Back to Journals » International Journal of Nanomedicine » Volume 12
Doxorubicin-loaded cell-derived nanovesicles: an alternative targeted approach for anti-tumor therapy
Authors Goh WJ, Lee CK, Zou S, Woon EC, Czarny B ![]() , Pastorin G
, Pastorin G
Received 6 January 2017
Accepted for publication 17 February 2017
Published 4 April 2017 Volume 2017:12 Pages 2759—2767
DOI https://doi.org/10.2147/IJN.S131786
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Wei Jiang Goh,1,2 Choon Keong Lee,2 Shui Zou,2 Esther CY Woon,2 Bertrand Czarny,2,3 Giorgia Pastorin1,2,4
1NUS Graduate School for Integrative Sciences and Engineering, Centre for Life Sciences (CeLS), 2Department of Pharmacy, National University of Singapore, 3School of Materials Science and Engineering (MSE) & Lee Kong Chian School of Medicine, Nanyang Technological University, 4NUSNNI-NanoCore, National University of Singapore, T-Lab, Singapore, Singapore
Abstract: Cell-derived nanovesicles (CDNs) are an emerging class of biological drug delivery systems (DDS) that retain the characteristics of the cells they were derived from, without the need for further surface functionalization. CDNs are also biocompatible, being derived from natural sources and also take advantage of the enhanced permeability and retention effect due to their nanodimensions. Furthermore, CDNs derived from monocytes were shown to have an in vivo targeting effect, accumulating at the tumor site in a previous study conducted in a mouse tumor model. Here, we report a systematic approach pertaining to various loading methods of the chemotherapeutic drug doxorubicin into our CDNs and examine the differential cellular uptake of drug-loaded CDNs in cancerous (HeLa) and healthy (HEK293) cell lines. Lastly, we proved that the addition of doxorubicin-loaded CDNs to the HeLa and HEK293 co-cultures showed a clear discrimination toward cancer cells at the cellular level. Our results further reinforce the intriguing potential of CDNs as an alternative targeted strategy for anticancer therapy.
Keywords: cell-derived nanovesicles, cell targeting, doxorubicin, antitumor therapy, extracellular vesicles, biomimetic, bionanotechnology, antitumor strategies
Introduction
Several antitumor strategies have been reported in the literature using drug delivery systems (DDS), with the goal of specific cell targeting to achieve efficient cancer cell death with minimal collateral damage. However, a common disadvantage of these synthetic DDS including liposomes,1 carbon nanotubes,2 dendrimers,3 and gold nanoparticles4 is the lack of inherent cell targeting without further surface functionalization. Rather, these DDS accumulate at the tumor site via the enhanced permeability and retention (EPR) effect due to their nanodimensions.5 This, in turn, may not necessarily release the therapeutic cargo only at the desired site but may also affect the neighboring healthy tissue. Moreover, the synthetic nature of these DDS may result in adverse immunogenic responses, such as the complement activation-related pseudoallergy (CARPA) effect upon liposomal administration, or unwanted accumulation in organs such as liver or kidneys.6 Lastly, several of these synthetic nanoparticles have poor in vivo circulation time due to recognition by the body’s immune system and premature elimination.3
Cell-derived nanovesicles (CDNs), on the other hand, are currently investigated as an alternative to the above-mentioned synthetic DDS. CDNs were inspired from endogenously produced exosomes, which were found to be suited for drug delivery purposes, being nanovesicles naturally involved in intercellular communication.7 In addition, exosomes already shuttle endogenous biological cargo, such as mRNAs, RNAs, and also proteins.8 However, a major drawback of using exosomes as a viable DDS is their relative low production yield and often complicated procedures.9
In comparison, a cost-effective, high-yield production of CDNs from cells has been recently published.9,10 As a result of their nanodimensions, CDNs are able to utilize the EPR effect, in a similar manner as synthetic DDS. CDNs also retain the surface membrane of the parent cell, maintaining the same properties as the parent cells.11 In this study, CDNs were derived from monocytes, which possess intrinsic targeting toward inflammation sites, and are therefore ideally suited for targeting tumors.12 Furthermore, CDNs are derived from natural sources, alleviating concerns of biocompatibility as well as adverse immunogenic responses. Lastly, CDNs are also amendable to further functionalization if required, due to the presence of several protein groups available for conjugation, in a manner similar to exosomes currently under investigations.13
Despite the allure of CDNs as a viable DDS, loading of therapeutics into CDNs has proven to be challenging. In this study, we examined various therapeutic loading conditions for CDNs. We chose the chemotherapeutic doxorubicin hydrochloride as the model drug due to its widespread clinical use in its liposomal formulation (Doxil®), and also due to its fluorescent properties, enabling ease of tracking and quantification.14
As CDNs were previously shown to be similar in several aspects to endogenously derived exosomes, including size range, key protein markers, and functional behavior both in in vitro and in vivo settings, therapeutic loading methods were similarly referenced from the literature on exosomes.15
In agreement with previous reports, we noticed a marked size increase in these cell-derived DDS after loading of doxorubicin. Moreover, doxorubicin was reported to further potentiate the aggregation of DDS, resulting in precipitation of the DDS and causing interferences during doxorubicin quantification.16 In this study, we also described a simple method of cleaning the unencapsulated doxorubicin from CDNs, to achieve an optimal balance between improved doxorubicin loading while maintaining the nanodimensions of CDNs.
Finally, we aimed to demonstrate the improved cellular uptake of doxorubicin-loaded CDNs by cancer cells vis-à-vis non-cancer cells in both monocultures and co-cultures to demonstrate a dose-sparing effect with optimal cancer cell killing and yet minimal collateral damage to healthy cells. This, in turn, demonstrates the potential of CDNs as a viable nanocarrier for chemotherapeutics in an anti-tumor strategy.
Experimental
Materials
U937 cells were a kind gift from Associate Professor Gigi Chiu, National University of Singapore. Spin columns were purchased from ThermoScientific and were supplied with 10-μm filters attached while hydrophilic 8- and 5-μm membrane filters were purchased from Merck Millipore and used as supplied. ThermoScientific microcentrifuge was used in the cell disruption process for the production of CDNs from U937 monocytes. Doxorubicin hydrochloride was purchased from Sigma-Aldrich. HeLa and HEK293 cell lines were purchased from American Type Tissue Culture (ATTC, Manassas, VA, USA).
This research was reviewed and approved by the National University of Singapore, Institutional Review Board (NUS–IRB) for human biomedical research.
Cell culture
U937 cells were grown in Roswell Park Memorial Institute medium (RPMI) 1640 culture medium supplemented with 10% fetal bovine serum (FBS). Cells were grown to 70%–80% confluence, before 2×107 cells were removed, centrifuged, and suspended in phosphate-buffered saline (PBS) twice and subsequently used for CDN production.
CDNs production
CDNs were produced as described in an earlier publication. Briefly, a serial extrusion method was adopted by fitting spin columns with 10- and 8-μm filter membranes and passing the cells through the membrane filters sequentially using a microcentrifuge. 2×107/mL U937 cells, suspended in PBS (pH 7.2), were added to each spin cup, corresponding to about 538 μg/mL of CDNs produced on a protein basis. After centrifuging at 14,000× g for 10 min in spin columns fitted with 10-μm membrane filters, followed by 8-μm membrane filters twice each, CDNs between 100 and 200 nm in size were produced. The subsequent dispersion is further purified using a Sephadex G50 size-exclusion column previously equilibrated with PBS, and the CDN fractions collected.
Characterization of CDNs
Size and zeta potential of CDNs were assayed by dynamic light scattering (DLS) using the Zetasizer Nano (Malvern Instruments). Protein concentration was determined using a standard protein assay kit (Pierce bicinchoninic acid [BCA] and Pierce microBCA kit).
CRYO-TEM imaging of CDNs
CDNs were fixed using 2.5% glutaraldehyde and placed onto a Quantifoil with 2-s blotting time. The samples were then plunged into liquid ethane using Gatan CP3 Cryo-Plunger 3 and viewed using JEOL JEM-2200FS.
Doxorubicin loading into CDNs
Loading of doxorubicin hydrochloride into CDNs was investigated using four different methods listed in Table 1 to investigate the loading efficiency and also size aggregation due to layering of doxorubicin onto the surface of the CDNs. Stirring rates were set at 200 rpm for methods A and B. Freeze–thaw cycles were performed by freezing at −80°C and thawing at room temperature thrice. The doxorubicin-loaded CDNs were removed of doxorubicin layered onto its exterior surface by passing through protein concentrators (MicroSep Centrifugal Devices 10k) at 2,000× g for 10 min. The size of the CDNs was checked via DLS to ensure that doxorubicin on the exterior surface of CDNs was sufficiently removed. Doxorubicin concentrations were determined by measuring its Ex/Em 480/590 nm against a set of known doxorubicin standards.
 | Table 1 Doxorubicin loading conditions investigated |
CDNs were normalized to a protein concentration of 500 μg/mL for all investigations. Doxorubicin hydrochloride was added to an equal volume of CDNs at 100, 200, and 500 μg/mL in the respective experiments.
Doxorubicin release studies
Doxorubicin release kinetics were performed by adding CDNs from loading conditions A and B to 1 L of stirring PBS set to 37°C, with a change of dialysis solution at 4-h time point to maintain sink conditions. The doxorubicin-loaded CDNs (CDNDox) were dialyzed using SnakeSkin™ 3.5 k molecular weight cutoff (MWCO) from ThermoFisher Scientific and 80 μL was collected at each point to assay the doxorubicin concentration using a prepared doxorubicin calibration curve.
Doxorubicin-loaded CDNs cellular uptake
CDNDox from loading condition A were added to HeLa cells grown to 70% confluency in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS. The cells were incubated for 1 and 6 h before being washed with sterile PBS, trypsinized, and assayed using flow cytometry (BD LSR Fortessa Flow Cytometry Analyser) to determine the extent of doxorubicin cellular uptake. For confocal microscopy imagery, HeLa cells were grown on glass bottom petri dishes under the same conditions, and doxorubicin-loaded CDNs were incubated with the cells for 6 h. Hoechst 33342 dye was added to stain the nuclei. The cells were washed twice with sterile PBS and fixed with 4% paraformaldehyde before imaging.
Doxorubicin-loaded CDNs cell viability assays
CDNDox from loading condition A were added to HeLa and HEK293 cells, both grown to 70% confluency in DMEM supplemented with 10% FBS. The cells were then incubated for 48 h at 37°C, 5% CO2, before removing the expended culture medium and washing twice with sterile PBS. The cells were then trypsinized, stained with trypan blue, and live cells were counted using a counting chamber. Free doxorubicin corresponding to the doxorubicin concentrations in CDNDox was used as the control.
Co-culture targeting experiments
1×104 HeLa and Hek293 cells were stained with CellTracker™ CM-DiI Dye and CellTracker™ Green CMFDA Dye, respectively, as per manufacturer’s recommendations. The cells were then washed, trypsinized, mixed, and added to a black 96-well plate format. The co-culture was allowed to incubate for 24 h before adding CDNdox, or an equivalent concentration of free doxorubicin. After incubation for 48 h, the co-culture was assayed using a standard plate reader. To quantify the percentage of targeting, the ratio of red to green fluorescence was then quantified with respect to the control (no treatment) after 48 h.
Statistical analysis
All statistical analyses were performed using IBM Statistical Package for the Social Sciences (SPSS) Version 21. Differences in cell viabilities for both mono-cultures and co-cultures were performed using one-way analysis of variance (ANOVA) statistical test, using Bonferroni post hoc tests. P-values <0.05 were considered to be significant.
Results and discussion
CDNs production and characterization
CDNs were prepared as described in an earlier publication. Briefly, U937 cells were sheared to nanodimensions by passing them through a series of 10 and 8 μm pore size membrane filters in a centrifuge at 14,000× g in a spin cup (Figure 1A and B).
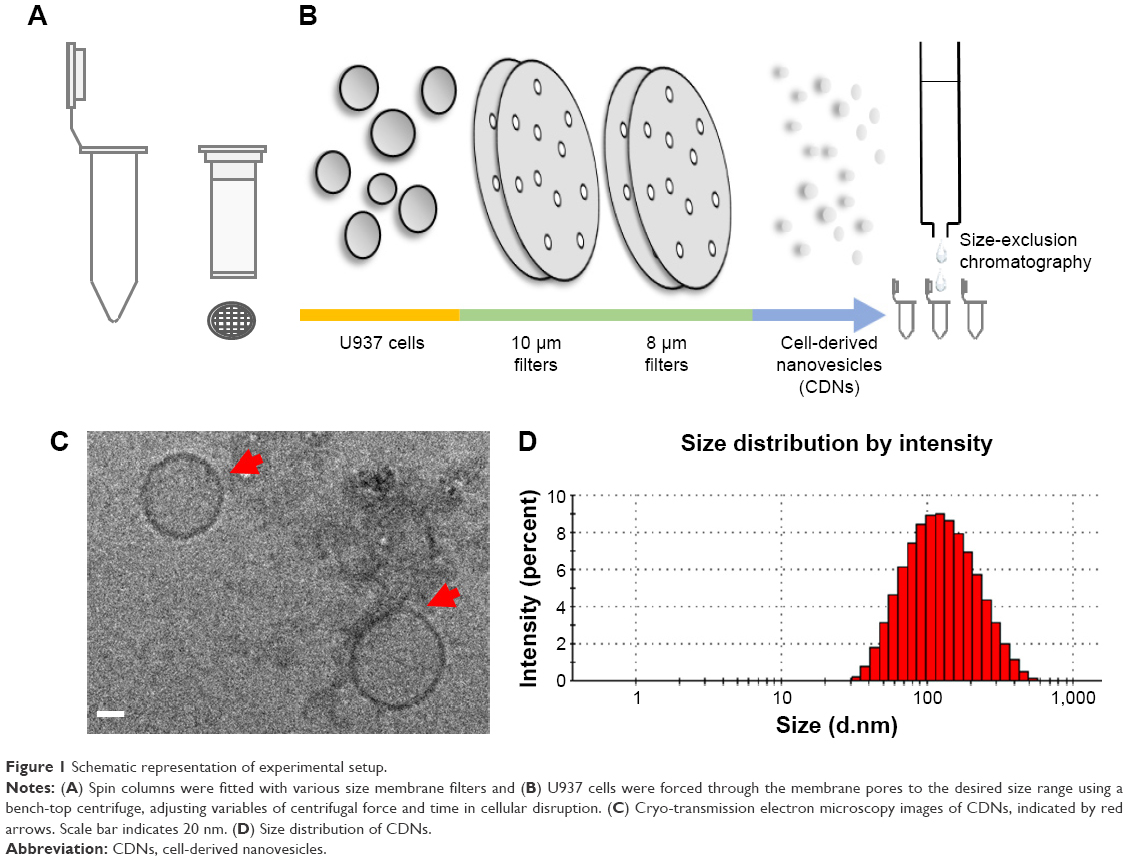 | Figure 1 Schematic representation of experimental setup. |
The CDNs were subsequently purified by size-exclusion chromatography and collecting the CDN fractions. The CDNs were then characterized by Cryo-transmission electron microscopy (TEM) (Figure 1C) to observe their morphological characteristics. The average size of the CDNs from this method was found to be about 93 nm, neutrally charged, and quantified by their protein concentration to be about 538 μg/mL (Figure 1D).
Doxorubicin loading of CDNs
Loading methods of CDNs were referenced from similar methods in the literature15 for loading of catalase into exosomes, as CDNs were previously shown to be similar in several aspects to exosomes. However, it was found from the preliminary loading of doxorubicin into CDNs that doxorubicin has a tendency to show signs of aggregation when added to CDNs. This, in turn, results in issues in quantifying the encapsulated doxorubicin concentration due to the tendency of doxorubicin to adhere to the surface of CDNs. Here, we demonstrated a cleaning protocol to remove doxorubicin that had adhered to the surface of CDNs, leaving behind only a solution of CDNs loaded with doxorubicin (Figure 2A). To our knowledge, this is an unprecedented improvement in the removal of unencapsulated small molecules from the surface of nanoparticles derived from natural sources, such as CDNs. The proposed cleaning protocol describes the use of protein concentrators to separate the adhered doxorubicin from CDNs by centrifuging at 2,000× g for 10 min, detaching the adhered doxorubicin from the CDNs surface and retaining the doxorubicin-loaded CDNs in the process.
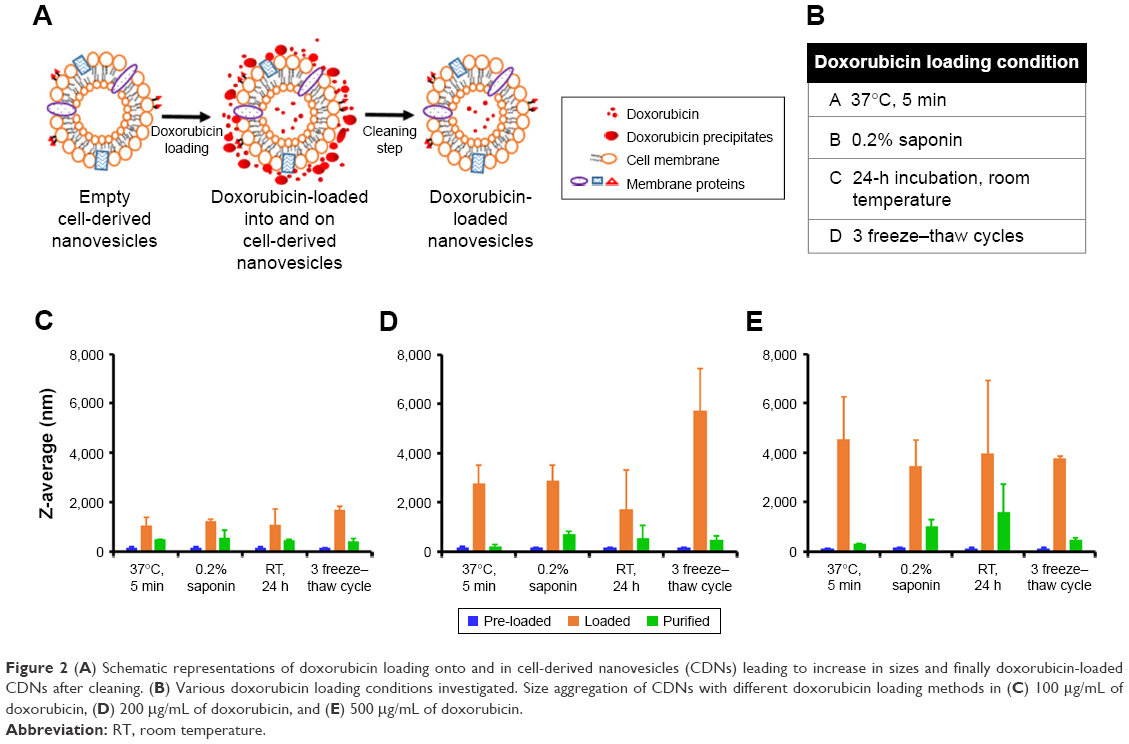 | Figure 2 (A) Schematic representations of doxorubicin loading onto and in cell-derived nanovesicles (CDNs) leading to increase in sizes and finally doxorubicin-loaded CDNs after cleaning. (B) Various doxorubicin loading conditions investigated. Size aggregation of CDNs with different doxorubicin loading methods in (C) 100 μg/mL of doxorubicin, (D) 200 μg/mL of doxorubicin, and (E) 500 μg/mL of doxorubicin. |
Four different loading methods of doxorubicin into CDNs were explored using three initial doxorubicin loading concentrations, namely 100, 200, and 500 μg/mL, as shown in Figure 2B. To assess the validity of our cleaning protocol, the average size of CDNs before loading, after loading, and after cleaning was reported (Figure 2C and E). The average size of CDNs showed a general increase in size after loading, but a marked decrease after cleaning, demonstrating that the adherence of doxorubicin to the CDNs surface contributed to its initial size increase and that our cleaning protocol significantly removed the adhered doxorubicin. The increased size after loading and its subsequent reduction due to removal of adhered doxorubicin were especially pronounced when a higher doxorubicin loading concentration (eg, 200 and 500 μg/mL) was used, further demonstrating the robustness of the cleaning method in removing adhered doxorubicin.
Loading efficiencies and release studies
Two important parameters were monitored after doxorubicin loading, namely the size of the CDNs and the encapsulation efficiencies of the four loading methods. Since the size of CDNs typically increased after drug loading, a balance between maximum drug loading and minimum size increase was sought after. The average sizes of CDNs after doxorubicin loading (Figure 3A) were compared against the encapsulation efficiencies of the respective loading methods (Figure 3B). While doxorubicin loading by addition of 0.2% saponin resulted in the highest loading efficiencies, possibly due to the increased permeabilization of the CDNs membrane, it also resulted in a corresponding marked increase in size. In comparison, doxorubicin loading by incubation at room temperature for 24 h on a shaker resulted in low loading efficiency but a large increase in size, suggesting that aggregation of CDNs occurred with doxorubicin acting as an intercalating agent. Interestingly, incubation of doxorubicin with CDNs at 37°C seemed to yield a balance between a marginal size increase, with a size of 210 nm after loading, and a modest drug loading efficiency of 16.5%, when a loading concentration of 200 μg/mL was used. Furthermore, it was observed that loading efficiencies increased when initial loading concentrations were increased from 100 to 200 μg/mL, but showed no further improvement when 500 μg/mL was used, suggesting an optimal loading concentration of 200 μg/mL.
 | Figure 3 Comparison of cell-derived nanovesicles (A) size aggregation with (B) doxorubicin loading efficiency among different loading conditions and doxorubicin concentrations. (C) Investigation of doxorubicin release profile of two formulations at 37°C for 36 h in phosphate-buffered saline. |
Hence, CDNs loaded with doxorubicin from two methods, namely incubation at 37°C (Method A) and with 0.2% saponin (Method B), were selected for further experimentation, to discriminate whether either the size of CDNs after doxorubicin loading or the loading efficiencies were more influential in the cellular uptake of doxorubicin loaded CDNs. Prior to in vitro experimentations, release studies of doxorubicin from the two formulations were performed in PBS at 37°C over 36 h to indicate the release profile of each formulation (Figure 3C). Free doxorubicin was used as a control, showing up to ~75% release due to some aggregation of the drug at the dialysis membrane. For both methods (A and B), the release profile that showed <50% of total drug is released over 36 h. This slow release profile should be considered as desirable in the context of drug delivery, as numerous DDS (eg, liposomes) are known to accumulate at the target site and subsequently release the therapeutics extracellularly. Conversely, our CDNs are expected to reach the diseased area and be taken up by the target cells together with the therapeutics at the cellular level. Hence, retaining the therapeutic within the CDNs could potentially mitigate off-target effects, reducing the possibility of adverse reactions in a clinical setting, especially in the case of doxorubicin, a drug known for its cardiotoxic side effect.
Although CDNs loaded with doxorubicin in the presence of 0.2% saponin (Method B) showed the highest loading efficiency, it was observed that only 20% of the total drug was released after 36 h. This was in contrast with CDNs loaded with doxorubicin upon incubation at 37°C (Method A), where 40% of the total drug was released by 36 h. Here, we postulate that the low drug release by CDNs loaded using Method B vis-à-vis Method A is due to two factors. First, the smaller size of CDNs from loading with Method A provided a larger surface area for doxorubicin to be released. Second, the large size of CDNs using Method B may be due to CDNs’ aggregation with doxorubicin acting as an intercalating agent. As a result of aggregation of CDNs from the addition of doxorubicin, the rate of release of doxorubicin through the dialysis membrane was decreased. Furthermore, doxorubicin is trapped between CDNs, aggregating CDNs together and may not be easily released. In addition, the large size (~700 nm) of CDNs associated with Method B may be an impediment for cell uptake in in vitro studies. Hence, CDNs loaded with doxorubicin by Method A may represent an optimal balance between the desired small size and high drug loading.
Cellular uptake studies
Using CDNs loaded with doxorubicin (CDNdox) from Method A, we were interested to examine if these drug-loaded nanovesicles were able to be taken up in an in vitro setting with some extent of selectivity. Here, cellular uptake of CDNdox by two cell lines, namely HeLa and HEK293, were examined. The cell lines were selected on the basis of the former being a cancer cell line, whereas the latter is a non-cancer cell line.
From the confocal microscopy images, it is clear that the CDNdox entered both cell lines and colocalized with the nuclei region (Figure 4A). Indeed, these images confirmed the properties of doxorubicin as described in the literature in its function as a DNA intercalator.17 Furthermore, the uptake profile of CDNdox and free doxorubicin in a Flow Activated Cell Sorting (FACS) assay seemed to indicate a similar uptake profile at both 1 and 6-h incubation time points, suggesting that CDNdox are taken up by the cells at a similar rate as free doxorubicin (Figure 4B). This is in contrast with the release study, where free doxorubicin crosses the semi-permeable membrane at almost twice the rate as CDNdox loaded from Method A. If CDNs were merely a synthetic DDS, a similar difference in cellular uptake would be expected. However, the cellular uptake of doxorubicin and CDNdox was almost similar at the two time points. Here, we postulate that the cell targeting properties of the CDNs were responsible for the improved cellular uptake.
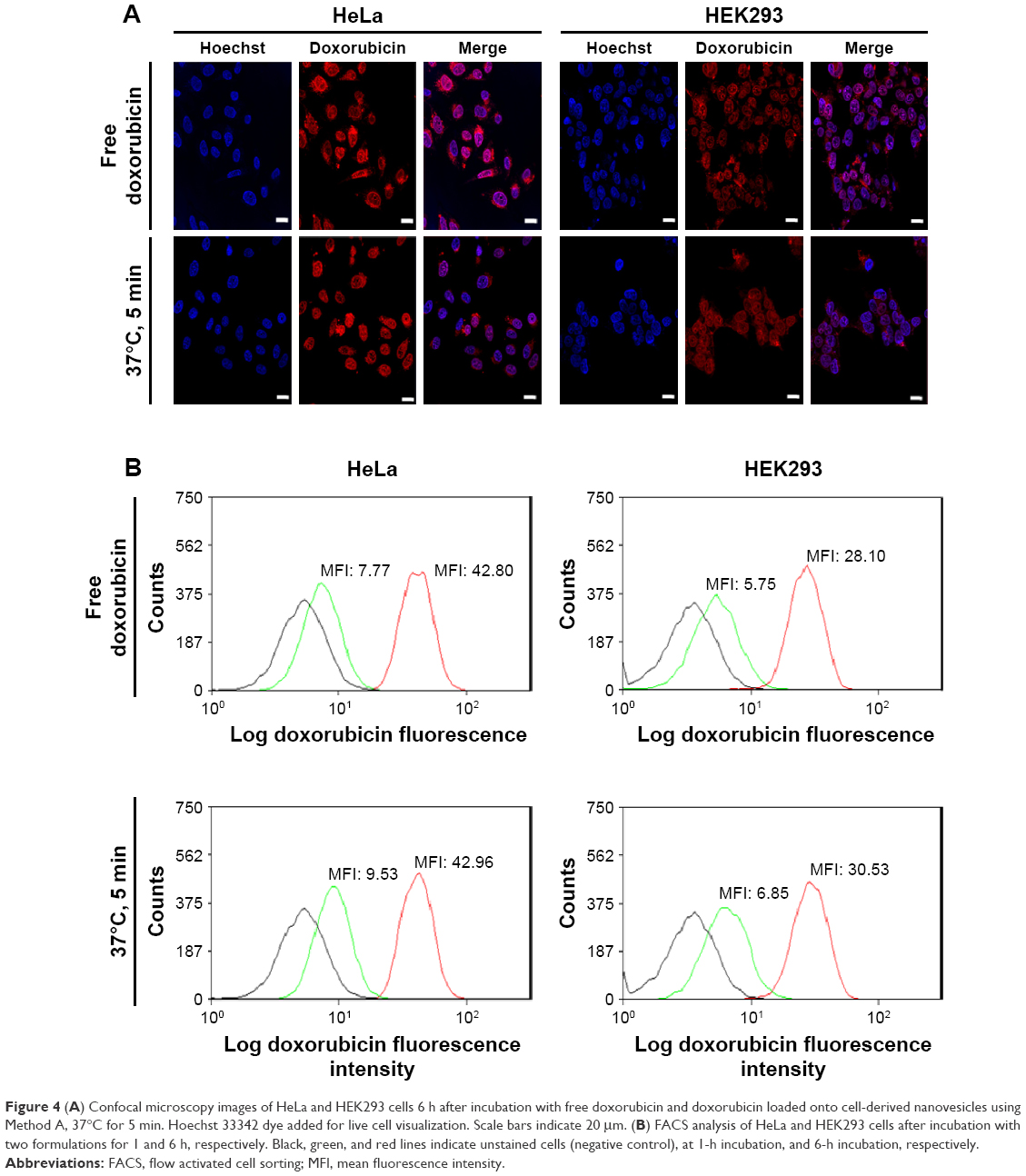 | Figure 4 (A) Confocal microscopy images of HeLa and HEK293 cells 6 h after incubation with free doxorubicin and doxorubicin loaded onto cell-derived nanovesicles using Method A, 37°C for 5 min. Hoechst 33342 dye added for live cell visualization. Scale bars indicate 20 μm. (B) FACS analysis of HeLa and HEK293 cells after incubation with two formulations for 1 and 6 h, respectively. Black, green, and red lines indicate unstained cells (negative control), at 1-h incubation, and 6-h incubation, respectively. |
Cell viability assays
We were also interested at assessing if the improved cellular uptake of CDNdox resulted in the required dose–response effect in terms of cell viability. This was elucidated by the addition of CDNdox to HeLa and HEK293 cell cultures at increasing concentrations. Free doxorubicin was added as a control. CDNs alone did not cause any cytotoxicity (data not shown), and all samples were normalized to the negative control of both cell lines.
As expected, the cell viability of both HeLa and HEK293 cells decreased with increasing doxorubicin concentration of both free doxorubicin and CDNdox (Figure 5A and B). However, a marked decreased in cell viability was observed at lower concentrations of CDNdox in HeLa but not in HEK293 cells as compared to free doxorubicin, where the cell viabilities of HeLa cells treated with free doxorubicin and CDNdox of the same concentration were almost similar. This suggests the preferential uptake by HeLa cells for CDNdox than HEK293, an effect that is especially pronounced when lower concentrations of CDNdox were used, indicating that a small dose of CDNdox may be used for an anticancer effect and yet leave non-cancerous cells almost unaffected. As HeLa cells are cancerous in origin whereas HEK293 cells are not, we hypothesize that the use of U937 monocyte-derived CDNs had a preferential uptake by cells targeted naturally by monocytes, such as cancer cells.
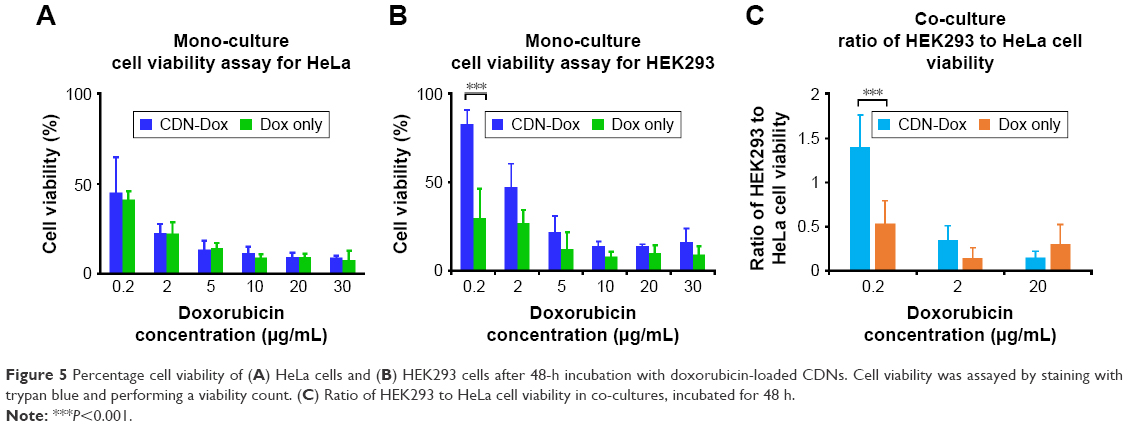 | Figure 5 Percentage cell viability of (A) HeLa cells and (B) HEK293 cells after 48-h incubation with doxorubicin-loaded CDNs. Cell viability was assayed by staining with trypan blue and performing a viability count. (C) Ratio of HEK293 to HeLa cell viability in co-cultures, incubated for 48 h. |
To further investigate this premise of preferential targeting of cancer cells, a co-culture of HeLa and HEK293 cells was grown and the CDNdox samples added (Figure 5C). Co-culture experiments were also used as a surrogate to proxy the in vivo tumor site, where the tumor is surrounded by a healthy tissue. Free doxorubicin of the same concentration was used as a control. The HeLa and HEK293 cells were previously stained with CellTracker CM-DiI Dye and CellTracker Green CMFDA Dye, respectively, to differentiate the cell types in a co-culture. It was observed that the CDNdox reduced the cell viability of HeLa, a cancer cell line, more than HEK293, a non-cancer cell line, with the greatest effect at the same dose of 0.2 μg/mL doxorubicin-loaded CDNs used in the monoculture experiments. This is in contrast to the effect of free doxorubicin, where the cell viability of both the cell lines are affected to a similar degree. Indeed, the results further exemplifies the cancer targeting potential of CDNs, in both monocultures and co-cultures, an effect we attribute to the retention of original monocytes’ membrane surface from which the CDNs were derived. This, in turn, suggests the potential of CDNs to be used as an antitumor strategy that targets the tumor cells while minimizing excessive deleterious effects on otherwise healthy tissue.
Conclusions
CDNs have being actively investigated as an alternative anticancer DDS. Here, we examined the various loading methods of a chemotherapeutic agent, doxorubicin hydrochloride, into CDNs. We also proposed an unprecedented cleaning protocol to remove unencapsulated doxorubicin or drugs of similar characteristics from the surface of biologically derived DDS. More importantly, we demonstrated the preferential targeting of CDNs derived from monocytes as a viable antitumor DDS.
Acknowledgments
This work was supported by the NUS, Department of Pharmacy (AcRF Tier 1 FRC grant R-148-000-213-112). Wei Jiang Goh gratefully acknowledges NUS Graduate School for Integrative Sciences and Engineering for financial support of graduate studies. Choon Keong Lee and Shui Zou acknowledges the Department of Pharmacy for support of graduate studies. The authors thank Ms Ang Kah Hwee for her contributions as a final year student. The authors also thank A/P Gigi Chiu of NUS for use of DLS equipment, and Associate Professor Ong Wei Yi and Mr Sim Aik Yong for support from the Electron Microscopy Unit (Yong Loo Lin School of Medicine, NUS).
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Estanqueiro M, Amaral MH, Conceição J, Sousa Lobo JM. Nanotechnological carriers for cancer chemotherapy: the state of the art. Colloids Surf B Biointerfaces. 2015;126:631–648. | ||
Sajid MI, Jamshaid U, Jamshaid T, Zafar N, Fessi H, Elaissari A. Carbon nanotubes from synthesis to in vivo biomedical applications. Inter J Pharm. 2016;501(1–2):278–299. | ||
Pérez-Herrero E, Fernández-Medarde A. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79. | ||
Zhu Y, Liao L. Applications of nanoparticles for anticancer drug delivery: a review. J Nanosci Nanotechnol. 2015;15(7):4753–4773. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Szebeni J, Storm G. Complement activation as a bioequivalence issue relevant to the development of generic liposomes and other nanoparticulate drugs. Biochem Biophys Res Commun. 2015;468(3):490–497. | ||
Fais S, Logozzi M, Lugini L. Exosomes: the ideal nanovectors for biodelivery. Biol Chem. 2013;394(1):1–15. | ||
Marcus ME, Leonard JN. FedExosomes: engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals (Basel). 2013;6(5):659–680. | ||
Jang SC, Kim OY, Yoon CM, et al. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano. 2013;7(9):7698–7710. | ||
Jo W, Kim J, Yoon J, Jeong D, et al. Large-scale generation of cell-derived nanovesicles. Nanoscale. 2014;6:12056–12064. | ||
Roth JC, Curiel DT, Pereboeva L. Cell vehicle targeting strategies. Gene Ther. 2008;15(10):716–729. | ||
Mahajan-Thakur S, Sostmann BD, Fender AC, et al. Sphingosine-1-phosphate induces thrombin receptor PAR-4 expression to enhance cell migration and COX-2 formation in human monocytes. J Leukoc Biol. 2014;96(4):611–618. | ||
Tian Y, Li S, Song J, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35(7):2383–2390. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Haney MJ, Klyachko NL, Zhao Y, et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Control Release. 2015;207:18–30. | ||
Fülöp Z, Gref R, Loftsson T. A permeation method for detection of self-aggregation of doxorubicin in aqueous environment. Int J Pharm. 2013;454(1):559–561. | ||
Arora HC, Jensen MP, Yuan Y, et al. Nanocarriers enhance doxorubicin uptake in drug-resistant ovarian cancer cells. Cancer Res. 2012;72(3):769–778. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.