electron microscopy further reveals prominent vacuolization inside affected tubular cells.102 Common agents that can result in osmotic nephrosis are intravenous immune globulin, hydroxyethyl starch, mannitol, and radiocontrast media.
Conclusion
Kidneys are frequently exposed to drugs and/or toxic metabolites and are therefore a common site for drug toxicity. Drug-induced nephrotoxicity is one of the leading causes of AKI worldwide. In addition to causing AKI, chronic drug toxicity can, in some cases, lead to CKD and eventual end-stage renal disease. Drugs can also interfere with normal transport mechanisms in the kidney, leading to a variety of electrolyte and acid-base disorders. When signs of renal dysfunction are first noted, the patient’s medication list should be carefully reviewed for potential nephrotoxic agents. In cases of drug-induced AIN, prompt withdrawal of the offending agent generally results in renal recovery. Although evidence is mixed and based on retrospective data, early use of steroids may be beneficial in some cases of severe AIN.103,104 In most situations, nephrotoxicity leads to discontinuation of the culprit drug, thereby limiting treatment options. Thus, prevention of nephrotoxicity should be at the forefront of the approaches employed to counteract drug-induced kidney failure. We have highlighted certain drug-specific preventive strategies throughout this article. A summary of general guidelines for avoiding drug-related renal injury is described in Table 5.
 | Table 5 General strategies to prevent drug-induced nephrotoxicity |
One of the biggest challenges lies in our ability to recognize nephrotoxicity in its early stages. Monitoring serum creatinine and blood urea nitrogen (BUN) levels is an ineffective method for the following reasons: 1) a rise in serum creatinine and BUN is a universal finding in AKI and does not reveal the etiology, which can delay or confound diagnosis; and 2) by the time a rise in these traditional markers is noticed, significant renal damage has likely already occurred. Recently, a variety of novel biomarkers have been identified, which allow early detection of kidney injury.105 Some biomarkers can also differentiate the location of injury (eg, glomerular injury versus tubular injury), thus allowing more precise identification of the culprit drug.105 Though clinical data are lacking for drug-induced nephrotoxicity, in a murine model of nephrotoxic kidney injury, the transmembrane tubular protein kidney injury molecule-1 (Kim-1) provided greater specificity and sensitivity compared to BUN and creatinine in detecting nephrotoxic injury from cisplatin, gentamicin, and cyclosporine.106 Ongoing research in the area of renal biomarkers may provide us with more sensitive and specific tools in the future to help detection of drug-induced nephrotoxicity in its early stages, thus allowing timely intervention.
Disclosure
The authors report no conflicts of interest in this work.
References
Mehta RL, Pascual MT, Soroko S, et al; Program to Improve Care in Acute Renal Disease. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int. 2004;66(4):1613–1621. | |
Uchino S, Kellum JA, Bellomo R, et al; Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294(7):813–818. | |
Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol. 2008;3(3): 844–861. | |
Perazella MA. Renal vulnerability to drug toxicity. Clin J Am Soc Nephrol. 2009;4(7):1275–1283. | |
Lee W, Kim RB. Transporters and renal drug elimination. Annu Rev Pharmacol Toxicol. 2004;44:137–166. | |
Khrunin AV, Moisseev A, Gorbunova V, Limborska S. Genetic polymorphisms and the efficacy and toxicity of cisplatin-based chemotherapy in ovarian cancer patients. Pharmacogenomics J. 2010;10(1): 54–61. | |
Anders MW. Metabolism of drugs by the kidney. Kidney Int. 1980;18(5): 636–647. | |
Lucena MI, Andrade RJ, Cabello MR, Hidalgo R, Gonzalez-Correa JA, Sanchez de la Cuesta F. Aminoglycoside-associated nephrotoxicity in extrahepatic obstructive jaundice. J Hepatol. 1995;22(2):189–196. | |
Patzer L. Nephrotoxicity as a cause of acute kidney injury in children. Pediatr Nephrol. 2008;23(12):2159–2173. | |
Carmody JB, Charlton JR. Short-term gestation, long-term risk: prematurity and chronic kidney disease. Pediatrics. 2013;131(6): 1168–1179. | |
Zappitelli M, Selewski DT, Askenazi DJ. Nephrotoxic medication exposure and acute kidney injury in neonates. Neoreviews. 2012;13(7): e420–e427. | |
Walsh SA, Creamer D. Drug reaction with eosinophilia and systemic symptoms (DRESS): a clinical update and review of current thinking. Clin Exp Dermatol. 2011;36(1):6–11. | |
Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124(7):588–597. | |
Augusto JF, Sayegh J, Simon A, et al. A case of sulphasalazine-induced DRESS syndrome with delayed acute interstitial nephritis. Nephrol Dial Transplant. 2009;24(9):2940–2942. | |
Pinto B, Dhir V, Krishnan S, Nada R. Leflunomide-induced DRESS syndrome with renal involvement and vasculitis. Clin Rheumatol. 2013;32(5):689–693. | |
Picard D, Janela B, Descamps V, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Sci Transl Med. 2010;2(46):46ra62. | |
Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med. 1999;106(5B):13S–24S. | |
Schneider V, Lévesque LE, Zhang B, Hutchinson T, Brophy JM. Association of selective and conventional nonsteroidal antiinflammatory drugs with acute renal failure: a population-based, nested case-control analysis. Am J Epidemiol. 2006;164(9):881–889. | |
Campbell WB, Gomez-Sanchez CE, Adams BV, Schmitz JM, Itskovitz HD. Attenuation of angiotensin II- and III-induced aldosterone release by prostaglandin synthesis inhibitors. J Clin Invest. 1979;64(6):1552–1557. | |
Kramer HJ, Glänzer K, Düsing R. Role of prostaglandins in the regulation of renal water excretion. Kidney Int. 1981;19(6):851–859. | |
Walker RM, Brown RS, Stoff JS. Role of renal prostaglandins during antidiuresis and water diuresis in man. Kidney Int. 1982;21(2): 365–370. | |
Denton KM, Fennessy PA, Alcorn D, Anderson WP. Morphometric analysis of the actions of angiotensin II on renal arterioles and glomeruli. Am J Physiol. 1992;262(3 Pt 2):F367–F372. | |
Palmer BF. Renal dysfunction complicating the treatment of hypertension. N Engl J Med. 2002;347(16):1256–1261. | |
Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med. 2013;369(20):1892–1903. | |
Lopez-Novoa JM, Quiros Y, Vicente L, Morales AI, Lopez-Hernandez FJ. New insights into the mechanism of aminoglycoside nephrotoxicity: an integrative point of view. Kidney Int. 2011;79(1):33–45. | |
Martínez-Salgado C, López-Hernández FJ, López-Novoa JM. Glomerular nephrotoxicity of aminoglycosides. Toxicol Appl Pharmacol. 2007;223(1):86–98. | |
Houghton DC, English J, Bennett WM. Chronic tubulointerstitial nephritis and renal insufficiency associated with long-term “subtherapeutic” gentamicin. J Lab Clin Med. 1988;112(6):694–703. | |
Casteels-Van Daele M, Corbeel L, Van de Casseye W, Standaert L. Gentamicin-induced Fanconi syndrome. J Pediatr. 1980;97(3):507–508. | |
Fujiwara K, Shin M, Matsunaga H, Saita T, Larsson LI. Light-microscopic immunocytochemistry for gentamicin and its use for studying uptake of the drug in kidney. Antimicrob Agents Chemother. 2009;53(8):3302–3307. | |
Levin ML. Aminoglycoside nephrotoxicity: keys to prevention. J Crit Illn. 1994;9(10):911–912, 915. | |
Hatala R, Dinh T, Cook DJ. Once-daily aminoglycoside dosing in immunocompetent adults: a meta-analysis. Ann Intern Med. 1996; 124(8):717–725. | |
Kastrup J, Petersen P, Bartram R, Hansen JM. The effect of trimethoprim on serum creatinine. Br J Urol. 1985;57(3):265–268. | |
Roy MT, First MR, Myre SA, Cacini W. Effect of co-trimoxazole and sulfamethoxazole on serum creatinine in normal subjects. Ther Drug Monit. 1982;4(1):77–79. | |
Alappan R, Perazella MA, Buller GK. Hyperkalemia in hospitalized patients treated with trimethoprim-sulfamethoxazole. Ann Intern Med. 1996;124(3):316–320. | |
Perazella MA, Mahnensmith RL. Trimethoprim-sulfamethoxazole: hyperkalemia is an important complication regardless of dose. Clin Nephrol. 1996;46(3):187–192. | |
Jick H, Derby LE. A large population-based follow-up study of trimethoprim-sulfamethoxazole, trimethoprim, and cephalexin for uncommon serious drug toxicity. Pharmacotherapy. 1995;15(4):428–432. | |
Fraser TN, Avellaneda AA, Graviss EA, Musher DM. Acute kidney injury associated with trimethoprim/sulfamethoxazole. J Antimicrob Chemother. 2012;67(5):1271–1277. | |
Hein R, Brunkhorst R, Thon WF, Schedel I, Schmidt RE. Symptomatic sulfadiazine crystalluria in AIDS patients: a report of two cases. Clinical Nephrol. 1993;39(5):254–256. | |
Simon DI, Brosius FC 3rd, Rothstein DM. Sulfadiazine crystalluria revisited. The treatment of Toxoplasma encephalitis in patients with acquired immunodeficiency syndrome. Arch Intern Med. 1990;150(11): 2379–2384. | |
Htike NL, Santoro J, Gilbert B, Elfenbein IB, Teehan G. Biopsy-proven vancomycin-associated interstitial nephritis and acute tubular necrosis. Clin Exp Nephrol. 2012;16(2):320–324. | |
van Hal SJ, Paterson DL, Lodise TP. Systematic review and meta-analysis of vancomycin-induced nephrotoxicity associated with dosing schedules that maintain troughs between and 20 milligrams per liter. Antimicrob Agents Chemother. 2013;57(2):734–744. | |
Chow AW, Azar RM. Glycopeptides and nephrotoxicity. Intensive Care Med. 1994;20 Suppl 4:S23–S29. | |
Oktem F, Arslan MK, Ozguner F, et al. In vivo evidences suggesting the role of oxidative stress in pathogenesis of vancomycin-induced nephrotoxicity: protection by erdosteine. Toxicology. 2005;215(3): 227–233. | |
Bird ST, Etminan M, Brophy JM, Hartzema AG, Delaney JAC. Risk of acute kidney injury associated with the use of fluoroquinolones. CMAJ. 2013;185(10):E475–E482. | |
Thorsteinsson SB, Bergan T, Oddsdottir S, Rohwedder R, Holm R. Crystalluria and ciprofloxacin, influence of urinary pH and hydration. Chemotherapy. 1986;32(5):408–417. | |
Stratta P, Lazzarich E, Canavese C, Bozzola C, Monga G. Ciprofloxacin crystal nephropathy. Am J Kidney Dis. 2007;50(2):330–335. | |
Barza M. The nephrotoxicity of cephalosporins: an overview. J Infect Dis. 1978;137 Suppl:S60–S73. | |
Pill MW, O’Neill CV, Chapman MM, Singh AK. Suspected acute interstitial nephritis induced by piperacillin-tazobactam. Pharmacotherapy. 1997;17(1):166–169. | |
Justo JA, Bosso JA. Adverse reactions associated with systemic polymyxin therapy. Pharmacotherapy. Epub 2014 September 30. | |
Becker BN, Schulman G. Nephrotoxicity of antiviral therapies. Curr Opin Nephrol Hypertens. 1996;5(4):375–379. | |
Sawyer MH, Webb DE, Balow JE, Straus SE. Acyclovir-induced renal failure. Clinical course and histology. Am J Med. 1988;84(6): 1067–1071. | |
Deray G, Martinez F, Katlama C, et al. Foscarnet nephrotoxicity: mechanism, incidence and prevention. Am J Nephrol. 1989;9(4):316–321. | |
Maurice-Estepa L, Daudon M, Katlama C, et al. Identification of crystals in kidneys of AIDS patients treated with foscarnet. Am J Kidney Dis. 1998;32(3):392–400. | |
Vela Moreno J, Pascual Catalán A, Ramos Paesa C, Arazo Garcés P, Aguirre Errasti JM. [Tetany caused by foscarnet in a female patient with acquired immunodeficiency syndrome]. An Med Interna. 1995;12(7): 358–359. Spanish. | |
Gearhart MO, Sorg TB. Foscarnet-induced severe hypomagnesemia and other electrolyte disorders. Ann Pharmacother. 1993;27(3): 285–289. | |
Zaidan M, Lescure FX, Brochériou I, et al. Tubulointerstitial nephropathies in HIV-infected patients over the past 15 years: a clinico-pathological study. Clin J Am Soc Nephrol. 2013;8(6):930–938. | |
Herlitz LC, Mohan S, Stokes MB, Radhakrishnan J, D’Agati VD, Markowitz GS. Tenofovir nephrotoxicity: acute tubular necrosis with distinctive clinical, pathological, and mitochondrial abnormalities. Kidney Int. 2010;78(11):1171–1177. | |
Tourret J, Deray G, Isnard-Bagnis C. Tenofovir effect on the kidneys of HIV-infected patients: a double-edged sword? J Am Soc Nephrol. 2013;24(10):1519–1527. | |
Cooper RD, Tonelli M. Renal disease associated with antiretroviral therapy in the treatment of HIV. Nephron Clin Pract. 2011;118(3): c262–c268. | |
Kalyesubula R, Perazella MA. Nephrotoxicity of HAART. AIDS Res Treat. 2011;2011:562790. | |
Mistro S, Maciel Ide M, de Menezes RG, Maia ZP, Schooley RT, Badaró R. Does lipid emulsion reduce amphotericin B nephrotoxicity? A systematic review and meta-analysis. Clin Infect Dis. 2012;54(12): 1774–1777. | |
Heyman SN, Stillman IE, Brezis M, Epstein FH, Spokes K, Rosen S. Chronic amphotericin nephropathy: morphometric, electron microscopic, and functional studies. J Am Soc Nephrol. 1993;4(1):69–80. | |
Sawaya BP, Briggs JP, Schnermann J. Amphotericin B nephrotoxicity: the adverse consequences of altered membrane properties. J Am Soc Nephrol. 1995;6(2):154–164. | |
Ciarimboli G, Ludwig T, Lang D, et al. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am J Pathol. 2005;167(6):1477–1484. | |
Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel). 2010;2(11):2490–2518. | |
Pabla N, Murphy RF, Liu K, Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2009;296(3):F505–F511. | |
Launay-Vacher V, Rey JB, Isnard-Bagnis C, Deray G, Daouphars M; European Society of Clinical Pharmacy Special Interest Group on Cancer Care. Prevention of cisplatin nephrotoxicity: state of the art and recommendations from the European Society of Clinical Pharmacy Special Interest Group on Cancer Care. Cancer Chemother Pharmacol. 2008;61(6):903–909. | |
Ciarimboli G, Holle SK, Vollenbröcker B, et al. New clues for nephrotoxicity induced by ifosfamide: preferential renal uptake via the human organic cation transporter 2. Mol Pharm. 2011;8(1):270–279. | |
Zamlauski-Tucker MJ, Morris ME, Springate JE. Ifosfamide metabolite chloroacetaldehyde causes Fanconi syndrome in the perfused rat kidney. Toxicol Appl Pharmacol. 1994;129(1):170–175. | |
Widemann BC, Adamson PC. Understanding and managing methotrexate nephrotoxicity. Ooncologist. 2006;11(6):694–703. | |
Thierry FX, Vernier I, Dueymes JM, et al. Acute renal failure after high-dose methotrexate therapy. Role of hemodialysis and plasma exchange in methotrexate removal. Nephron. 1989;51(3):416–417. | |
Saland J, Leavey P, Bash R, Hansch E, Arbus G, Quigley R. Effective removal of methotrexate by high-flux hemodialysis. Pediatr Nephrol. 2002;17(10):825–829. | |
Buchen S, Ngampolo D, Melton RG, et al. Carboxypeptidase G2 rescue in patients with methotrexate intoxication and renal failure. Br J Cancer. 2005;92(3):480–487. | |
Glezerman IG, Pietanza MC, Miller V, Seshan SV. Kidney tubular toxicity of maintenance pemetrexed therapy. Am J Kidney Dis. 2011;58(5):817–820. | |
Vootukuru V, Liew Y, Nally JV Jr. Pemetrexed-induced acute renal failure, nephrogenic diabetes insipidus, and renal tubular acidosis in a patient with non-small cell lung cancer. Med Oncol. 2006;23(3): 419–422. | |
Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11): 1129–1136. | |
Muallem S, Moe OW. When EGF is offside, magnesium is wasted. J Clin Invest. 2007;117(8):2086–2089. | |
Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int. 2008;74(11):1385–1393. | |
Kyle RA, Yee GC, Somerfield MR, et al; American Society of Clinical Oncology. American Society of Clinical Oncology 2007 clinical practice guideline update on the role of bisphosphonates in multiple myeloma. J Clin Oncol. 2007;25(17):2464–2472. | |
Murphy SW, Barrett BJ, Parfrey PS. Contrast nephropathy. J Am Soc Nephrol. 2000;11(1):177–182. | |
Mehran R, Nikolsky E. Contrast-induced nephropathy: definition, epidemiology, and patients at risk. Kidney Int Suppl. 2006(100):S11–S15. | |
Brar SS, Hiremath S, Dangas G, Mehran R, Brar SK, Leon MB. Sodium bicarbonate for the prevention of contrast induced-acute kidney injury: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2009;4(10):1584–1592. | |
ACT Investigators. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: main results from the randomized Acetylcysteine for Contrast-induced Nephropathy Trial (ACT). Circulation. 2011;124(11):1250–1259. | |
Tepel M, van der Giet M, Schwarzfeld C, Laufer U, Liermann D, Zidek W. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med. 2000;343:180–184. | |
Miner SE, Dzavik V, Nguyen-Ho P, et al. N-acetylcysteine reduces contrast-associated nephropathy but not clinical events during long-term follow-up. Am Heart J. 2004;148:690–695. | |
Weisbord SD, Gallagher M, Kaufman J, et al. Prevention of contrast-induced AKI: a review of published trials and the design of the prevention of serious adverse events following angiography (PRESERVE) Trial. Clin J Am Soc Nephrol. 2013;8(9):1618–1631. | |
Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol. 2009;4(2):481–508. | |
Zarifian A, Meleg-Smith S, O’Donovan R, Tesi RJ, Batuman V. Cyclosporine-associated thrombotic microangiopathy in renal allografts. Kidney Int. 1999;55(6):2457–2466. | |
Nankivell BJ, Borrows RJ, Fung CL, O’Connell PJ, Allen RDM, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med. 2003;349(24):2326–2333. | |
Myers BD, Sibley R, Newton L, et al. The long-term course of cyclosporine-associated chronic nephropathy. Kidney Int. 1988;33(2): 590–600. | |
Marples D, Christensen S, Christensen EI, Ottosen PD, Nielsen S. Lithium-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla. J Clin Invest. 1995;95(4):1838–1845. | |
Markowitz GS, Radhakrishnan J, Kambham N, Valeri AM, Hines WH, D’Agati VD. Lithium nephrotoxicity: a progressive combined glomerular and tubulointerstitial nephropathy. J Am Soc Nephrol. 2000;11(8):1439–1448. | |
Tam VK, Green J, Schwieger J, Cohen AH. Nephrotic syndrome and renal insufficiency associated with lithium therapy. Am J Kidney Dis. 1996;27(5):715–720. | |
Bendz H, Sjödin I, Toss G, Berglund K. Hyperparathyroidism and long-term lithium therapy – a cross-sectional study and the effect of lithium withdrawal. J Intern Med. 1996;240(6):357–365. | |
Brewster UC, Perazella MA. Proton pump inhibitors and the kidney: critical review. Clin Nephrol. 2007;68(2):65–72. | |
Simpson IJ, Marshall MR, Pilmore H, et al. Proton pump inhibitors and acute interstitial nephritis: report and analysis of 15 cases. Nephrology (Carlton). 2006;11(5):381–385. | |
Blakely P, McDonald BR. Acute renal failure due to acetaminophen ingestion: a case report and review of the literature. J Am Soc Nephrol. 1995;6(1):48–53. | |
Jones AF, Vale JA. Paracetamol poisoning and the kidney. J Clin Pharm Ther. 1993;18(1):5–8. | |
Chapman MJ, Carrie A. Mechanisms of statin-induced myopathy: a role for the ubiquitin-proteasome pathway? Arterioscler Thromb Vasc Biol. 2005;25(12):2441–2444. | |
Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292(21):2585–2590. | |
van Zyl-Smit R, Firth JC, Duffield M, Marais AD. Renal tubular toxicity of HMG-CoA reductase inhibitors. Nephrol Dial Transplant. 2004;19(12):3176–3179. | |
Dickenmann M, Oettl T, Mihatsch MJ. Osmotic nephrosis: acute kidney injury with accumulation of proximal tubular lysosomes due to administration of exogenous solutes. Am J Kidney Dis. 2008;51(3):491–503. | |
Clarkson MR, Giblin L, O’Connell FP, et al. Acute interstitial nephritis: clinical features and response to corticosteroid therapy. Nephrol Dial Transplant. 2004;19(11):2778–2783. | |
González E, Gutiérrez E, Galeano C, et al; Grupo Madrileño De Nefritis Intersticiales. Early steroid treatment improves the recovery of renal function in patients with drug-induced acute interstitial nephritis. Kidney Int. 2008;73(8):940–946. | |
van Meer L, Moerland M, Cohen AF, Burggraaf J. Urinary kidney biomarkers for early detection of nephrotoxicity in clinical drug development. Br J Clin Pharmacol. 2014;77(6):947–957. | |
Vaidya VS, Ozer JS, Dieterle F, et al. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol. 2010;28(5):478–485. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.