Back to Journals » Cancer Management and Research » Volume 11
Downregulated MCOLN1 Attenuates The Progression Of Non-Small-Cell Lung Cancer By Inhibiting Lysosome-Autophagy
Authors Yin C, Zhang H, Liu X, Zhang H, Zhang Y, Bai X, Wang L, Li H, Li X, Zhang S, Zhang L, Zhang Y
Received 21 May 2019
Accepted for publication 6 September 2019
Published 23 September 2019 Volume 2019:11 Pages 8607—8617
DOI https://doi.org/10.2147/CMAR.S216538
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Video abstract presented by Yin C.
Views: 152
Chuntong Yin,1,* Han Zhang,1,* Xin Liu,2 Haiying Zhang,2 Yue Zhang,2 Xue Bai,2 Lei Wang,2 Huimin Li,2 Xia Li,2 Shuqian Zhang,2 Linyou Zhang,1 Yong Zhang2
1Department of Thoracic Surgery, The Second Affiliated Hospital of Harbin Medical University, Harbin 150086, People’s Republic of China; 2Department of Pharmacology (The State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), College of Pharmacy, Harbin Medical University, Harbin 150081, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Linyou Zhang
Department of Thoracic Surgery, The Second Affiliated Hospital of Harbin Medical University, Harbin 150086, People’s Republic of China
Email [email protected]
Yong Zhang
Department of Pharmacology (The State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), College of Pharmacy, Harbin Medical University, Harbin 150081, People’s Republic of China
Email [email protected]
Objectives: Autophagy plays various roles in non-small-cell lung cancer (NSCLC). MCOLN1, a reactive oxygen species sensor, can regulate autophagy via lysosomal Ca(2+); however, the role of MCOLN1 in NSCLC is largely unknown. This study aimed to explore the effects of MCOLN1 on proliferation, invasion and migration in NSCLC and the underling mechanisms.
Materials and methods: The tissues of NSCLC patients were collected, then MCOLN1 expression in tumor and adjacent tissues was measured and its relationship with pathological staging was analyzed. The Cell Counting Kit-8 (CCK-8) assay, wound healing assay and transwell migration assay were used to evaluate the proliferation, migration and invasion ability, respectively. Live-cell imaging and transmission electron microscopy (TEM) were used to observe autophagic flux and autolysosomes.
Results: It was found that MCOLN1 expression was significantly decreased in human NSCLC tissues compared with normal lung tissues while more MCOLN1 in stage III–IV was shown than stage I–II, indicating that MCOLN1 increased along with the progression of NSCLC. Furthermore, CCK-8 assay, wound healing assay and transwell migration assay confirmed that the inhibition of MCOLN1 suppressed NSCLC cells proliferation migration and invasion. Overexpression of MCOLN1 promoted autophagy in A549 and H1299 cells with increased LC3-II/I, lamp1 expression and autolysosomes as well as autophagic flux shown by live-cell imaging and TEM.
Conclusion: Our study shows that downregulated MCOLN1 reduced lysosome-autophagy activity contributing to inhibited tumor progression, which reveals a novel role of MCOLN1 in NSCLC, and targeting MCOLN1 may be a therapeutic potential for NSCLC.
Keywords: MCOLN1, NSCLC, autophagy, lysosome, progression
Introduction
Non-small-cell lung cancer (NSCLC) accounts for the majority of lung cancer, which to date remains the primary cause of cancer death worldwide.1 However, the mechanisms that contribute to NSCLC development have yet to be fully elucidated. Autophagy is an evolutionally conserved lysosomal degradation approach that plays a crucial role in tissue homeostasis and disease.2 Dysfunctional autophagy contributes to many diseases, as well as cancer.3 In cancer, autophagy can be neutral, tumor-suppressive or tumor-promoting in different contexts.4 As autophagy is a process in which the cytoplasmic proteins or organelles of the body engulf itself into vesicles and fuse with lysosomes to form autophagic lysosomes to degrade the contents, it is clear that lysosomes give an essential role in autophagic clearance. Lysosomes are organelles that break down biomacromolecules, such as proteins, nucleic acids and polysaccharides. They are located at the end of the endocytic pathway and participate in many key biological processes, including autophagy.5 The function of lysosome is critically dependent on both soluble and endolysosomal (ES) membrane proteins such as ion channels and transporters. Transient receptor potential (TRP) channel superfamily, namely TRPML channels (MCOLN), is an important member of cation channels in ES. Non-selective ES two-pore cation channels have been found to facilitate cancer migration,6,7 however, the exact effects of MCOLN on cancer progression are still unclear.
The mammalian target of rapamycin (mTOR), a nutrient sensor that resides on lysosomal membranes, is regarded as the master regulator of growth.8 Under starvation conditions, inhibition of mTOR results in a decrease in the phosphorylation of transcription factor (TF)EB, a master transcriptional regulator of both autophagy and lysosomal biogenesis.5,9–11 For another, AMPK (AMP-activated protein kinase) directly activate Ulk1 through phosphorylation of Ser 317 and Ser 777, then the activated ULK phosphorylates Beclin, DENND3 and ATG9 to initiate autophagy.12–14 Under nutrient sufficiency, TFEB is phosphorylated in Ser 211 in an mTOR-dependent manner, and high mTOR activity prevents Ulk1 activation by phosphorylating Ulk1 Ser 757 and disrupting the interaction between Ulk1 and AMPK.15 Recently, it has been proved that an elevation of ROS levels induces MCOLN1 activation and lysosomal Ca2+ release.16 Lysosomal Ca2+ leads to TFEB-nuclear translocation. This progression promotes autophagosome biogenesis and lysosome biogenesis. Similar as mTOR, MCOLN1 may also directly influence lysosome biogenesis and autophagosome–lysosome fusion via regulating lysosomal Ca2+. To sum up, we considered that MCOLN1 is a ROS sensor localized on the lysosomal membrane which arranges lysosomal autophagy to alleviate oxidative stress in cells. Moreover, defective autophagy is implicated in human pathology, as disruption of protein and organelle homeostasis enables disease-promoting mechanisms, such as toxic protein aggregation, oxidative stress, genomic damage, and inflammation.17 Thus, we hypothesize that MCOLN1 could mediate lysosomal autophagy to reduce intracellular ROS activity and promote the progression of NSCLC. In this study, we firstly identified the influence of MCOLN1 on the proliferation, migration and invasion of NSCLC cells using Cell Counting Kit-8 (CCK-8) assay, wound healing assay and transwell migration assay. Then by knocking-down or overexpression of MCOLN1, the relationship between MCOLN1 and lysosome-autophagy was evaluated. This study aims to explore the effects of MCOLN1 on progression of NSCLC, which may potentially lay the foundation for designing targeted drugs that have clinical value for NSCLC.
Materials And Methods
Tumor And Normal Tissue Samples
Tumor and normal tissue samples were collected according to guidelines approved by the Ethics and Scientific Committees of Harbin Medical University. From August 2017 to July 2018, 52 fresh NSCLC specimens and corresponding normal specimens confirmed by pathology were collected from the Second Affiliated Hospital of Harbin Medical University. All patients had received informed consent before tissue collection. No patients had received radiotherapy or chemotherapy before surgery. The clinicopathological information of patients is summarized in Table 1.
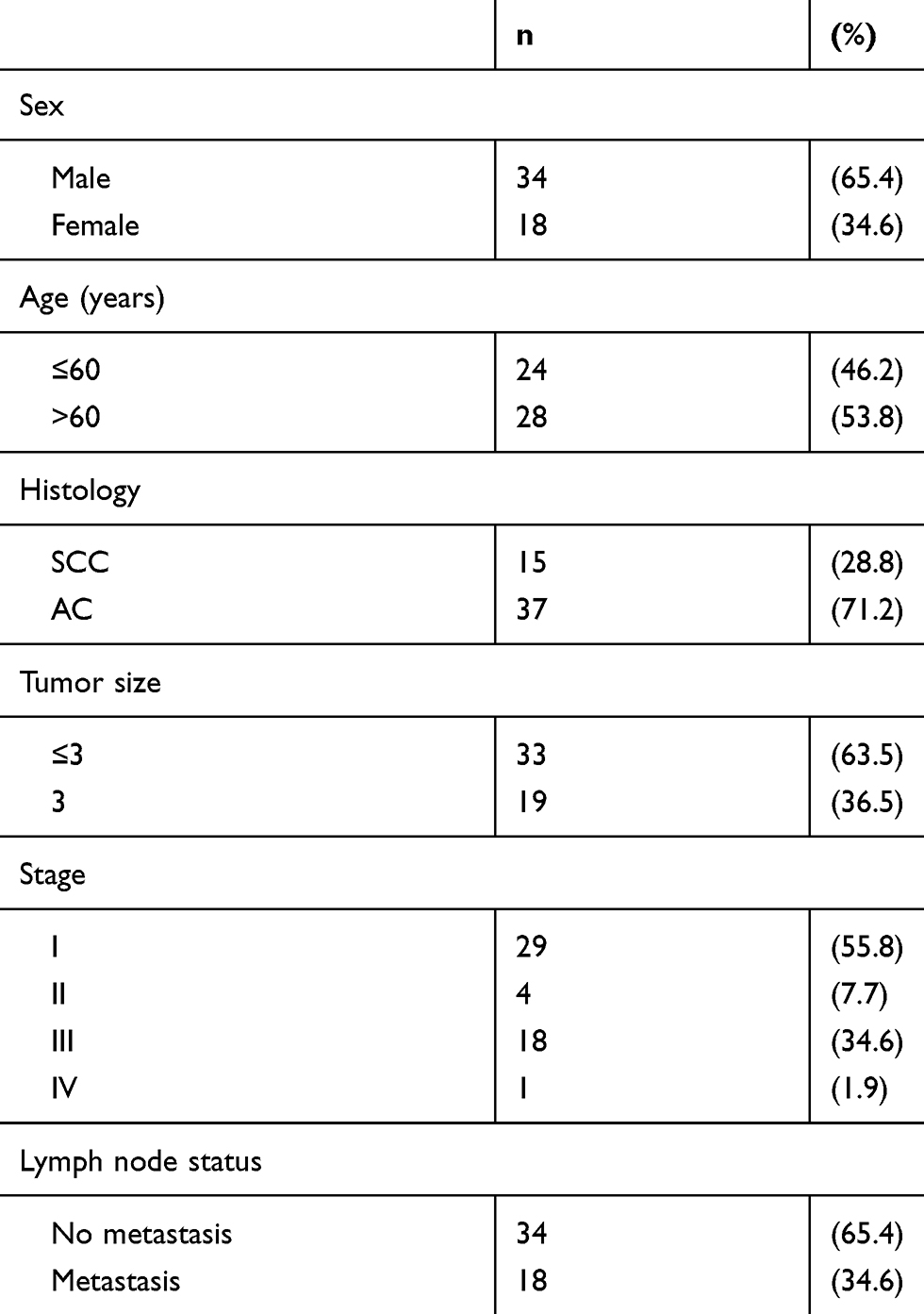 |
Table 1 Clinicopathological Parameters In 52 Fresh Samples Of Lung Cancers |
Cell Culture And Treatment
The human NSCLC cell line A549 and H1299 cell lines were from the American Type Culture Collection (Manassas, VA, USA). A549 cells were grown in DMEM containing 10% FBS at 37°C in 5% CO2. H1299 cells were grown in Roswell Park Memorial Institute medium containing 10% FBS at 37°C in 5% CO2. The cells were starved in serum-free medium for 24 hrs, and then transiently transfected with MCOLN1 mimic, MCOLN1 inhibitor, negative control and plasmid (abcam). We used X-treme RNA transfection reagent (Invitrogen, Ltd, America) for cell transfection.
RNA Extraction And Reverse Transcription‑Quantitative Polymerase Chain Reaction (RT‑qPCR)
Total RNA was harvested from tissues and cells using TRIzol reagent (Invitrogen, CA, USA). The levels of MCOLN1 mRNA were detected by qRT-PCR. RNA levels were quantitatively analyzed by SYBR Green Master Mix Kit (TOYOBO). GAPDH was selected as an internal control for MCOLN1. The following primers were used: MCOLN1 forward primer, 5′-TATGCGTATGTCCGTGGTGG-3′; MCOLN1 reverse primer, 5′-CCAAGAGGGTGAGATCGTCG-3′; GAPDH forward primer, 5′-AAGAAGGTGGTGAAGCAGGC-3′; GAPDH reverse primer, 5′ -TCCACCACCCAGTTGCTGTA-3′.
Cell Proliferation
Cell Counting Kit-8 (CCK-8) (Dojindo, Kumamoto, Japan) assay was used to measure NSCLC cell proliferation. Briefly, H1299 and A549 cells were transfected with MCOLN1 overexpressed plasmid or si-RNA and sent up an empty control group, then cultured in a 96-well plate for 24 hrs. Then, cells were incubated at 37°C for 1 hr in 10% CCK-8 reagent. Finally, the absorbance at 450 nm was recorded.
Cell Migration And Invasion
Wound healing assay and transwell migration assay were used to measure NSCLC cell migration and invasion. For wound healing assay, cells were seeded in six-well plates with medium to analyze wound healing. Twenty-four hours after transfection, A549 and H1299 cells were seeded into 6-well plates. Twenty-four-hours later, the cell monolayer was scratched with a plastic pipette tip. Cells were washed by PBS and cultured with serum-free medium for 24 hrs. Use a microscope to observe the scratches and take pictures. Image software (1.47v; National Institutes of Health, Bethesda, MD, USA) was used to calculate cell migration distance. For transwell assay, 8-μm pore size chambers (Corning, NY, USA) with an insert coated Matrigel (BD Bioscience) were used. Forty-eight hours after transfection, 200 μL cell suspension was added to the upper chamber, and 500 L culture medium containing FBS was added to the lower chamber. The number of migrated cells was detected after 48 hrs of culture under a phase-contrast microscope (Olympus, Japan).
Western Blot Analysis
Protein samples were lysed in RIPA buffer supplemented with protease inhibitor and the concentration was tested by BCA method (Beyotime, Shanghai, People’s Republic of China). A quantity of 100 μg protein samples were separated by SDS-PAGE gel electrophoresis (10% and 12%) then transferred to PVDF membranes. Primary antibodies against MCOLN1, P62, LC3-2A/B, Lamp (Abcam, Cambridge, MA, USA) were used, and GAPDH (anti-GAPDH from Kangcheng Inc., Shanghai, People’s Republic of China) as an internal control. The blotted proteins were detected and quantified using an Odyssey Infrared Imaging System (LI-COR, Lincoln, USA).
Immunofluorescence Staining And Confocal Microscopy
For immunofluorescence staining, tissues were fixed with 4% paraformaldehyde in PBS. Primary antibody against MCOLN1 (1:150) was incubated at 4°C overnight, after that secondary antibody was added for 1 hr at room temperature, DAPI (1:100) staining for 20 mins. The samples were then examined and analyzed by laser confocal microscopy (FV300, Olympus, Japan).18
Live-Cell Imaging For Autophagic Flux
The mRFP-GFP-LC3 adenovirus particles (Shanghai, People’s Republic of China) were used to infect cells and cultured for another 24 hrs. Imaging was performed by the VoX 3D living cell imaging system. Image analysis was conducted by Volocity Demo 5.4 software.
Transmission Electron Microscopy
After transfection for 24 hrs, the cells were fixed in 2.5% glutaraldehyde 0.1mol/L PBS (pH 7.4) for 2 hrs. The specimens were then washed with buffer solution, fixed with 1% OsO4 for 1–2 hrs, stained with uranyl acetate, dehydrated in ethanol, and embedded in epoxy resin according to the standard procedures. Ultrathin sections were stained with electron and observed under electron microscope (JEM-1220, JEOL Ltd., Tokyo, Japan).
Immunohistochemical Analysis
Tissue specimens were fixed in 4% buffered formalin for 8–24 hrs and embedded in paraffin. Specimens were dehydrated by an ascending series of ethanol and cleared with xylene. All sections were immunostained with primary antibodies against MCOLN1 at 4°C overnight. After incubation with secondary antibodies, the sections were stained with diaminobenzidine. Then, specimens were examined by light microscopy.19
Statistical Analysis
The data are expressed in the form of mean±SEM Student’s t-test was used to compare the two groups. Multiple groups were assessed by one-way ANOVA accompanied by Bonferroni’s multiple comparison test. A two-tailed P<0.05 was considered as significant difference. Data were dealt with the GraphPad Prism 5.0 software.
Results
Downregulated MCOLN1 In NSCLC
To study the effect of MCOLN1 on tumorigenesis of NSCLC, we first evaluated MCOLN1 expression in NSCLC tissues and matched normal lung tissues. Western blot results showed that compared with matched tissues, MCOLN1 was obviously downregulated in NSCLC tissues (Figure 1A). Likewise, with qRT-PCR, we got the same tendency (Figure 1B). However, the expression of MCOLN1 mRNA was increased in higher pathological staging (Figure 1C). We then tested MCOLN1 expression in several NSCLC cell lines (H460, H1299, A549, H1915, PC9) and normal bronchial epithelial cells (HBE). Compared with HBE, A549 and H1299 cells had the most significant upregulation of MCOLN1 mRNA (Figure 1D). Thus, A549 and H1299 cell lines were focused in subsequent experiments. For further observation, we also assessed protein levels with immunofluorescence and immumohistochemical staining, showing reduced MCOLN1 expression in NSCLC than in normal lung tissues (Figure 1E and F).
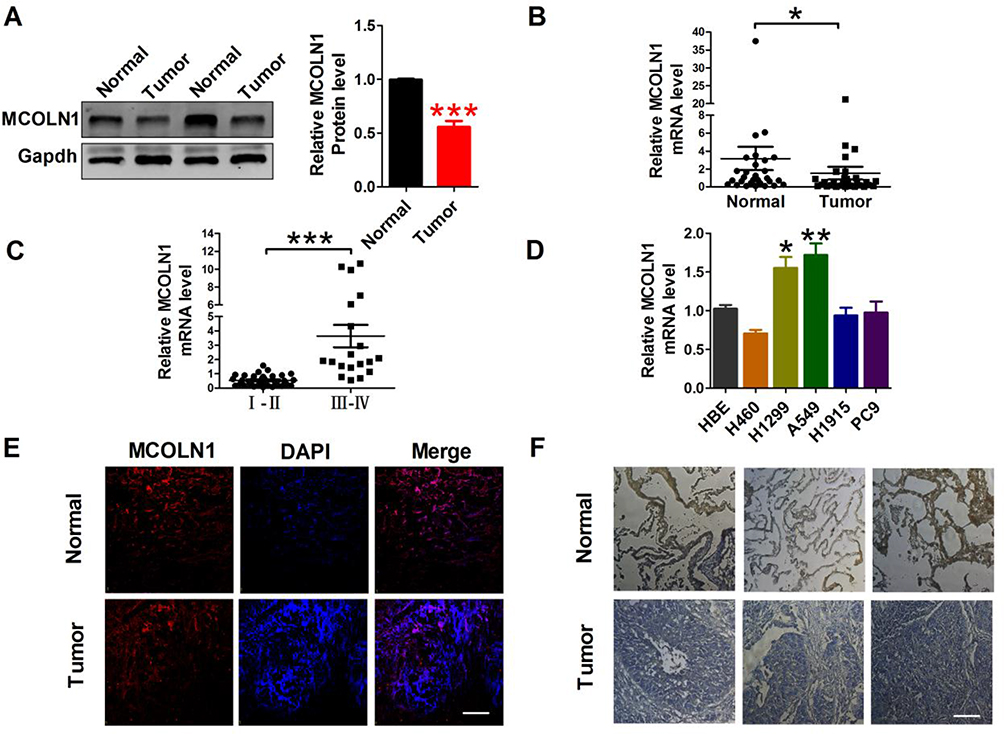 |
Figure 1 MCOLN1 expression in NSCLC tissues and cell lines. (A) The expression of MCOLN1 was analyzed by Western blot. ***P<0.001 vs normal. (B, C) The relative quantities of MCOLN1 mRNA in both NSCLC tissues and normal lung specimens were detected by qRT-PCR. *P< 0.05, ***P<0.001 vs normal or I–II stage group. (D) Expression levels of MCOLN1 mRNA in NSCLC cell lines and normal bronchial epithelial cells (HBE) were determined by qRT-PCR. *P<0.05, **P<0.01 vs HBE. (E) Immunofluorescence images (×400) showed the expression of MCOLN1 in normal lung specimens (control) and NSCLC tissues. blue: nuclear staining (DAPI); red: MCOLN1 staining; violet: merged images. (F) MCOLN1 expression of consecutive sections of lung specimens in control and NSCLC tissues assessed by immunohistochemical staining. Scale bar indicates 100 μm. Data are presented as the mean±SEM. |
Inhibition Of MCOLN1 Suppresses Cell Migration, Proliferation And Invasion In NSCLC Cells
To further investigate the functional role of MCOLN1 in migration, proliferation and invasion in NSCLC, we first knocked down MCOLN1 in A549 and H1299 cells. We used qRT-PCR assay to detect transfection efficiency (Figure 2A). CCK-8 assay was used to detect the activity of tumor cells. As shown in Figure 2B, siMCOLN1 suppressed cell viability in both cell lines. Besides, wound healing assay was performed to evaluate NSCLC cells migration and invasion. The results demonstrated that cells transfected with siMCOLN1 were not easy to heal compared with the NC group (Figure 2C and D). Consistent with wound healing assay, transwell migration assay showed that MCOLN1 inhibition led to a visibly decrease in NSCLC cell migration (Figure 2E and F). To sum up, these results indicated that MCOLN1 might act as a new target for tumor inhibition in NSCLC.
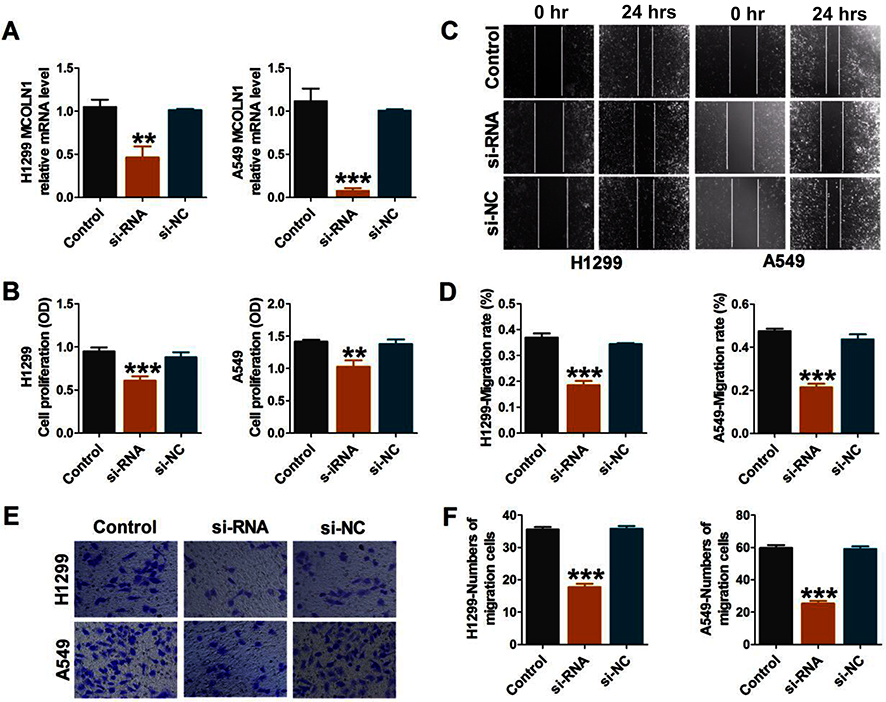 |
Figure 2 Inhibition of MCOLN1 suppresses cell migration, proliferation and invasion in NSCLC cells. (A) Cells were transfected with siMCOLN1, and the expression of MCOLN1 was analyzed by qRT-PCR. (B) A549 and H1299 cells were transfected with siMCOLN1 for 48 hrs and cell viability was measured with a CCK-8 assay. (C,D) Wound healing assay showed that MCOLN1 inhibitor resulted in a slower closing of scratch wounds. Scale bar 200 μm. (E, F) The effects of MCOLN1 on invasion of A549 and H1299 cells were assessed by transwell assay. The data are expressed as mean±SEM of five independent experiments. **P< 0.01, ***P<0.001 vs control. |
Overexpression Of MCOLN1 Promotes Migration, Proliferation And Invasion In NSCLC Cells
A549 and H1299 cells were transfected with MCOLN1 plasmid for MCOLN1 overexpression (Figure 3A). CCK-8 assay showed that the overexpression of MCOLN1 increased the viability of A549 and H1299 cells (Figure 3B). In addition, MCOLN1 overexpression accelerated the scratch closing (Figure 3C and D) and promoted NSCLC cell invasion (Figure 3E and F). Collectively, these findings indicated that MCOLN1 might contribute to the progression of NSCLC.
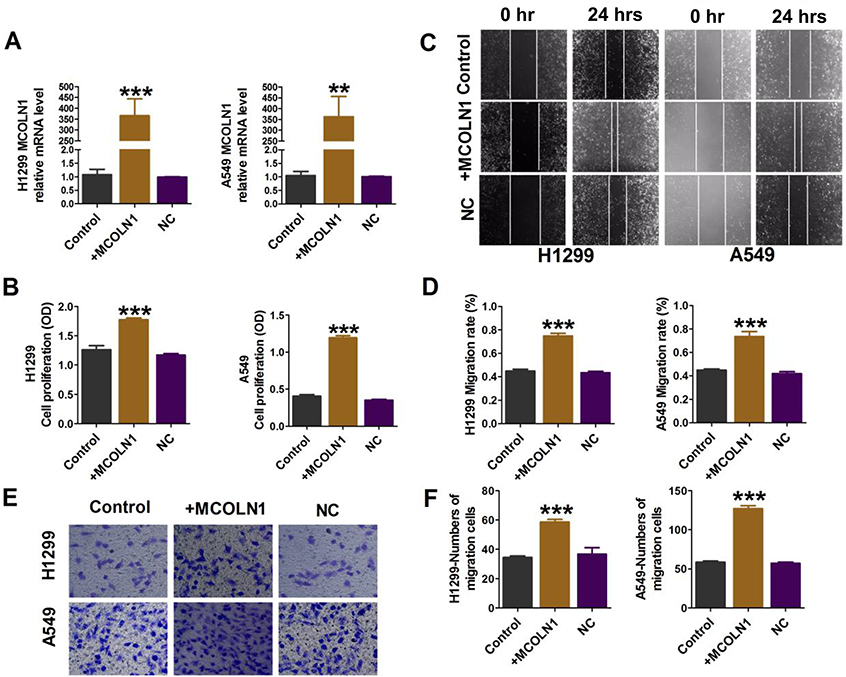 |
Figure 3 Overexpression of MCOLN1 promotes cell migration, proliferation and invasion in NSCLC cells. (A) Cells were transfected with MCOLN1-mimic, and the expression of MCOLN1 was analyzed by qRT-PCR. (B) The effects of MCOLN1 on cell viability were measured with a CCK-8 assay. A549 and H1299 cells were transfected with MCOLN1-mimic for 48 hrs. (C, D) Wound healing assay showed that MCOLN1 resulted in a faster closing of scratch wounds. Scale bar 200 μm. (E, F) The effects of MCOLN1 transfection on the invasion of A549 and H1299 cells were assessed by transwell assay. The quantitative presentation of the number of invasive cells. The data are expressed as mean±SEM of five independent experiments. **P< 0.01, ***P<0.001 vs control. |
MCOLN1 Increased Expression Of Autophagic Markers In NSCLC Cells
Autophagy plays vital roles in tumor cells. In order to clarify the sub-cellular mechanisms of MCOLN1 in NSCLC cells, autophagic markers were then investigated. As evidenced by Western blot analysis (Figure 4A and B), both H1299 and A549 cell lines with MCOLN1 siRNA had decreased conversion of LC3-I to LC3-II (the lipidated, autophagosome-associated form of LC3) and SQSTM1/p62 degradation. The protein expression level of lysosomal-associated membrane protein 1 (lamp1) was also decreased following transfection with MCOLN1 inhibitor. In contrast, the level of endogenous LC3-II and lamp1 were significantly increased by MCOLN1 overexpression in NSCLC cells, while the level of p62 was markedly decreased (Figure 4C and D). These results suggested that MCOLN1 facilitates lysosome function and autophagy level in NSCLC cells.
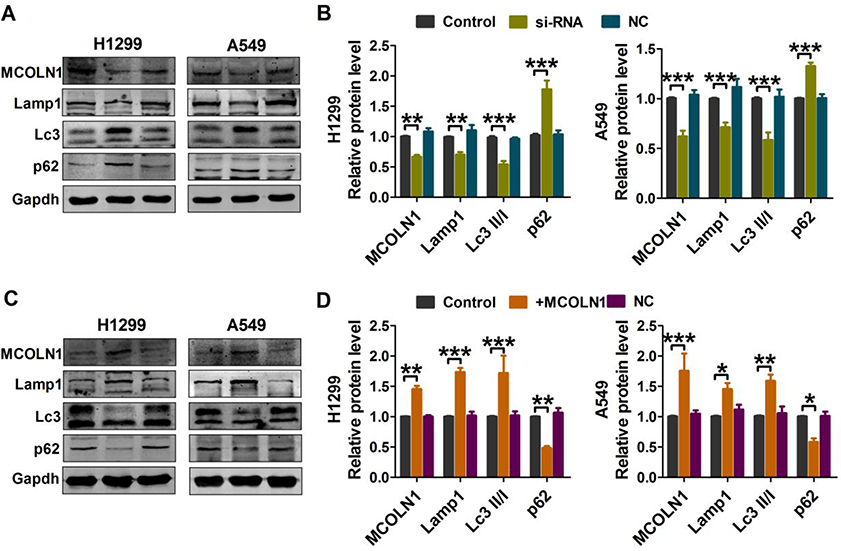 |
Figure 4 MCOLN1 affects lysosome function and autophagy level. (A, B) A549 and H1299 cells were transfected with siMCOLN1 for 48 hrs, and then Western blot was used for the analysis of autophagy and lysosomal function-related protein levels in NSCLC cells. (C, D) A549 and H1299 cells were transfected with MCOLN1-mimic for 48 hrs. Western blot analysis of autophagy and lysosomal function-related protein levels in NSCLC cells. The data are expressed as mean±SEM of five independent experiments. *P< 0.05, **P < 0.01, ***P<0.001 vs control. |
MCOLN1 Reinforces Autophagy In NSCLC Cells
To further confirm that MCOLN1 could enhance autophagy in NSCLC, cells were transfected with mRFP-GFP-LC3 adenovirus to monitor autophagic flux, a specific marker of autophagosomes (the yellow dots) and autolysosomes (the red dots), representing ongoing autophagy (Figure 5A). Cells treated with plasmid displayed significant increases in autophagic flux compared with the control group. On the contrary, cells transfected with MCOLN1 inhibitor, autophagic flux decreased compared with the control group (Figure 5B). In addition, transmission electron microscopy (TEM) revealed that overexpressing MCOLN1 increased number of autophagosomes and autolysosomes in NSCLC cells (Figure 5C). These data suggest that MCOLN1 could strengthen the autophagic flux in NSCLC cells. Altogether, autophagy may play a multifactorial role on the initiation and progression of NSCLC. Autophagy provides an anticarcinogenic function in normal tissues to maintain homeostasis. In established tumors, however, autophagy may promote tumor cell growth (Figure 6). As our results demonstrated, MCOLN1 is downregulated in NSCLC tissues while increased in higher pathological staging. Downregulation of MCOLN1 remarkably attenuates NSCLC cells progression by modulating the fuse of autophagosomes and lysosomes.
 |
Figure 5 MCOLN1 reinforces autophagic flux. (A, B) Representative images of fluorescent LC3 puncta after mRFP-GFP-LC3 adenovirus transduction (24 hrs). Mean number of autophagosomes represented by yellow puncta and autolysosomes represented by red puncta in merged images per cell. Scale bar indicates 10 μm. Results represent the mean value from 3 independent experiments, ***P<0.001. (C) Representative electronic micrograph images were shown. Scale bar indicates 500 nm. The arrows depict autolysosome, and the nucleus is denoted by N. n = 3. |
 |
Figure 6 Schematic diagram: autophagy may play a multifactorial role on the initiation and progression of NSCLC. Autophagy provides an anticarcinogenic function in normal tissues to maintain homeostasis. In established tumors, however, autophagy may promote tumor cell growth. |
Discussion
Autophagy is a process of phagocytosis of cytoplasmic proteins or organelles and endocytosis into vesicles and fusion with lysosomes to form autophagic lysosomes and degrade the enclosing contents, so as to achieve cell metabolism and the renewal of some organelles to maintain intracytoplasmic homeostasis.20–22 Emerging evidence suggests that autophagy exerts complex functions in human diseases that may include both protective and potentially deleterious processes.23 During the occurrence and development of tumors, the relevant genetic changes, including both the activation or upregulation of oncogenes and the deletion, mutation or inactivation of tumor suppressor genes, all have negative regulatory effects on autophagy.24 Cancer is both the first disease that existed a deficiency in autophagy and, studies of the role of autophagy in tumors are somewhat contradictory, for which clinical trials are being carried out in patients to inhibit autophagy.25 The contradictory results may be explained by the different roles played by autophagy at different stages of tumorigenesis. On the one hand, tumors formed by autophagy-deficient cells display genomic instability and DNA damage, which is in part mediated by ROS.26 On the other hand, there is increasing evidence that autophagy may be a necessary condition for tumor progression.27,28 Autophagy provides a survival advantage to tumor cells by enabling them to overcome the metabolic stress that is inherently present in the tumor microenvironment. The prevailing current view is that autophagy as a pro-survival pathway that helps tumor cells endure metabolic stress. Importantly, similar to our findings, inhibition of autophagy enhanced the therapeutic efficacy in NSCLC cells.19 Since MCOLN1 is a ROS sensor in lysosomes that regulates autophagy, we focused on the effect of MCOLN1 on NSCLC. Autophagy is an important part of tumor self-protection mechanism, meanwhile the loss of autophagy function is also a necessary condition for the occurrence of tumor.29 Consistent with our results, MCOLN1 was remarkably downregulated in NSCLC tissues, while the expression of MCOLN1 was increased in higher pathological staging to maintain metabolic and energy balance. MCOLN1 may play a multifactorial role on the initiation and progression of NSCLC. MCOLN1 provides an anticarcinogenic function in normal tissues by safeguarding against metabolic stress,16 and our results showed more MCOLN1 in normal lung tissues than NSCLC tissues. Genetic deletion of MCOLN1 may cause cells dysfunction, increased oxidative stress, and susceptibility to inflammatory that permit DNA damage and thus lead to genetic instability and tumorigenesis. In established tumors, however, MCOLN1 may confer a survival advantage on tumor cells. In our study, we identified MCOLN1 mediates lysosomal autophagy function to enhance the proliferation, migration and invasion of NSCLC cells.
Our data also showed that the ability of tumor cells to migrate and invade was significantly reduced when MCOLN1 was downregulated. P62 is a protein associated with autophagosomes. After the fusion of autophagosome and lysosome, it will be degraded in lysosomes.30 P62 accumulation suggests the inhibition of autophagy. As a lysosomal marker, lamp1 is a lysosomal membrane protein capable of maintaining lysosomal acidification.31 In our experiment, upregulated MCOLN1 induced significant increase of both lamp1 and LC3-II/I but decrease of P62, which indicates positive regulatory effects of autophagy. These data provide evidence that fate decision of MCOLN1 might depend on its influence on autophagy in NSCLC cells. At the same time, we further confirmed that MCOLN1 triggers increased autophagic flux in NSCLC cells. In order to further verify the ability of MCOLN1 to promote autophagy in NSCLC cells, we infected NSCLC cells with mRFP-GFP-LC3 adenovirus and detected the autophagy flux rate by living cell imaging. We found that in NSCLC cells treated with MCOLN1 plasmid, the abnormal expression of mRFP-GFP-LC3 was red and yellow spots, which was higher than that in cells simulated with MCOLN1, indicating that MCOLN1 mediated the accumulation of autophagosomes. TEM revealed that overexpressing MCOLN1 increased number of autophagosomes and autolysosomes in NSCLC cells. Since increased autophagosome formation does not lead to an increase in autophagosome flux, a MCOLN1-dependent mechanism must exist to regulate lysosome. These data suggest that MCOLN1 induces autophagic flux, whereas silencing MCOLN1 suppresses the autophagic flux. Given the established roles of MCOLN1, which both affects lysosomal trafficking and biogenesis and operates on mitigated oxidative stress through TFEB, we identified MCOLN1 as a factor in both initiation and maturation of autophagy.32 Only when MCOLN1 can be activated normally, autophagosomes and lysosomes can fuse normally and the autophagy process can proceed smoothly. As shown in our mRFP-GFP-LC3 adenoviral autophagy flow results, when MCOLN1 is downregulated in NSCLC cells, autophagosomes in cells fail to fuse with lysosomes indicating autophagy blocking. Therefore, the co-localization of green fluorescence and red fluorescence mixtures in cells, that is why yellow fluorescence occurs. When MCOLN1 is upregulated, autophagosomes fuse with lysosomes and autophagy is activated. The green fluorescence signal of GFP will be quenched after entering the lysosome due to the decrease of pH value and the cells are mainly red fluorescence.
Recent years, some researchers have focused tumors on autophagy and immune response. Autophagy has been proved to help tumors to adapt to harsh microenvironment and to reduce the sensitivity of tumor cells to radiotherapy and chemotherapy.33,34 Enhanced autophagy in tumor cells can be induced by radiotherapy, chemotherapy or other stimulating factors, including hypoxia and nutrient starvation.35 Autophagy will be occurred when treating with chemotherapy drugs in tumor cells. Dying tumor cells caused by chemotherapy drugs always release ATP and recruit both dendritic cells and T cells to enter tumor regions, which cause immunogenic cell death in tumor cells. On the other hand, autophagy inhibitors can inhibit the release of ATP by dying tumor cells. In autophagy-deficient tumor cell lines, the extracellular ATP degradation enzyme can be inhibited to increase the extracellular ATP concentration, and immune cells can be recruited again to restore the therapeutic effect of chemotherapy drugs.36 In addition, other studies have revealed that autophagy is closely related to cell senescence.37 The results of this study show, for the first time, that increase of MCOLN1 will boost autophagy in NSCLC cells and promote tumor progression. As we all know, aging individuals are often associated with impaired lysosomal function. Considering that MCOLN1 plays a role in regulating lysosomal function, whether MCOLN1 regulates senescence in worm and mouse models is particularly interested and further studies will be focused on if increasing MCOLN1 will open up novel paths to extending lifespan.
Taken together, we report the novel role of MCOLN1 in autophagy regulation. Our results demonstrate that MCOLN1 is downregulated in NSCLC tissues while increased in higher pathological staging. Downregulation of MCOLN1 remarkably attenuates NSCLC cells progression by modulating autophagy. In summary, our results revealed a new mechanism of MCOLN1 by influence autophagy to play an anti-tumor role in NSCLC cells, which might be a potential therapeutic for NSCLC treatment.
Ethics
The Ethics and Scientific Committees of Harbin Medical University approved this study, and that all patients provided written informed consent, in accordance with the Declaration of Helsinki.
Acknowledgments
This study was supported by the National Nature Science Foundation of China (81773735) and the Postdoctoral Science Foundation of China (2018M641880).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387
2. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi:10.1016/j.cell.2011.10.026
3. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi:10.1056/NEJMra1205406
4. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–1930. doi:10.1101/gad.287524.116
5. Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477. doi:10.1126/science.1174447
6. Grimm C, Butz E, Chen CC, Wahl-Schott C, Biel M. From mucolipidosis type IV to ebola: TRPML and two-pore channels at the crossroads of endo-lysosomal trafficking and disease. Cell Calcium. 2017;67:148–155. doi:10.1016/j.ceca.2017.04.003
7. Nguyen ON, Grimm C, Schneider LS, et al. Two-pore channel function is crucial for the migration of invasive cancer cells. Cancer Res. 2017;77(6):1427–1438. doi:10.1158/0008-5472.CAN-16-0852
8. Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283–296. doi:10.1038/nrm3565
9. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–914. doi:10.4161/auto.19653
10. Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5(228):ra42. doi:10.1126/scisignal.2002790
11. Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J. 2012;31(5):1095–1108. doi:10.1038/emboj.2012.32
12. Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750. doi:10.1038/ncb2757
13. Xu J, Fotouhi M, McPherson PS. Phosphorylation of the exchange factor DENND3 by ULK in response to starvation activates Rab12 and induces autophagy. EMBO Rep. 2015;16(6):709–718. doi:10.15252/embr.201440006
14. Papinski D, Schuschnig M, Reiter W, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53(3):471–483. doi:10.1016/j.molcel.2013.12.011
15. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:10.1038/ncb2152
16. Zhang X, Cheng X, Yu L, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109. doi:10.1038/ncomms12109
17. Ozpolat B, Benbrook DM. Targeting autophagy in cancer management - strategies and developments. Cancer Manag Res. 2015;7:291–299. doi:10.2147/CMAR.S34859
18. Tohme S, Simmons RL, Tsung A. Surgery for cancer: a trigger for metastases. Cancer Res. 2017;77(7):1548–1552. doi:10.1158/0008-5472.CAN-16-1536
19. Lu X, Zhou D, Hou B, et al. Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer. Cancer Manag Res. 2018;10:1231–1241. doi:10.2147/CMAR.S156530
20. Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90(4):1383–1435. doi:10.1152/physrev.00030.2009
21. Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109(2):151–160. doi:10.1161/CIRCRESAHA.110.237339
22. Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45–59. doi:10.1146/annurev-physiol-021909-135757
23. Ryter SW, Choi AM. Autophagy in lung disease pathogenesis and therapeutics. Redox Biol. 2015;4:215–225. doi:10.1016/j.redox.2014.12.010
24. Vera-Ramirez L, Vodnala SK, Nini R. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nature Communications. 2018;9(1):1944. doi:10.1038/s41467-018-04070-6
25. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10(9):1533–1541. doi:10.1158/1535-7163.MCT-11-0047
26. Mathew R, Kongara S, Beaudoin B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21(11):1367–1381. doi:10.1101/gad.1545107
27. Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–730. doi:10.1038/nrd3802
28. Liu J, Debnath J. The evolving, multifaceted roles of autophagy in cancer. Adv Cancer Res. 2016;130:1–53. doi:10.1016/bs.acr.2016.01.005
29. Nassour J, Radford R, Correia A, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. 2019;565(7741):659–663. doi:10.1038/s41586-019-0885-0
30. Gu X, Han M, Du Y, et al. Pb disrupts autophagic flux through inhibiting the formation and activity of lysosomes in neural cells. Toxicol In Vitro. 2019;55:43–50. doi:10.1016/j.tiv.2018.11.010
31. Cook NR, Row PE, Davidson HW. Lysosome associated membrane protein 1 (Lamp1) traffics directly from the TGN to early endosomes. Traffic. 2004;5(9):685–699. doi:10.1111/j.1600-0854.2004.00212.x
32. Wang W, Gao Q, Yang M, et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci U S A. 2015;112(11):E1373–E1381. doi:10.1073/pnas.1419669112
33. Ramakrishnan R, Huang C, Cho HI, et al. Autophagy induced by conventional chemotherapy mediates tumor cell sensitivity to immunotherapy. Cancer Res. 2012;72(21):5483–5493. doi:10.1158/0008-5472.CAN-12-2236
34. Liang ZG, Lin GX, Yu BB, et al. The role of autophagy in the radiosensitivity of the radioresistant human nasopharyngeal carcinoma cell line CNE-2R. Cancer Manag Res. 2018;10:4125–4134. doi:10.2147/CMAR.S176536
35. Weiner LM, Lotze MT. Tumor-cell death, autophagy, and immunity. N Engl J Med. 2012;366(12):1156–1158. doi:10.1056/NEJMcibr1114526
36. Arroyo DS, Gaviglio EA, Peralta Ramos JM, Bussi C, Rodriguez-Galan MC, Iribarren P. Autophagy in inflammation, infection, neurodegeneration and cancer. Int Immunopharmacol. 2014;18(1):55–65. doi:10.1016/j.intimp.2013.11.001
37. Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi:10.1016/j.cell.2011.07.030
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.