Back to Journals » International Journal of Nanomedicine » Volume 12
Doxorubicin-loaded poly (lactic-co-glycolic acid) nanoparticles coated with chitosan/alginate by layer by layer technology for antitumor applications
Authors Chai F, Sun L, He X, Li J, Liu Y, Xiong F, Ge L, Webster TJ ![]() , Zheng C
, Zheng C
Received 16 December 2016
Accepted for publication 23 January 2017
Published 3 March 2017 Volume 2017:12 Pages 1791—1802
DOI https://doi.org/10.2147/IJN.S130404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Israel Rubinstein
Fujuan Chai, 1,* Linlin Sun, 2,3,* Xinyi He, 1 Jieli Li, 1 Yuanfen Liu, 4 Fei Xiong, 5 Liang Ge, 1 Thomas J Webster, 2,3 Chunli Zheng 1
1Department of Pharmaceutics, China Pharmaceutical University, Nanjing, 2Wenzhou Institute of Biomaterials and Engineering, Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 3Department of Chemical Engineering, Northeastern University, Boston, MA, USA; 4Department of Pharmacy, Jiangsu Jiankang Vocational College, 5State Key Laboratory of Bioelectronics, Jiangsu Laboratory for Biomaterials and Devices, School of Biological Science and Medical Engineering, Southeast University, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Abstract: Natural polyelectrolyte multilayers of chitosan (CHI) and alginate (ALG) were alternately deposited on doxorubicin (DOX)-loaded poly (lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) with layer by layer self-assembly to control drug release for antitumor activity. Numerous factors which influenced the multilayer growth on nano-colloidal particles were studied: polyelectrolyte concentration, NaCl concentration and temperature. Then the growth regime of the CHI/ALG multilayers was elucidated. The coated NPs were characterized by transmission electron microscopy, atomic force microscopy, X-ray diffraction and a zeta potential analyzer. In vitro studies demonstrated an undesirable initial burst release of DOX-loaded PLGA NPs (DOX-PLGA NPs), which was relieved from 55.12% to 5.78% through the use of the layer by layer technique. The release of DOX increased more than 40% as the pH of media decreased from 7.4 to 5.0. More importantly, DOX-PLGA (CHI/ALG) 3 NPs had superior in vivo tumor inhibition rates at 83.17% and decreased toxicity, compared with DOX-PLGA NPs and DOX in solution. Thus, the presently formulated PLGA-polyelectrolyte NPs have strong potential applications for numerous controlled anticancer drug release applications.
Keywords: layer by layer, chitosan/alginate, poly(lactic-co-glycolic acid) nanoparticles, multilayer growth influencing factors, burst release, pH-dependent
Corrigendum for this paper has been published
Introduction
Cancer remains a major cause of death in the world, killing approximately 570,000 people in the USA in 2011 alone.1 Over the past several decades, significant advancements have been made to understand cancer biology. However, most current anticancer agents lead to significant systemic toxicity and side effects (such as bone marrow suppression, cardiomyopathy, nephrotoxicity, neurotoxicity and multidrug resistance), which limits their clinical use.2
Recently, nanoscale drug delivery systems have been gaining in popularity. Entrapping drugs in nanocarriers can bypass the above impediments by modifying drug biodistribution and controlling drug release.3–5 Santhi et al demonstrated that methotrexate-loaded bovine serum albumin nanoparticles (NPs) caused a significant increase in drug distribution to the spleen, liver and lungs, in comparison to the drug power.6 Feng et al developed paclitaxel-loaded poly (lactic-co-glycolic acid) (PLGA) nanospheres to achieve prolonged drug release profiles and better therapeutic effects.7 Moreover, NPs not only effectively protect drugs from ionic or enzymatic degradation, but also increase their efficacy and stability and decrease their toxicity.4,8
PLGA copolymers are degraded in vivo by hydrolysis of the ester linkage to glycolic acid and lactic acid, which are then easily metabolized by the Krebs cycle and excreted as water and carbon dioxide.9,10 Owing to good biocompatibility properties, biodegradability, nontoxicity, nonimmunogenicity and sustained release properties, PLGA NPs have been widely used as drug delivery carriers for anticancer drugs.11–15 PLGA is approved by the Food and Drug Administration and the European Medicines Agency for various drug delivery systems for human applications.11 Several drugs (including dexamethasone, 5-fluorouracil and paclitaxel) have been successfully incorporated into PLGA.16 In addition, PLGA-based nanomedicine products have been investigated in clinical trials.17,18 However, the obvious initial burst release phenomenon of PLGA NPs is unfavorable for anticancer drugs with significant systemic toxicity, thus indicating that drug release needs to be better controlled.19,20
Recently, some strategies to control initial drug burst release have received increasing attention. Adibkia et al demonstrated that naproxen–eudragit® RS100 NPs showed a slow drug release rate with reduced burst release compared to the free drug.21 Ud Din et al developed hydrogel-loaded solid lipid NPs for rectal administration of flurbiprofen to reduce the initial burst effect.22 Although these NPs could decrease the initial burst release to some degree, there were still some disadvantages, including the poor biodegradability of these polymers, the use of surfactants and so on. In addition, the classical methods for controlling the release of drugs from PLGA particles are inconvenient, such as using various additives23,24 and preparing particles mixed with other polymers,25 limiting their wide application. Therefore, a simple and effective method is required for achieving lower initial drug burst release and well-controlled drug release from PLGA NPs.
Layer by layer (LbL) assembled NPs are promising strategies for incorporating polyelectrolytes onto the surfaces of nanocarriers to provide enhanced stability, cellular uptake, regulation of drug release and targeting capabilities,26–29 and have recently shown great value in gene and drug delivery.28,30–32 With multilayer alternative deposition of oppositely charged polyelectrolytes, drug-loaded carriers and drug microcrystals could decrease initial drug burst release and sustain drug release.33–35 The release of several drugs, including dexamethasone, artemesinin and ampicillin, has been prolonged by the use of LbL NPs.36–38 Although LbL self-assembly has been widely used recently to control initial burst release, research on the use of LbL for controlling drug release over a long period of time from NPs is limited. The elucidation of factors that influence multilayer growth on nanoscale particles is also largely absent, which plays an important role in multilayer growth.
In this study, multilayers of the bio-polyelectrolytes chitosan (CHI) and alginate (ALG) were successfully deposited by LbL on the surface of doxorubicin-loaded PLGA NPs (DOX-PLGA NPs; Figure 1). Factors influencing the CHI/ALG coating were investigated by monitoring the coating weight of the LbL multilayers, and the growth regime of the CHI/ALG multilayers on nano-colloidal particles was demonstrated as well. A systematic characterization of coated and bare NPs, including transmission electron microscopy (TEM), atomic force microscopy, zeta potential and X-ray diffraction (XRD), was performed here, and the effects of the different layers and medium pH on the controlled release of doxorubicin (DOX) were also studied. Finally, the in vivo antitumor efficiency and toxicity of DOX-loaded NPs were evaluated.
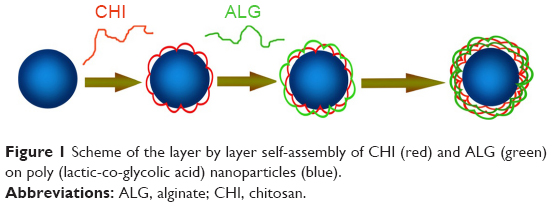 | Figure 1 Scheme of the layer by layer self-assembly of CHI (red) and ALG (green) on poly (lactic-co-glycolic acid) nanoparticles (blue). |
Materials and methods
Materials
DOX (99.41% purity) was purchased from HuaFeng United Technology Co. Ltd (Beijing, People’s Republic of China). PLGA 50:50 (molecular weight 15–20 kDa) was obtained from the Institute of Medical Instruments (Shandong, People’s Republic of China). ALG (200 MPa·s), CHI (molecular weight 49 kDa) and fluorescein isothiocyanate (FITC) were purchased from Sigma Aldrich. FITC-labeled ALG (FITC-ALG) was synthesized as reported in the literature.39 All other materials were of analytical grade.
Preparation of DOX-PLGA NPs
DOX-PLGA NPs were prepared by the double water-in-oil-in-water (W1/O/W2) emulsion-solvent evaporation technique.40 Briefly, 0.25 mL of a 2 mg/mL DOX solution was emulsified in a mixture of dichloromethane (1 mL) and acetone (1 mL) containing 50 mg of PLGA by sonication for 15 min (100 W) in an ice bath. The resulting primary emulsion was added to 8 mL of 2% (w/v) Poloxamer188 solution and was sonicated at 300 W for 10 min in an ice bath to form the double emulsion. The resulting double emulsion was diluted with a 0.3% Poloxamer188 aqueous solution and was stirred at 150 rpm for 4 h at room temperature to evaporate the organic solvents. The NPs were collected by ultracentrifugation at 12,000 rpm for 20 min, and washed with distilled water repeatedly to remove the residual Poloxamer188 and excess DOX. The PLGA NPs were resuspended in solution with a DOX concentration of 0.183 mg/mL for further study.
LbL self-assembly on DOX-PLGA NPs
Polyelectrolyte multilayers of CHI and ALG were deposited on the DOX-PLGA NPs through LbL self-assembly in water. All factors influencing the LbL process were investigated and optimized as summarized in Table 1 (when the main factor was studied in the experiment and other factors were selected at level 3).
 | Table 1 Factors influencing the multilayer growth |
The first layer was deposited by adding 4 mL of a CHI solution (pH 3.5, 2 mg/mL) to 2 mL of DOX-PLGA NPs. The mixture was incubated under gentle shaking to allow for the adsorption of CHI onto the negatively charged NPs. Excess CHI was removed by three cycles of centrifugation (12,000 rpm, 10 min), washing and redispersing with water. The following FITC-ALG layer was deposited with an FITC-ALG solution (pH 6–8) and the processes of centrifugation/washing were performed as noted above. A bilayer coating was then finished. The two polyelectrolytes were deposited alternatively until the desired number of LbL coating layers was reached. Finally, the PLGA-polyelectrolyte NPs were redispersed in water.
To determine the adsorption time and the amount of adsorbed FITC-ALG on the PLGA NPs, the fluorescence intensity of the supernatant after adsorption was measured with an RF-5301 PC spectrofluorophotometer (Shimadzu Co. Ltd., Tokyo, Japan) at each end of the CHI/FITC-ALG adsorption cycle. The excitation wavelength was 495 nm and the emission wavelength was 515 nm.
Characterization
Particle size and ζ-potential
Particle sizes and ζ-potential of DOX-PLGA NPs and CHI/ALG-coated DOX-PLGA NPs after every layer of deposition were measured by laser light scattering (ZetaPlus; Brookhaven Instrument Corp., Holtsville, NY, USA) at a scattering angle of 90°C at 25°C. Each sample was measured in triplicate.
Transmission electron microscopy
TEM measurements for the coated and uncoated NPs were performed with a JEM-1010 electron microscope operating at an accelerating voltage of 80 kV. Samples were prepared by depositing the NP suspension on a copper grid. The suspension was then air-dried before all measurements.
Powder XRD measurement
Powder X-ray diffractograms of DOX, blank PLGA NPs, DOX-PLGA NPs with the physical mixture of DOX and blank PLGA NPs were obtained with an X-ray diffractometer (D8 Advance, Bruker AXS). All samples were used with lyophilized powder. Typically, the diffractogram was recorded in the 2θ range of 3–40.
In vitro drug release
Triplicate samples of DOX-PLGA, DOX-PLGA (CHI/ALG)1, DOX-PLGA (CHI/ALG)2 and DOX-PLGA (CHI/ALG)3 NPs were respectively suspended in 1 mL of isotonic phosphate-buffered saline (PBS; 0.05 M, pH 7.4) in a microcentrifuge tube. The samples were then incubated on a shaker at a fixed speed of about 100 rpm (SHY-2A, Jin Tan, People’s Republic of China) at 37°C. At defined time intervals, the NPs were centrifuged, the supernatant was removed, and fresh phosphate buffer was replaced. The concentration of each supernatant sample was measured by an ultraviolet spectrophotometer (Agilent 8453; Agilent Technologies, Santa Clara, CA, USA) at 480 nm. The cumulative percentage of the drug released in the first hour was expressed as the initial burst release. The time at which half of the final theoretically encapsulated drug released was expressed as the half release time (t1/2).
Triplicate samples of DOX-PLGA (CHI/ALG)3 NPs were suspended in 1 mL PBS at different pH values (pH 7.4, pH 6.8 and pH 5.0) in a microcentrifuge tube, and other measurements were performed as described above.
In vivo study
Animals
Male mice (4–6 weeks old, 20–25 g) bearing mouse sarcoma 180 (S180) tumors were supplied by Origin Biosciences Inc. (Nanjing, People’s Republic of China). All studies were conducted in accordance with the principles of Laboratory Animal Care and were approved by the Department of Laboratory Animal Research at China Pharmaceutical University. The usage of animals was agreed upon by the China Pharmaceutical University Animal Management and Ethics Committee.
Antitumor efficacy of DOX-PLGA (CHI/ALG)3 NPs
The effect of the developed formulations on tumor growth was assessed by daily measurements of tumor size with a digital caliper. The mice were randomly divided into five groups with six males in each group and intravenously administered one of the following treatments through the tail vein: 1) saline (the control group), 2) blank PLGA NPs, 3) DOX solution, 4) DOX-PLGA NPs and 5) DOX-PLGA (CHI/ALG)3 NPs with a DOX dose of 5 mg/kg on days 0, 2, 4 and 6.
After administration, the animals were monitored carefully, and the body weight and tumor size were measured at given time intervals. Tumor volumes were calculated as follows:
V = a × b2/2, |
where a represents the length of the tumor and b represents the width.41
After 8 days of observation, the mice were sacrificed by cervical dislocation, and tumor inhibition rate was calculated by the equation:
|
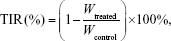
where Wtreated and Wcontrol are the average tumor weight of the treated group and the normal saline group, respectively, and TIR is the tumor inhibition rate.
Statistical analysis
All the results were presented as mean ± standard deviation (SD) and were analyzed by one-way analysis of variance. Statistical significance was set at *P<0.05 and **P<0.01 throughout the study.
Results and discussion
Preparation of DOX-PLGA NPs
PLGA NPs with a DOX concentration of 0.183 mg/mL were successfully prepared. As shown in Table 2, the resulting colloidal solution was a translucent pale blue liquid with ζ-potential −30.5 mV. The particle size and the polydispersity index of DOX-PLGA NPs were 172.3±3.9 nm and 0.156±0.002, respectively.
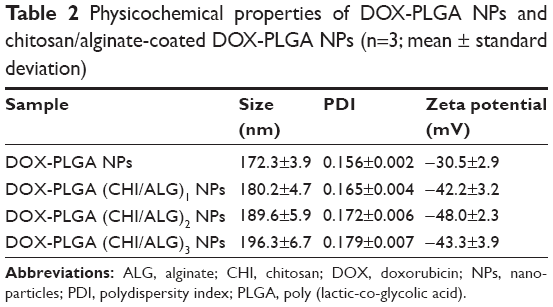 | Table 2 Physicochemical properties of DOX-PLGA NPs and chitosan/alginate-coated DOX-PLGA NPs (n=3; mean ± standard deviation) |
The XRD patterns are shown in Figure 2. DOX showed one intense peak at 2θ of 4.5° and numerous small peaks between 16.75° and 27° (Figure 2A). The XRD thermogram of DOX-PLGA NPs (Figure 2D) was similar to that of the blank PLGA NPs (Figure 2B), while the characteristic crystalline peaks of DOX disappeared in the NPs compared with the physical mixture of DOX and blank PLGA NPs (Figure 2C), indicating that DOX was encapsulated in DOX-PLGA NPs successfully.
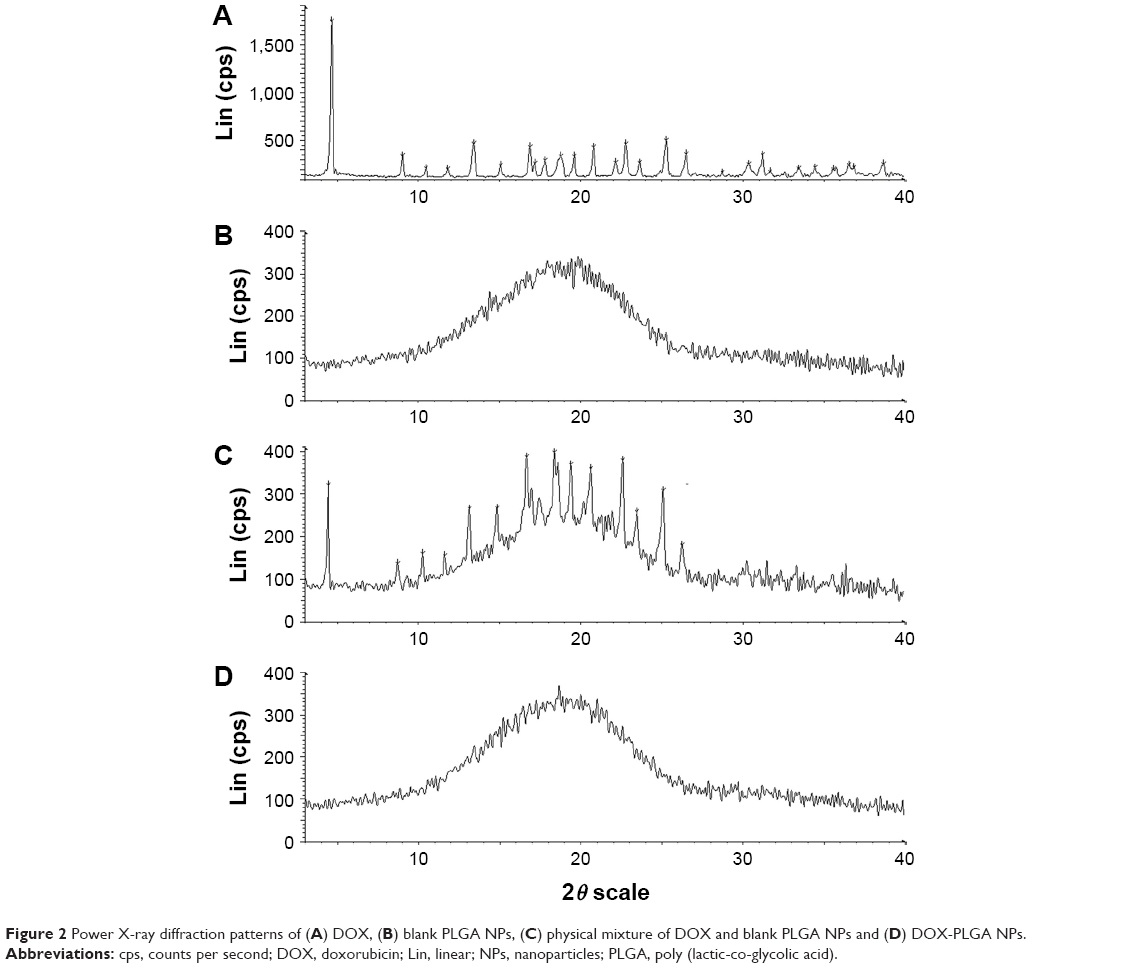 | Figure 2 Power X-ray diffraction patterns of (A) DOX, (B) blank PLGA NPs, (C) physical mixture of DOX and blank PLGA NPs and (D) DOX-PLGA NPs. |
Factors affecting the growth of the CHI/ALG multilayer
Saturation adsorption time
The same amounts of DOX-PLGA NPs and polyelectrolyte were added to each tube, and then the FITC-ALG of the supernatant was determined after 5, 10, 15, 20 and 30 min. The results are shown in Figure 3. Initially, the concentration of FITC-ALG decreased remarkably from 1.31 mg/mL at 0 min to 0.47 mg/mL at 10 min, and then achieved a balance at 20 min after centrifugation. It was found that the adsorption of polyelectrolyte on the colloidal particles reached a maximum after 20 min of incubation. Therefore, the following experiments were performed at this level (20 min).
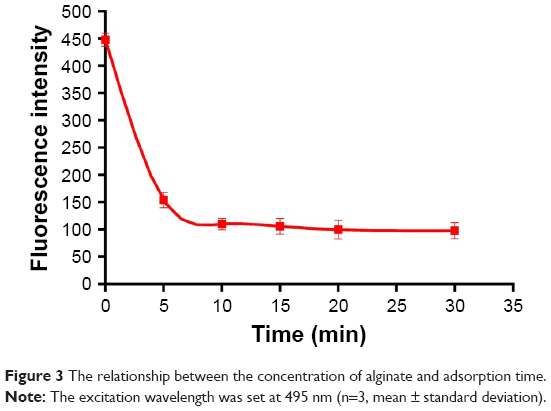 | Figure 3 The relationship between the concentration of alginate and adsorption time. |
Effect of polyelectrolyte concentration
Three concentrations of FITC-ALG were used for (CHI/ALG)3 deposition. Growth of the polyelectrolyte multilayer was monitored by determining the fluorescence intensity of the supernatant after each adsorption cycle of FITC-ALG. The results (Figure 4A) showed that the coating weight of the multilayer increased with increased concentration of the polyelectrolyte. This could be explained by a change in polymer conformation. At low polyelectrolyte concentrations, the conformation of the absorbed chain was rather flat without irregular structure because each polyelectrolyte chain could adsorb on many binding sites.42 In contrast, there were not enough binding sites for polyelectrolyte chains and each could adsorb on just a few binding sites at high polyelectrolyte concentrations, which resulted in more polymer chains absorbed and an increase of the coating weight of the multilayer.42,43
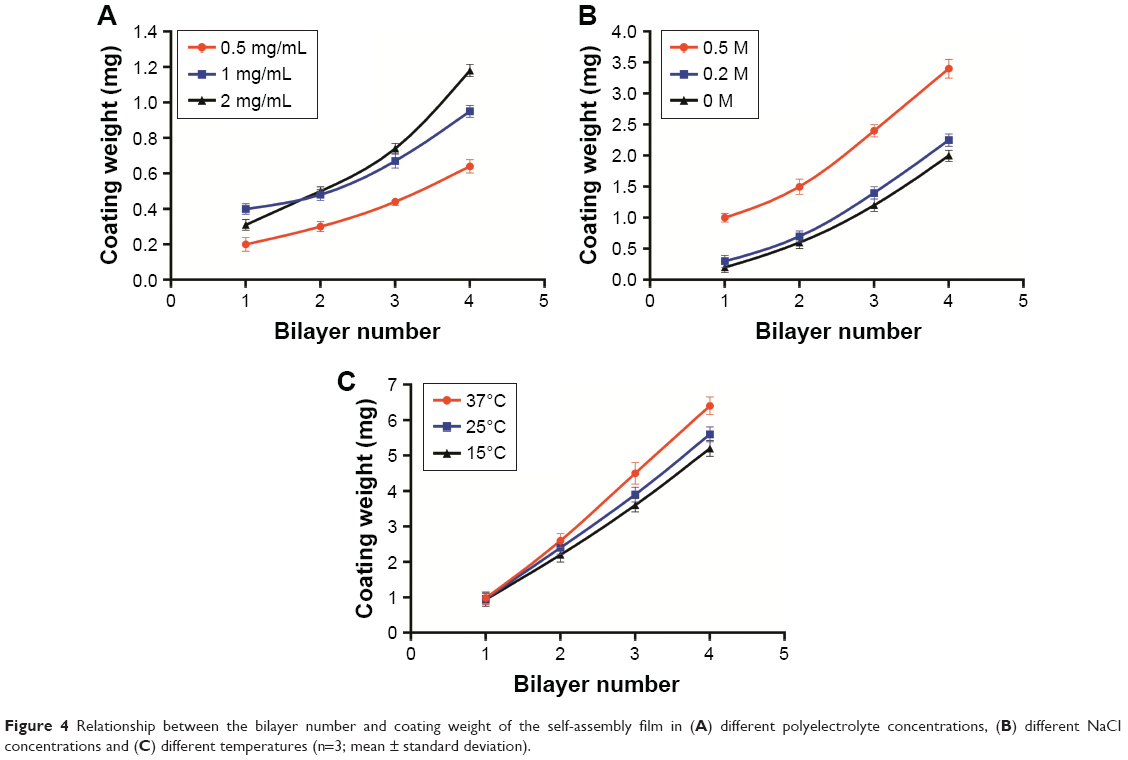 | Figure 4 Relationship between the bilayer number and coating weight of the self-assembly film in (A) different polyelectrolyte concentrations, (B) different NaCl concentrations and (C) different temperatures (n=3; mean ± standard deviation). |
Moreover, the intersection of the curves with 1 and 2 mg/mL was probably due to the viscosity and film-forming properties of the polyelectrolyte.44,45 Initially, the growth rate of the multilayer with 2 mg/mL was very slow, which was limited by the high viscosity of the polyelectrolyte solution. After several adsorption cycles, the growth of the multilayer was more stable and regular with little effect of viscosity on polyelectrolyte adsorption. So, the growth of the multilayer increased with polyelectrolyte concentration, which was also verified by Voigt et al.46
During the coating process, irreversible particle aggregation occurred due to poor dispersion of the polyelectrolyte solution at concentrations beyond 2 mg/mL. Tan et al found that thicker multilayers could significantly reduce the burst release of the drug from a nanogel.47 In order to obtain a satisfactory release profile, 2 mg/mL of the polyelectrolyte was selected as the optimal concentration for subsequent experiments.
Effect of NaCl concentration
During (CHI/ALG)3 deposition, growth of the polyelectrolyte multilayer was determined as described above. The results observed in Figure 4B revealed that with increasing NaCl concentration, the coating weight of the multilayer film increased, and a similar trend was also seen in the modifications of (CHI/ALG)1, (CHI/ALG)2 and (CHI/ALG)4 for the PLGA NPs. This could be explained by the fact that ions in the solution can shield the charge from the long polymer chain and reduce repulsion between charged groups in polymer chains which became curled to form a thicker multilayer.48 Ye et al reported that the average thickness of the CHI/ALG multilayer was maintained when the added NaCl concentration exceeded 0.5 mg/mL.49 In our experiment, it was also found that the coating weight profiles of the multilayer film became similar when the NaCl concentration was 0.5 mg/mL. Therefore, 0.5 mg/mL NaCl was chosen as the optimal condition in our study.
Effect of temperature
Three temperatures were investigated when the CHI/ALG was deposited, as the growth of the polyelectrolyte multilayer was monitored. The findings (Figure 4C) suggested that the increase in temperature caused a gradual increase in the multilayer coating weight at two or more bilayers of PLGA NP, and that temperature had no significant influence on the multilayer coating weight of one bilayer of NPs. High temperature was supposed to increase the thermal motion of the polymer long chain, which improved the fluidity of the multilayer.47 Therefore, more polyelectrolytes can infiltrate into the layer, which increases the thickness of the film. The glass transition temperature of PLGA used in our study was about 45°C;50 therefore, the temperature was fixed at 37°C to avoid PLGA phase changes.
Growth regime of CHI/ALG multilayer on three-dimensional PLGA NPs
As shown in Table 3, the fitting equations for growth curves at different polyelectrolyte concentrations, NaCl concentrations (>0) and adsorption temperatures were exponential, power and power, respectively, showing that the multilayer growth was asymmetrical. The asymmetrical-like growth behavior could be attributed to the lower charge density of bio-polyelectrolyte chains, resulting in weakened electrostatic repulsion between charged segments and highly coiled conformations of polymer chains.46 The adsorption of highly coiled polymer chains led to a thicker multilayer with a rough surface. This result indicated that bio-polyelectrolyte multilayers on three-dimensional colloidal particles grew asymmetrically, which was similar to the results on two-dimensional silicon substrates.51,52 Compared with a bi-dimensional environment, three-dimensional colloidal particles could be used to investigate the coating process quantitatively by monitoring the coating weight of the LbL multilayers, which could reflect the film-forming process more accurately.
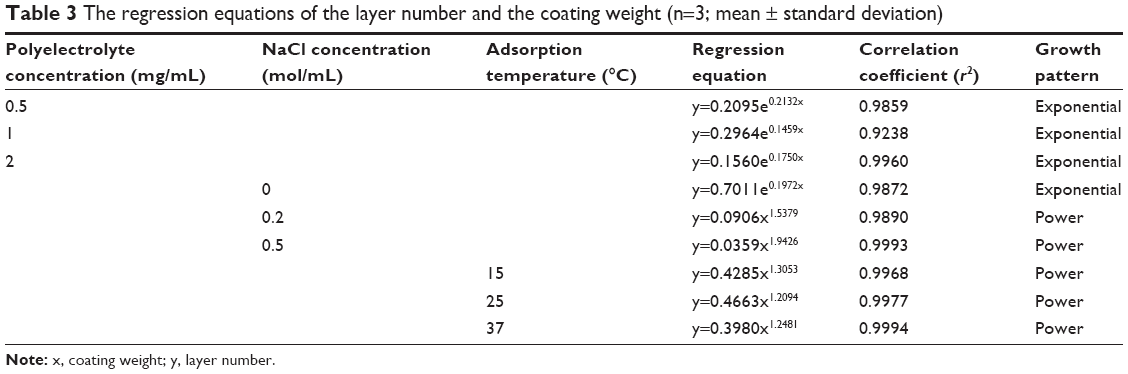 | Table 3 The regression equations of the layer number and the coating weight (n=3; mean ± standard deviation) |
With these factors influencing multilayer growth and the asymmetrical-like growth of the CHI/ALG multilayers, the CHI/ALG coating could be adjusted to reduce a burst release and achieve a satisfied and sustained release of anticancer drugs.
Characterization
Particle size and ζ-potential characterization
The particle sizes, polydispersity indexes and zeta potentials of DOX-PLGA NPs and CHI/ALG-coated DOX-PLGA NPs are presented in Table 3. The size and the polydispersity index of DOX-PLGA (CHI/ALG)3 NPs prepared in optimal conditions were 196.3±6.7 nm and 0.179±0.007, respectively. With the coating of a polyelectrolyte, the sizes of the PLGA-polyelectrolyte NPs gradually increased. Figure 5 shows the ζ-potential as a function of the layer number of polyelectrolytes for the DOX-PLGA NPs coated with CHI/ALG layers. The value of ζ-potential for the first polyelectrolyte layer (36.4 mV) corresponds to the deposition of CHI, the second (−42.2 mV) corresponds to the deposition of ALG, the third (36.7 mV) corresponds to another layer of CHI and the fourth (−48.0 mV) corresponds to a new layer of ALG NPs. The obvious switching of ζ-potential indicated a successful self-assembly on the DOX-PLGA NPs, and there was no significant difference between the zeta potentials of the different bilayers of the PLGA NPs. Moreover, the negative surface charge also reflected the physical stability of the DOX-PLGA (CHI/ALG)3 NPs.
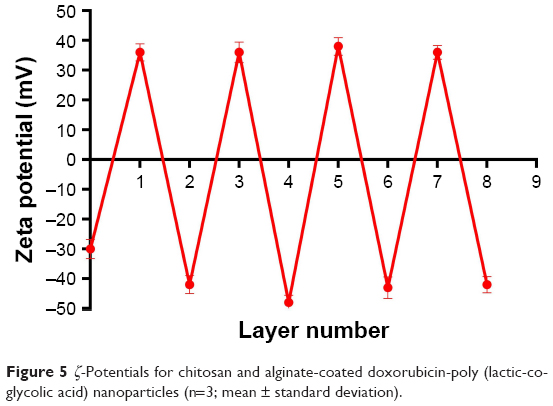 | Figure 5 ζ-Potentials for chitosan and alginate-coated doxorubicin-poly (lactic-co-glycolic acid) nanoparticles (n=3; mean ± standard deviation). |
Morphology characterization
TEM was used to examine the morphology of the NPs. As seen in Figure 6, the DOX-PLGA NPs were found to be spherical and uniform with sizes of <150 nm (Figure 6A). With LbL self-assembly of CHI and ALG, a core–shell structure was observed, and the DOX-PLGA (CHI/ALG)3 NP periphery showed nonuniform polyelectrolyte anisomerous textures (Figure 6B). This phenomenon provided further proof of formation of polyelectrolyte multilayers and was a reflection of the nonuniform asymmetrical-like growth of the multilayer. Meanwhile, the average sizes of the DOX-PLGA NPs and DOX-PLGA (CHI/ALG)3 NPs, as preliminarily determined by dynamic light scattering (172.3±3.9 and 196.3±6.7 nm, respectively), were larger than those determined from TEM images (118.7±7.3 and 179.4±4.2 nm, respectively). This could be attributed to the fact that the intensity of the scattered light is proportional to the sixth power of the droplet size, and dynamic light scattering overdepends on larger size fractions.53
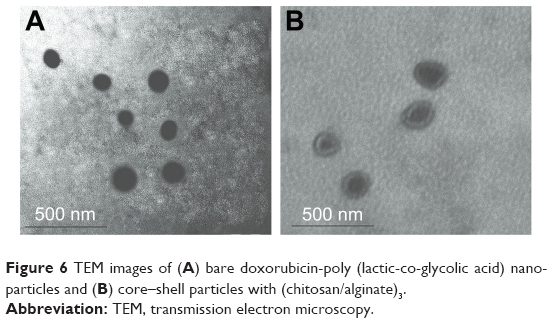 | Figure 6 TEM images of (A) bare doxorubicin-poly (lactic-co-glycolic acid) nanoparticles and (B) core–shell particles with (chitosan/alginate)3. |
In vitro drug release
Effect of different layer numbers on the drug release
The in vitro release behavior of DOX from the NPs with different bilayers was investigated. As shown in Figure 7 and Table 4, the initial burst release was obviously reduced, compared with the bare DOX-PLGA NPs. The release rate and release percentage of DOX decreased with an increase of CHI/ALG layer numbers on the surface of the NPs. With CHI/ALG coating, the initial burst release from the DOX-PLGA (CHI/ALG)3 NPs was reduced from 55.12% to 5.78%, while the half release time (t1/2) was prolonged from 0.78 to 61.58 h, which could be attributed to the increased layer thickness that made DOX hard to permeate. Zhou et al developed poly (acrylic acid) and polyethyleneimine-coated PLGA NPs, and it was also found that the more layers, the slower the drug release rate.54 Based on the above results, it could be concluded that polyelectrolyte multilayers coated on NP surfaces can not only prolong drug release time, but also relieve initial burst release, which may decrease systemic adverse reactions toward drug resistance, diminish their toxicity and improve their therapeutic efficacy. In comparison to common PLGA NPs, based on these results, DOX-PLGA (CHI/ALG)3 NPs may present higher safety profiles and better antitumor efficacy.
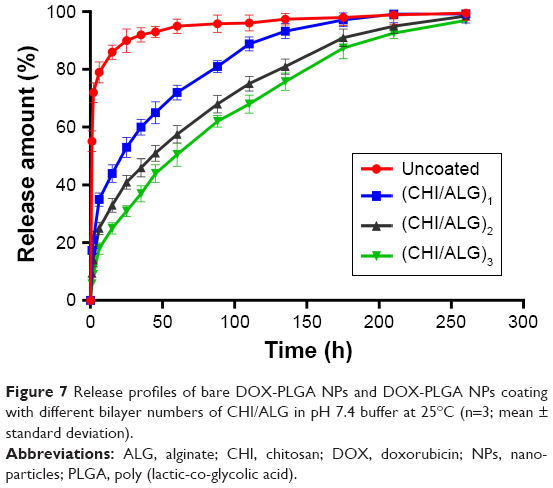 | Figure 7 Release profiles of bare DOX-PLGA NPs and DOX-PLGA NPs coating with different bilayer numbers of CHI/ALG in pH 7.4 buffer at 25°C (n=3; mean ± standard deviation). |
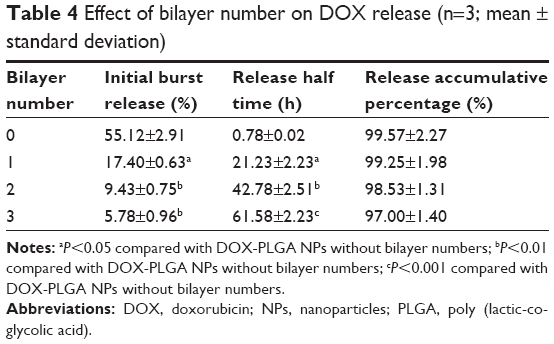 | Table 4 Effect of bilayer number on DOX release (n=3; mean ± standard deviation) |
Effect of medium pH on the release of DOX-PLGA (CHI/ALG)3 NPs
Figure 8 and Table 5 show the release behavior of DOX-PLGA (CHI/ALG)3 NPs for different values of isotonic PBS (pH 5.0, pH 6.8 and pH 7.4). A significant decrease in half release time (t1/2) and an increase in the release rate of DOX were observed at a decreased medium pH, from a t1/2 of 61.58 h at pH 7.4 to 20.5 h at pH 5.0, similar to earlier reports.55,56 This could be attributed to the fact that DOX, a basic drug, had a strongly pH-dependent solubility with decreased solubility at basic pH values, and the medium with low pH could facilitate drug release.57 In addition, acidic conditions are conducive to enhancing the hydrolysis of the ester bonds in the polymer chains, resulting in the degradation of the polymer and further acceleration of DOX release.58 Typically, the environment of stromal cells in the tumor tissue is weakly acidic (pH <7), while the pH of endosomes and lysosomes in tumor cells is even lower (pH 4–6).59–65 These results indicated that there would be superiority of antitumor therapy to some degree and DOX-loaded PLGA-polyelectrolyte NPs would be appropriate for antitumor therapy in acidic tumor environments.
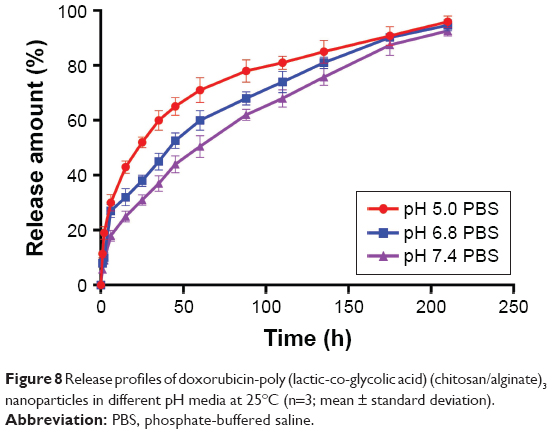 | Figure 8 Release profiles of doxorubicin-poly (lactic-co-glycolic acid) (chitosan/alginate)3 nanoparticles in different pH media at 25°C (n=3; mean ± standard deviation). |
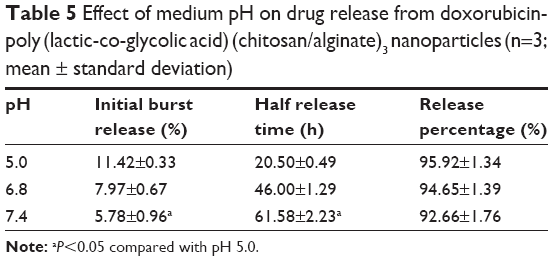 | Table 5 Effect of medium pH on drug release from doxorubicin-poly (lactic-co-glycolic acid) (chitosan/alginate)3 nanoparticles (n=3; mean ± standard deviation) |
In vivo antitumor efficacy
To confirm the antitumor potential of DOX-PLGA (CHI/ALG)3 NPs in vivo, the antitumor efficiency was evaluated in S180 tumor-bearing mice.66 As shown in Figure 9A, all DOX formulations effectively inhibited tumor growth, compared with the saline group and blank PLGA NPs (P<0.01). Among all groups, DOX-PLGA (CHI/ALG)3 NPs exhibited the greatest tumor growth inhibition, followed by DOX-PLGA NPs and the DOX solution, and a significant difference was found between DOX-PLGA (CHI/ALG)3 NPs and DOX-PLGA NPs (P<0.05). Moreover, tumor inhibition rates for the different treatments based on tumor weights (Table 6) were in agreement with the results of the tumor volume experiment. DOX-PLGA (CHI/ALG)3 NPs exhibited the best antitumor efficiency (83.17%), followed by DOX-PLGA NPs (61.35%) and DOX solution (23.77%). CHI and ALG could increase the hydrophilicity of the PLGA NPs, which shields the NPs from clearance by the mononuclear phagocytic system/reticuloendothelial system. In addition, the enhanced permeability and retention affect has the ability to passively target NPs to the tumor sites.67,68 NPs with particle sizes of 20–200 nm tend to concentrate in tumors by the enhanced permeability and retention phenomenon.69,70
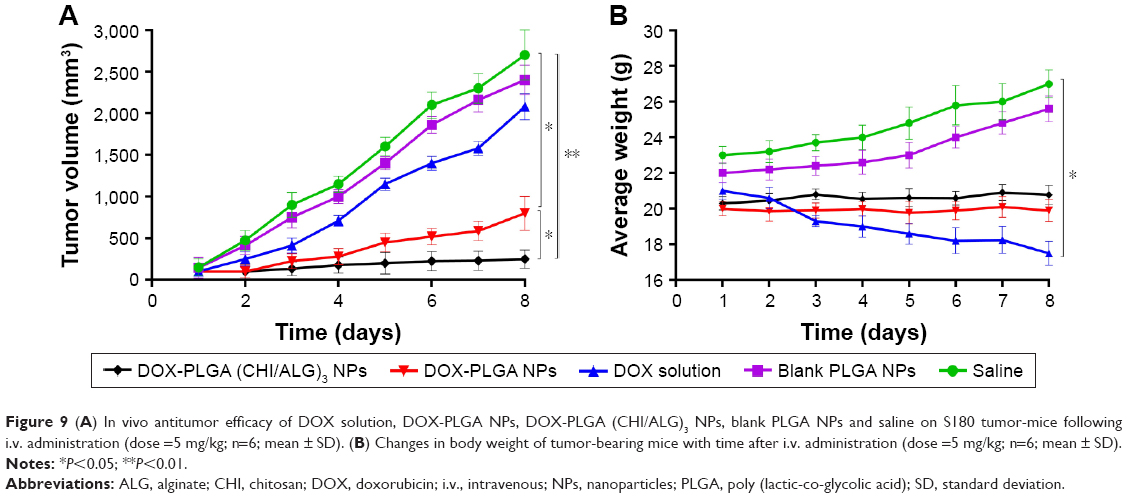 | Figure 9 (A) In vivo antitumor efficacy of DOX solution, DOX-PLGA NPs, DOX-PLGA (CHI/ALG)3 NPs, blank PLGA NPs and saline on S180 tumor-mice following i.v. administration (dose =5 mg/kg; n=6; mean ± SD). (B) Changes in body weight of tumor-bearing mice with time after i.v. administration (dose =5 mg/kg; n=6; mean ± SD). |
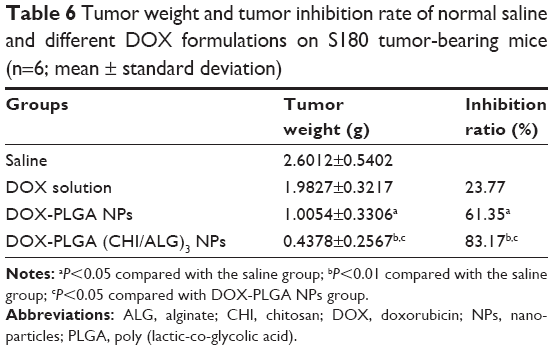 | Table 6 Tumor weight and tumor inhibition rate of normal saline and different DOX formulations on S180 tumor-bearing mice (n=6; mean ± standard deviation) |
In order to estimate the in vivo toxicity of the DOX formulations, the body weight of the mice was measured. Obvious body weight loss after drug administration is usually associated with drug toxicity. As presented in Figure 9B, there was a significant decrease in body weight for the DOX solution treated group (16.67%). However, this phenomenon was not found after administration of the DOX-PLGA (CHI/ALG)3 NPs, indicating that DOX-PLGA (CHI/ALG)3 NPs could significantly decrease the toxicity of the free DOX and could be well tolerated at the tested dose level (5 mg DOX/kg). It could be easily envisioned that the lower drug burst release decreased toxicity. The integrated results verified that the DOX-PLGA (CHI/ALG)3 NPs were much safer and could effectively improve antitumor efficacy, compared with the DOX solution and the DOX-PLGA NPs.
Conclusion
DOX-PLGA NPs coated with CHI and ALG were successfully prepared by the LbL self-assembly process, and factors influencing the multilayer growth on the PLGA NPs were investigated. DOX-PLGA NPs sized below 200 nm were formed. The undesired initial burst release of DOX was significantly reduced from 55.12% to 5.78% through the LbL approach, and the half release time (t1/2) increased from 0.78 to 61.58 h with increasing layer number. Meanwhile, the NPs coated with CHI and ALG showed pH-dependent characteristics and the release of DOX increased with a decrease in medium pH. Compared with the DOX solution and DOX-PLGA NPs, DOX-PLGA (CHI/ALG)3 NPs exhibited a superior in vivo tumor inhibition rate of 83.17% and decreased toxicity.
To conclude, PLGA NPs coated with CHI/ALG by LbL is a novel and effective approach to minimize initial burst release, sustain release of DOX and decrease toxicity. These findings indicate that PLGA-polyelectrolyte NPs have potential numerous applications for controlled anticancer drug release.
Acknowledgments
This work was financially supported by the Natural Science Foundation of China (no 81402878, 81572978 and 81473160) and the Scientific Foundation of Jiangsu Provincial Commission of Health (no Z201508). The abstract of this paper was presented at the American Institute of Chemical Engineers Conference on Nov 16, 2016 as a poster presentation with preliminary findings. The poster’s abstract was published in “Poster Abstracts” on the website http://www3.aiche.org/proceedings/Abstract.aspx?PaperID=471267.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ward E, Brawley O, Jemal A. The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61(4):212-236. | ||
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157-170. | ||
Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116(4):2602-2663. | ||
Elzoghby AO, Samy WM, Elgindy NA. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157(2):168-182. | ||
Arvizo RR, Miranda OR, Moyano DF, et al. Modulating pharmacokinetics, tumor uptake and biodistribution by engineered nanoparticles. PloS One. 2011;6(9):e24374. | ||
Santhi K, Dhanaraj SA, Koshy M, Ponnusankar S, Suresh B. Study of biodistribution of methotrexate-loaded bovine serum albumin nanospheres in mice. Drug Dev Ind Pharm. 2000;26:1293-1296. | ||
Feng SS, Mu L, Chen BH, Pack D. Polymeric nanospheres fabricated with natural emulsifiers for clinical administration of an anticancer drug paclitaxel (Taxol®). Mater Sci Eng C. 2002;20(1):85-92. | ||
Merisko-Liversidge EM, Liversidge GG. Drug nanoparticles: formulating poorly water-soluble compounds. Toxicol Pathol. 2008;36(1):43-48. | ||
Jain RA. The manufacturing techniques of various drug loaded biodegradable poly (lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475-2490. | ||
Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly (DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16(10):1217-1226. | ||
Thamake SI, Raut SL, Gryczynski Z, Ranjan AP, Vishwanatha JK. Alendronate coated poly-lactic-co-glycolic acid (PLGA) nanoparticles for active targeting of metastatic breast cancer. Biomaterials. 2012;33(29):7164-7173. | ||
Hrkach J, Von Hoff D, Ali MM, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4(128):128-139. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505-522. | ||
Dawidczyk CM, Russell LM, Searson PC. Nanomedicines for cancer therapy: state-of-the-art and limitations to pre-clinical studies that hinder future developments. Frontiers Chem. 2014;2:69. | ||
Tosi G, Bortot B, Ruozi B, et al. Potential use of polymeric nanoparticles for drug delivery across the blood-brain barrier. Curr Med Chem. 2013;20(17):2212-2225. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Delivery Rev. 2011;63(3):170-183. | ||
Ganju A, Yallapu MM, Khan S, Behrman SW, Chauhan SC, Jaggi M. Nanoways to overcome docetaxel resistance in prostate cancer. Drug Resist Updat. 2014;17(1):13-23. | ||
Yallapu MM, Gupta BK, Jaggi M, Chauhan SC. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J Colloid Interface Sci. 2010;351(1):19-29. | ||
Gomes C, Moreira RG, Castell-Perez E. Poly (DL-lactide-co-glycolide) (PLGA) nanoparticles with entrapped trans-cinnamaldehyde and eugenol for antimicrobial delivery applications. J Food Sci. 2011;76(2):N16-N24. | ||
Seju U, Kumar A, Sawant KK. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: in vitro and in vivo studies. Acta Biomater. 2011;7(12):4169–4176. | ||
Adibkia K, Javadzadeh Y, Dastmalchi S, Mohammadi G, Niri FK, Alaei-Beirami M. Naproxen–Eudragit® RS100 nanoparticles: preparation and physicochemical characterization. Colloid Surf B. 2011;83(1):155-159. | ||
Ud Din F, Mustapha O, Kim DW, et al. Novel dual-reverse thermosensitive solid lipid nanoparticle-loaded hydrogel for rectal administration of flurbiprofen with improved bioavailability and reduced initial burst effect. Eur J Pharm Biopharm. 2015;94:64–72. | ||
Rojas J, Pinto-Alphandary H, Leo E, Pecquet S, Couvreur P, Fattal E. Optimization of the encapsulation and release of β-lactoglobulin entrapped poly (DL-lactide-co-glycolide) microspheres. Int J Pharm. 1999;183(1):67-71. | ||
Sheikh Hasan A, Socha M, Lamprecht A, et al. Effect of the microencapsulation of nanoparticles on the reduction of burst release. Int J Pharm. 2007;344(1):53-61. | ||
Matsumoto A, Matsukawa Y, Suzuki T, Yoshino H, Kobayashi M. The polymer-alloys method as a new preparation method of biodegradable microspheres: principle and application to cisplatin-loaded microspheres. J Control Release. 1997;48(1):19-27. | ||
Poon Z, Lee JB, Morton SW, Hammond PT. Controlling in vivo stability and biodistribution in electrostatically assembled nanoparticles for systemic delivery. Nano Lett. 2011;11(5):2096-2103. | ||
Poon Z, Chang D, Zhao X, Hammond PT. Layer-by-layer nanoparticles with a pH-sheddable layer for in vivo targeting of tumor hypoxia. ACS Nano. 2011;5(6):4284-4292. | ||
Hammond PT. Polyelectrolyte multilayered nanoparticles: using nanolayers for controlled and targeted systemic release. Nanomedicine. 2012;7(5):619-622. | ||
Morton SW, Poon Z, Hammond PT. The architecture and biological performance of drug-loaded LbL nanoparticles. Biomaterials. 2013;34(21):5328-5335. | ||
Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2012;64:61–71. | ||
Lavan DA, Lynn DM, Langer R. Moving smaller in drug discovery and delivery. Nat Rev Drug Discov. 2002;1(1):77–84. | ||
Hammond PT. Building biomedical materials layer-by-layer. Mater Today. 2012;15(5):196–206. | ||
Li H, Zhu X, Xu J, Peng W, Zhong S, Wang Y. The combination of adsorption by functionalized halloysite nanotubes and encapsulation by polyelectrolyte coatings for sustained drug delivery. RSC Adv. 2016;6(59):54463-54470. | ||
Amancha KP, Balkundi S, Lvov Y, Hussain A. Pulmonary sustained release of insulin from microparticles composed of polyelectrolyte layer-by-layer assembly. Int J Pharm. 2014;466(1):96-108. | ||
Park MR, Seo BB, Song SC. Dual ionic interaction system based on polyelectrolyte complex and ionic, injectable, and thermosensitive hydrogel for sustained release of human growth hormone. Biomaterials. 2013;34(4):1327-1336. | ||
Pargaonkar N, Lvov YM, Li N, Steenekamp JH, de Villiers MM. Controlled release of dexamethasone from microcapsules produced by polyelectrolyte layer-by-layer nanoassembly. Pharm Res. 2005;22(5):826-835. | ||
Anal AK, Stevens WF. Chitosan–alginate multilayer beads for controlled release of ampicillin. Int J Pharm. 2005;290(1):45-54. | ||
Chen Y, Lin X, Park H, Greever R. Study of artemisinin nanocapsules as anticancer drug delivery systems. Nanomed Nanotechnol Biol Med. 2009;5(3):316-322. | ||
Lamprecht A, Schäfer UF, Lehr C. Characterization of microcapsules by confocal laser scanning microscopy: structure, capsule wall composition and encapsulation rate. Eur J Pharm Biopharm. 2000;49(1):1-9. | ||
Dördelmann G, Kozlova D, Karczewski S, Lizio R, Knauer S, Epple M. Calcium phosphate increases the encapsulation efficiency of hydrophilic drugs (proteins, nucleic acids) into poly (d, l-lactide-co-glycolide acid) nanoparticles for intracellular delivery. J Mater Chem B. 2014;2(41):7250-7259. | ||
Zhen X, Wang X, Xie C, Wu W, Jiang X. Cellular uptake, antitumor response and tumor penetration of cisplatin-loaded milk protein nanoparticles. Biomaterials. 2013;34(4):1372-1382. | ||
Fleer G, Stuart MC, Scheutjens JMHM, Cosgrove T, Vincent B. Polymers at Interfaces. Cambridge, UK: Chapman & Hall; 1993. | ||
Klitzing RV, Kolarić B, Jaeger W, Brandt A. Structuring of poly (DADMAC) chains in aqueous media: a comparison between bulk and free-standing film measurements. Phys Chem Chem Phys. 2002;4(10):1907-1914. | ||
Picart C, Lavalle P, Hubert P, et al. Buildup mechanism for poly (L-lysine)/hyaluronic acid films onto a solid surface. Langmuir. 2001;17(23):7414-7424. | ||
Boulmedais F, Ball V, Schwinte P, Frisch B, Schaaf P, Voegel JC. Buildup of exponentially growing multilayer polypeptide films with internal secondary structure. Langmuir. 2003;19(2):440-445. | ||
Voigt U, Jaeger W, Findenegg GH, Klitzing RV. Charge effects on the formation of multilayers containing strong polyelectrolytes. J Phys Chem B. 2003;107(22):5273-5280. | ||
Tan HL, McMurdo MJ, Pan G, Van Patten PG. Temperature dependence of polyelectrolyte multilayer assembly. Langmuir. 2003;19(22):9311-9314. | ||
Lösche M, Schmitt J, Decher G, Bouwman WG, Kjaer K. Detailed structure of molecularly thin polyelectrolyte multilayer films on solid substrates as revealed by neutron reflectometry. Macromolecules. 1998;31(25):8893-8906. | ||
Ye S, Wang C, Liu X, Tong Z. Multilayer nanocapsules of polysaccharide chitosan and alginate through layer-by-layer assembly directly on PS nanoparticles for release. J Biomater Sci Polym Ed. 2005;16(7):909-923. | ||
Muthu M. Nanoparticles based on PLGA and its co-polymer: an overview. Asian J Pharm. 2009;3(4):266-273. | ||
Nazaran P, Bosio V, Jaeger W, Anghel DF, Klitzing RV. Lateral mobility of polyelectrolyte chains in multilayers. J Phys Chem B. 2007;111(29):8572-8581. | ||
Poptoshev E, Schoeler B, Caruso F. Influence of solvent quality on the growth of polyelectrolyte multilayers. Langmuir. 2004;20(3):829-834. | ||
Calzolai L, Gilliland D, Garcìa CP, Rossi F. Separation and characterization of gold nanoparticle mixtures by flow-field-flow fractionation. J Chromatogr A. 2011;1218(27):4234-4239. | ||
Zhou J, Moya S, Ma L, Gao C, Shen J. Polyelectrolyte coated PLGA nanoparticles: templation and release Bbehavior. Macromol Biosci. 2009;9(4):326-335. | ||
Feng W, Zhou X, He C, et al. Polyelectrolyte multilayer functionalized mesoporous silica nanoparticles for pH-responsive drug delivery: layer thickness-dependent release profiles and biocompatibility. J Mater Chem B. 2013;1(43):5886-5898. | ||
Zheng J, Tian X, Sun Y, Lu D, Yang W. pH-sensitive poly (glutamic acid) grafted mesoporous silica nanoparticles for drug delivery. Int J Pharm. 2013;450(1):296-303. | ||
Gautier J, Munnier E, Soucé M, Chourpa I, Eyrolles LD. Analysis of doxorubicin distribution in MCF-7 cells treated with drug-loaded nanoparticles by combination of two fluorescence-based techniques, confocal spectral imaging and capillary electrophoresis. Anal Bioanal Chem. 2015;407(12):3425-3435. | ||
Nakayama M, Akimoto J, Okano T. Polymeric micelles with stimuli-triggering systems for advanced cancer drug targeting. J Drug Target. 2014;22(7):584-599. | ||
Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441-1454. | ||
Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752-3756. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Part 1):6387-6392. | ||
Popović Z, Liu W, Chauhan VP, et al. A nanoparticle size series for in vivo fluorescence imaging. Angew Chem. 2010;122(46):8831-8834. | ||
Gallagher FA, Kettunen MI, Day SE, et al. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C-labelled bicarbonate. Nature. 2008;453(7197):940-943. | ||
Urano Y, Asanuma D, Hama Y, et al. Selective molecular imaging of viable cancer cells with pH-activatable fluorescence probes. Nat Med. 2008;15(1):104-109. | ||
Lee ES, Kim D, Youn YS, Oh KT, Bae YH. A Virus-mimetic nanogel vehicle. Angew Chem Int Ed. 2008;47(13):2418-2421. | ||
Hu M, Shen Y, Zhang L, Qiu L. Polymersomes via self-assembly of amphiphilic β-cyclodextrin-centered triarm star polymers for enhanced oral bioavailability of water-soluble chemotherapeutics. Biomacromolecules. 2016;17(3):1026-1039. | ||
Storm G, Belliot SO, Daemen T, Lasic DD. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Adv Drug Deliv Rev. 1995;17(1):31-48. | ||
Steichen SD, Caldorera-Moore M, Peppas NA. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci. 2013;48(3):416-427. | ||
Lane D. Designer combination therapy for cancer. Nat Biotechnol. 2006;24(2):163-164. | ||
Shi S, Shi K, Tan LW, et al. The use of cationic MPEG-PCL-g-PEI micelles for co-delivery of Msurvivin T34A gene and doxorubicin. Biomaterials. 2014;35(15):4536-4547. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.