Back to Journals » Cancer Management and Research » Volume 12
Double Agent: SPDEF Gene with Both Oncogenic and Tumor-Suppressor Functions in Breast Cancer
Authors Ye T ![]() , Feng J, Wan X, Xie D, Liu J
, Feng J, Wan X, Xie D, Liu J ![]()
Received 25 December 2019
Accepted for publication 25 April 2020
Published 25 May 2020 Volume 2020:12 Pages 3891—3902
DOI https://doi.org/10.2147/CMAR.S243748
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Ting Ye,* Jia Feng,* Xue Wan, Dan Xie, Jinbo Liu
Department of Laboratory Medicine, The Affiliated Hospital of Southwest Medical University, Sichuan 646000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jinbo Liu
Department of Laboratory Medical, The Affiliated Hospital of Southwest Medical University, 25 Taiping Street, Luzhou 646000, Sichuan, People’s Republic of China
Tel +86830 3165730
Email [email protected]
Abstract: The dichotomy of cancer-regulatory genes into “oncogenes (OCGs)” and “tumor-suppressor genes (TSGs)” has greatly helped us in learning molecular details of tumor biology. SPDEF, known as the prostate-derived ETS factor, is reported to play a pivotal role in normal cell development and survival, which has also been endowed with dual characteristics in cancers. Breast cancer (BC) is a highly heterogeneous disease which becomes the leading reason for cancer-related fatality among women worldwide. The involvement of SPDEF in many aspects of BC has been postulated, whereas the mechanism governing the regulation of the pro- and anti-oncogenic activities of SPDEF in BC state remains poorly defined. In this review, we summarized SPDEF as the double agent involving in expression profiles, the regulatory mechanism in BC progression, as well as the role in diagnosis, treatment and prognosis of BC. The understanding of SPDEF duality has contributed to gain insight into the tumor biology and also add a new dimension to the new therapy targets for BC.
Keywords: SPDEF, double agent, oncogenes, tumor-suppressor genes, transcription factor, breast cancer
Introduction
As one of the most prevalent cancers, BC is the leading reason for cancer-related fatality among women worldwide.1 Moreover, BC is a highly heterogeneous disease which not only brings difficulty to mechanisms research but also hinders the development of molecularly targeted drugs in clinical practice. In order to organize this heterogeneity, multiple BC classification systems have been developed. Especially, BC can be clustered into four subtypes including triple-negative, human epidermal growth factor receptor 2 positive (Her2+), luminal A and luminal B tumors according to gene expression profiling.2 Triple-negative breast cancer (TNBC) is characterized by the absence of estrogen receptor (ER), progesterone receptor (PR) and Her2, which exhibits commonly more aggressive and invasive. Since luminal A and luminal B are both typically ER+, the major difference of the two types is the higher ki-67 expression in luminal B BC.3 As for Her2+ BC, it is driven by overexpression of Her2 and genes associated with related pathways or the Her2 amplicon on chromosome 17q12,4 which has been shown to have a worse outcome than ER+ BC.5
More recently, the double-agent nature of genes has gradually been discovered to be one of the main reasons behind the heterogeneity, as well as the main mechanism underlying these in-tumor adversities.6 Targeted treatment for one gene or gene product may be effective for certain cancer cells, but it may also promote the survival or progression of other cells leading to tumorigenesis. Therefore, understanding the duality genes will further help us in selecting target molecules for the treatment of different BC sub-populations. Dichotomy of cancer-regulatory genes into two opposed classes in the tumorigenesis: “oncogenes (OCGs)” and “tumor-suppressor genes (TSGs)”. OCGs can induce a series of gene expressions related to cell growth and differentiation, leading to uncontrolled growth of normal cells, and eventually become cancer cells. On the contrary, TSGs protect normal cells from degradation into cancer cells. They appear to be two opposite gene classes in the tumorigenesis. Paradoxically, some certain genes exhibit carcinogenic and tumor-suppressing functions at the same time.7–10 For instance, p53 is recognized as a tumor suppressor which inhibits the malignant growth of tumor cells.11–15 Conversely, germline mutation of p53 causes Li-Fraumeni syndrome, which is a familial cancer syndrome including BC, soft tissue sarcoma and various other types of cancer.16 And mutant p53 (p53-R248Q) functions as an oncogene in promoting endometrial cancer by up-regulating REGγ.17 KMT2D epigenetic regulator in B cell lymphoma acts as a tumor suppressor,18 while its knockdown slows the growth and sensitizes to PI3K inhibitor treatment in BC cells.19 Further, ARID1A is hypothesized to be tumor suppressive, but the same protein has opposing and stage-dependent roles within the same tissue, as Arid1a deletion can both impair20 and promote21 the initiation of liver tumorigenesis and accelerates progression and metastasis in established disease.21 In order to promote the rapid development of cancer research and ensure the emergence of new concepts that is conducive to research, it is necessary to provide a highly flexible data classification such as “double-agent genes”.
SPDEF gene is vital for normal cell development and survival, the role of which in BC has been controversial over the years. Firstly, current studies have shown that the dual function of SPDEF is mainly dependent on molecular subtypes of BC. In TNBC, SPDEF is confirmed as a TSG. The invasive inhibition of SPDEF mediated is related to the regulation of downstream target genes, such as decreasing the expression of epithelial-mesenchymal transformation (EMT) related gene urokinase-type plasminogen activator (uPA)22 and SLUG,23 and elevating the expression of cell cycle regulation-related protein p21.24 In Her2+ subtype, SPDEF can promote proliferation, migration and invasion of SK-BR-3 cells by AR-SPDEF pathway25 or SPDEF-CEACAM6 oncogenic axis26 as an OCG. Noteworthy, for luminal BC, SPDEF has dual behavior by promoting oncogenesis and progression through ER/FOXA1/GATA3 network,27 and growth suppressed in MCF 7 cells28,29 as a TSG. In addition, the researchers have also found SPDEF to be a biomarker of poor prognosis in ER+ primary BC.27,30 Secondly, both the pro- and anti-oncogenic activities of SPDEF have been demonstrated and are stage-dependent. From benign breast to ductal carcinoma in situ (DCIS), the protein expression of SPDEF gradually increased,31 and then lost in invasive BC (IBC).32,33 Given all that, SPDEF mainly plays the role of OCG in luminal BC and Her2+ BC, plays the role of TSG in TNBC, and even shows a dual role switch during the malignant progression of BC. Therefore, the purpose of this review is to provide compelling evidence that SPDEF can be presented as both TSG and OCG in BC.
SPDEF introduction
The Structure of SPDEF
SPDEF gene is located at chromosome 6p21.31 and encodes for 6 exons with the length of the coding sequence of 1005 nucleotides (Figure 1A). Alternatively, spliced transcript variants encoding different isoforms have been found for SPDEF gene, and exon 4 skipping is one predominant alternative splicing event (Figure 1B). Two isoforms have been produced by alternative splicing so far. Isoform 1 has been chosen as the canonical sequence with the missing of the amino acid sequence from 212 to 227 of isoform 2. And the function of isoform 2 has not been described in the published literature in detail. SPDEF protein is composed of 335 amino acids (Figure 1C). Unlike other ETS proteins, SPDEF mainly contains a pointed domain and a conserved 88 amino acid ETS domain. Moreover, the ETS domain of SPDEF protein prefers binding to GGAT to binding to GGAA core compared with other ETS TFs.34
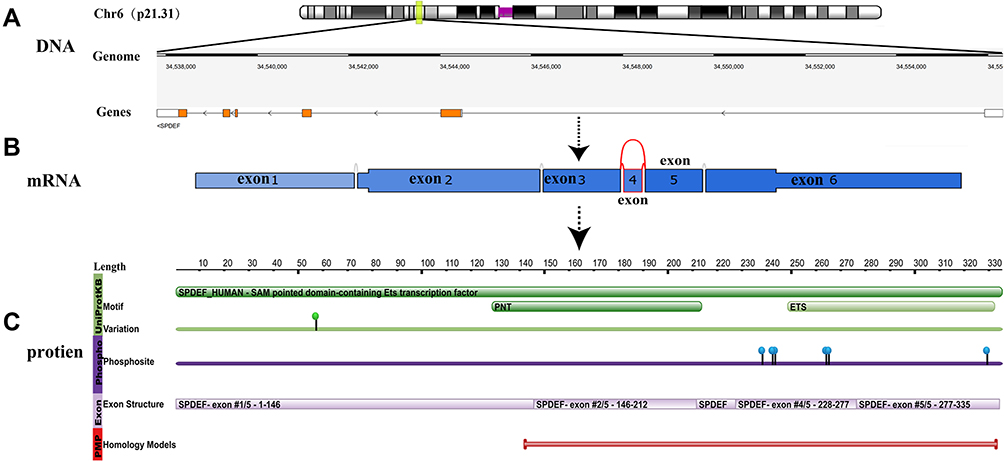 |
Figure 1 Schematic diagram of the SPDEF structure at the DNA, mRNA and protein level. Notes: (A) The gene track represents the gene-structure on the genome: white boxes represent untranslated regions; orange: protein-coding regions; the black lines connecting boxes represent introns; (B) Exon 4 skip yield the major isoform of SPDEF at the mRNA level; (C) The green bar shows the motif of SPDEF mainly including EST and PNT. And the green point displays the variation data (sourced from UniProt) with non-genetic variation. Data in purple show phosphorylation sites; Data in lilac represent the genomic exon structure; Data in red indicate combined ranges of homology models. (A and C) are obtained from the RCSB PDB database, (B) is obtained from the TCGA SpliceSeq database. |
Phosphorylation Sites
ETS family members are regulated generally by phosphorylation and rarely by other post-translational modifications.35,36 Potential phosphorylation sites present in SPDEF include a protein kinase C site, two tyrosine kinase phosphorylation sites, two AKT phosphorylation sites, and eight MAPK phosphorylation sites,34 whereas are little verified yet. One is the activation of extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) through Her2 and colony-stimulating factor 1 receptor/colony-stimulating factor 1 (CSF-1R/CSF-1), which may regulate SPDEF phosphorylation, and thus promote MCF-10A movement and invasion.37 The other is that the cell cycle kinase CDK11p5 directly interacts with and phosphorylates SPDEF on serine residues, leading to subsequent ubiquitination and degradation of SPDEF via the proteasome pathway. Then, the migration and invasion of prostate cancer cells increased due to the loss of SPDEF protein.38 Future proofs will be needed to discover the full extent of SPDEF phosphorylation and the role phosphorylation plays in the regulation of SPDEF activity.
Expression Profiles of SPDEF
Unlike the majority of ETS family members, another feature of SPDEF is expressed exclusively in tissues with a high epithelial content such as prostate, breast, colon, and trachea.31,34,39,40 Such limited expression suggests that SPDEF plays a crucial role in the normal development and/or function of these tissues. Contrary to normal tissues where the limited tissue-specific SPDEF expression is generally accepted, the controversial expression of SPDEF in BC is not easy to understand. In general, analysis of the expression characteristics of SPDEF in tumors arising from basal versus luminal epithelial lineage showed widespread SPDEF expression in tumors arising from the luminal epithelial lineage, and these included luminal, Her2+, and apocrine subtypes of luminal BC. In contrast, little SPDEF expression is observed in TNBC.30 Table 1 summarizes the expression profiles of SPDEF in different BC cell lines and tissues reported over the years.
 |
Table 1 Differential Expression of SPDEF in Normal and Breast Tumor Specimens |
The following points are summed up based on Table 1: (i) SPDEF mRNA and protein are over-expressed in early low-grade malignant tumors and cell lines compared with normal tissues and cell lines;26,27,31,39-41 (ii) SPDEF mRNA and protein expression levels are too low or even undetectable in the highly malignant, advanced IBC and cell lines compared with early low-grade malignant tumors and cell lines;32 (iii) mRNA levels are not necessarily related to protein levels;29 (iv) In IBC, a weak SPDEF staining is detected mainly in the cytosol.42
Taken together, a large number of literatures have revealed that SPDEF expression alone is not enough to induce invasive activity in BC. However, an increased SPDEF expression levels may initiate transformation activity and/or sensitize these cells, resulting in other outcomes such as receptor tyrosine kinase (RTK) amplification or mutation activation to promote tumor progression.37 SPDEF can promote progression in all aspects of the tumor when it works with oncogenes and/or carcinogenic-related genes, including increased cellular mobility, invasiveness, and non-anchored growth of breast epithelial cells. It is revealed that SPDEF may involve in the occurrence or development of early BC40 and may facilitate phenotypic effects in different stages of tumorigenesis.37 Additionally, SPDEF mRNA expresses lower in advanced tumors than early tumors, which is usually consistent with the down-regulation of SPDEF and the detection of low protein in the highly malignant, advanced tumors, and also reconciles with a scheme of epithelium to mesenchymal transition to neoplasia.40 In short, SPDEF expression tends to be reduced or lost during tumor progression,32,43 suggesting that SPDEF may have diverse functions in different stages of BC development, which further supports the dual function of SPDEF. Moreover, mRNA levels are not always associated with protein levels. Test studies indicated that low SPDEF mRNA expression still translates into high protein nuclear detection in normal breasts.32 Instead, high SPDEF mRNA expression can be detected in some BC cell lines, with barely detectable protein, like HCC-1428.40 It may be because of a post-translation modification mechanism such as miRNAs regulation29 or the rapid degradation of SPDEF.34 It is worth noting that low SPDEF mRNA expression can be connected with sizeable protein expression such us MDA-MB-231.29 Furthermore, a high nuclear SPDEF stain is usually reflected in most tumor tissues, but the cytoplasmic pool of SPDEF expression indicates that there may be non-functioning SPDEF protein or may represent the stored SPDEF for use in the nuclei when needed. However, since many members of the ETS family still express in the cytoplasm, non-specific reactions cannot always be excluded.
The Role of Double-Sided Gene SPDEF in the Progress of BC
Over the past few years, several studies have shown that the highly conservative ETS family of TFs regulate a variety of biological processes including cell proliferation, differentiation, apoptosis, transformation, migration, and invasion and are thought to play a momentous role in oncogenesis.44–47 SPDEF, as one of these ETS TFs, has been observed to be involved in these biological processes in BC in many studies. All of these studies concluded that SPDEF has dual functions. Researchers usually describe OCGs and TSGs with dominant mutation activation of proto-oncogenes and silent mutation inactivation of TSGs, respectively. However, SPDEF gene activation and inactivation mutations are rarely described in hundreds of BC that have been sequenced so far.48 According to the studies about the effect of SPDEF on the biological behavior of BC cells, as well as the related underlying mechanisms of it, a summary of the dual function of SPDEF can be found in Figure 2.
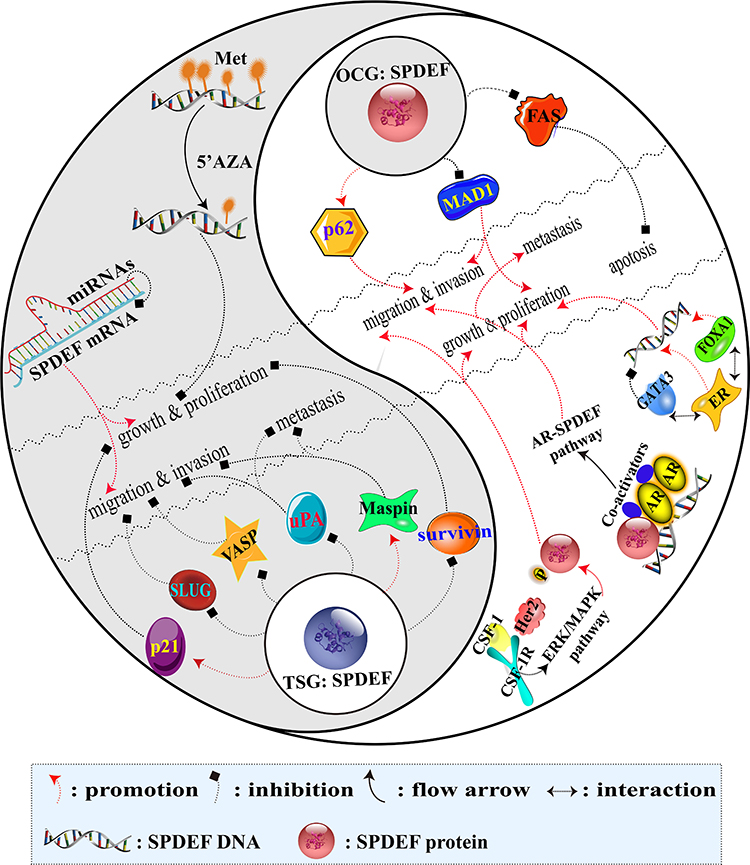 |
Figure 2 Regulatory mechanism diagram of double-agent SPDEF in BC. Notes: The darker part of the Tai Chi diagram illustrates the network of SPDEF involved in tumor-inhibiting effect as a TSG. Conversely, the lighter part of the Tai Chi diagram shows the network of SPDEF involved in tumor-promoting effect as an OCG. |
SPDEF, an OCG: Promoting Growth and Proliferation
SPDEF appears to be a critical factor in regulating tumor growth and proliferation, and the molecular mechanism underlying the oncogenic effect of it on BC cells clarified below.
A research showed that SPDEF levels strongly correlate with ER+ luminal BC, and ER is recruited at the SPDEF gene locus in an estrogen-dependent manner. ER-cooperating factors, FOXA1 and GATA3 are also directly recruited at SPDEF gene locus. It is confirmed that SPDEF is a direct target of ER, FOXA1 and GATA3 in ER+BC cells. The researchers further reported that SPDEF is co-expressed with ER and regulated by GATA3 and FOXA1. GATA3 inhibits ER-mediated SPDEF expression and plays a negative role in the regulation of SPDEF expression. However, FOXA 1 promotes ER-mediated expression of SPDEF and further promotes cell growth.27
Cao et al have proposed that androgen receptor (AR) can directly up-regulate the expression of SPDEF when AR and SPDEF are co-expressed, resulting in activation of SPDEF in ER−AR+BC cells. Activated SPDEF directly down-regulates the expression of MAD1, a transcriptional repressor of oncogene MYC, and promotes the degradation of MAD1 and the separation of MAD1 from MAX. In the absence of MAD1 competition, MAX and MYC form the largest heterodimer, which in turn induces MYC mediated gene transcription and promotes the proliferation, migration and invasion of ER−BC cells in vitro. Rescue experiment has also verified that the up-regulation of MAD1 expression significantly inhibits SPDEF-induced proliferation, migration and invasion of SKBR-3 cells. Moreover, pulmonary metastasis has been found in the mice inoculated with SPDEF-overexpressing cells instead of in the mice inoculated with MAD1-overexpressing cells.25
SPDEF, an OCG: Promoting Migration, Invasion and Metastasis
The pro-cancer function of SPDEF can not only promote the survival of cancer cells but also accelerate the malignant transformation of cancer cells. Studies have demonstrated that the signal adaptor p62 is overexpression in metastatic BC. SPDEF upregulates p62 transcription by directly binding to p62 promoter at least two sites, so SPDEF may act as p62 co-activating factor and motivate the overexpression of p62 in BC.49,50 Although no studies have confirmed the effect of SPDEF-upregulated p62 on the biological behavior of BC cells so far, it has been proved that p62 promotes the invasive phenotypes of BC cells in vitro by interacting with intermediate filament vimentin. It also has been proved that p62 depletion inhibits BC metastasis and reduces the tumorigenicity in vivo.50 In TNBC, patients with p62 overexpression have a higher risk of lymph node-positive and lymphoid metastasis.51
Gunawardane et al have illustrated that with the exception of MDA-MB-231 cells, SPDEF can induce migration in normal and cancer cell lines including non-metastatic BC cell lines and melanoma cell lines. Although SPDEF independent expression induced limited motility of MCF-10A cells, the co-expression of SPDEF with the RTK Her2 and CSF-1R/CSF-1 significantly enhanced MCF-10A motility and anchorage-independent growth and induced a dramatic invasive phenotype in three-dimensional cultures. Then, Her2 and CSF-1R/CSF-1 may activate ERK/MAPK, which can cooperate with SPDEF to promote motility and invasion.37 However, the specific molecular mechanism remains largely undiscovered.
SPDEF, an OCG: Inhibition of Apoptosis
To date, many researchers have revealed that the pro-apoptotic genes including FAS directly are regulated by SPDEF in BC. However, the mechanism by which SPDEF inhibits the apoptosis pathway to stimulate the progression of BC remains unclear. FAS receptor, a member of the tumor necrosis family involved in the extrinsic apoptosis pathway,52 is a direct SPDEF transcriptional target. High-level expression of FAS is detected in non-transformed mammary epithelial cell lines, but the expression of it is reduced in several BC cell lines.53 SPDEF suppresses the apoptosis of MCF-7 cells by down-regulating the expression of the FAS gene, especially under stress conditions such as hormone depletion.27
SPDEF, a TSG: Inhibition of Growth and Proliferation
As it is mentioned above, many researchers have confirmed the role of SPDEF in promoting cancer in BC, but some studies have shown that SPDEF exhibits a clear tumor-suppressor function. The effects of SPDEF on the growth and proliferation of BC cells through the regulation of target genes and methylated modification are elucidated as follows.
A study has indicated that SPDEF directly binds to cyclin-dependent kinase (CDK) inhibitor p21 promoter and then up-regulates the expression levels of p21 in PyV-mT (polyoma virus middle-T) mouse mammary tumor cell line. Elevated p21 leads to the reduction of CDK2 activity, which ultimately results in decreased PyV-mT cells growth and proliferation under non-stress/damaged conditions both in vitro and in vivo. SPDEF expression partially blocks the cell cycle progression in G1/S phase without an effect on apoptosis has been reflected using cell cycle analysis. Nevertheless, p21 silencing eliminates the SPDEF growth inhibition in vitro and in vivo.24
Another study has also proved that the inhibitor of apoptosis survivin is the direct transcriptional target of SPDEF in prostate cancer,54 and ectopic SPDEF expression can down-regulate survivin promoter activity and endogenous survivin expression in BC. The down-regulated survivin can inhibit the growth of MCF 7 BC cells in vitro and xenograft tumor formation in vivo. Conversely, SPDEF silencing can up-regulate survivin expression, which triggers the growth of MCF 7 BC cells in vitro and xenograft tumor formation in vivo.28
DNA methylation is a specific change in tumorigenesis. SPDEF with DNA methylation as an epigenetic alteration in MDA-MB-468 cells, indicating that the functions of SPDEF are selective in BC cells. Increased SPDEF expression shows a dose-dependent manner following DNA methylation inhibitor 5ʹ azacytidine (5ʹAZA) treatment decreased or the growth or proliferation rate of BC cells slowed. Meanwhile, an enhanced p21 expression is found following the same expression pattern as SPDEF.55
SPDEF, a TSG: Inhibition of Migration, Invasion and Metastasis
Many direct downstream target genes of SPDEF have been found during these years, which are participated in the negative regulation of migration, invasion and metastasis of BC including but not limited to uPA, Maspin, VASP and SLUG.
uPA ligand and its membrane-bound receptor uPAR are responsible for tumor development such as cell proliferation, migration and adhesion.56,57 Plenty of related researches have shown that uPA is a direct transcriptional target of SPDEF-negative regulation.22,54,58 The negative regulation of uPA by SPDEF can change the migration ability of the cancer cells through a variety of possible mechanisms.22 First of all, uPA is activated on the surface of MDA-MB-231 cells59 and can convert surface plasminogen into plasmin. The plasmin can not only directly degrade various extracellular matrix (ECM) components,60 but also directly or indirectly activate matrix metalloprotease (MMP) that can further enhance the degradation of ECM.61 The process of degradation of the basement membrane might be inhibited by SPDEF-downregulated uPA. Second, in IBC cells, the SPDEF-downregulated uPA leads to compensatory increased expression of uPAR mRNA. Intriguingly, increased soluble uPAR may damage many functions of urokinase system such as proteolysis and tumor growth to limit the potential of metastasis in BC cells.58 Third, the uPA/uPAR system is also very important in intracellular signal transduction, including interaction with tyrosine kinase,62 EGFR signaling pathway63 and signal-related integration family members.64 SPDEF-downregulated uPA may reduce the binding of uPAR and change the intracellular signal pattern and then inhibit cell migration.
Maspin is often down-regulated in the development of BC.32,65 It has been revealed that SPDEF can regulate Maspin promoter28,32 and positively regulate the expression of Maspin.32 In IBC, the decrease of Maspin expression is largely due to the deletion of SPDEF, which may contribute to the invasion and metastasis of tumor cells. Moreover, several studies have mentioned that Maspin is a type II TSG, which can inhibit tumor growth, motility, invasion and metastasis when expressed in some cancers, including BC.66–68
VASP, an action-binding protein, participates in linking signaling pathways to the remodel actin cytoskeleton.69 Bioinformatics initially analysis shows that VASP is a presumptive target gene for SPDEF. Turner et al subsequently have verified that VASP is up-regulated directly by SPDEF in vitro.22 Multiple lamellipodia can be produced when the up-regulated VASP is located on the cell membrane, which can retard cell migration and its phenotype is similar to the re-expression of SPDEF.70
SLUG is a member of the SNAIL superfamily, and its high expression associates with the aggressive basal phenotype in breast tumors.71 Besides, SLUG is also inversely correlated with E-cadherin expression and is a critical event–promoting EMT in many tumor types.72 As the study identified that SPDEF is able to regulate downstream targets of SLUG in both SLUG-dependent and -independent manners, suggesting a critical role for regulating EMT.23 E-cadherin is also known to be a transcriptional target of SLUG during carcinogenesis. Low expressed SPDEF is able to relieve the repression of E-cadherin through direct inhibition of SLUG, which is a critical interaction in inhibiting the migratory phenotype.23
miRNAs are endogenous 19–25 nucleotide noncoding RNAs that also have dual functions in tumor progression.73 To date, although there have been many studies on miRNAs in BC, only few studies have focused on the miRNAs that interact with SPDEF mRNA. Fortunately, Findlay et al have discovered that SPDEF is directly regulated by two kinds of miRNAs (miRNA-204 and miRNA-510), which can prevent the translation of SPDEF mRNA, resulting in the loss of SPDEF protein expression and promoting tumors to gain a more aggressive phenotype. In addition, extrinsic SPDEF expression can inhibit the overexpression phenotype of miRNAs.29
The Role of Dual-Functional SPDEF in Diagnosis and Prognosis of BC
Oncologists have looking for new BC related molecules as the markers for early diagnosis or prognosis. Dual-functional SPDEF is one of the most potential candidate markers at present. As described above, SPDEF is necessary for ER+BC survival, and there is a strong positive correlation between the expression of SPDEF and ER. In order to evaluate the sensitivity and specificity at which SPDEF expression could predict an association of the ER positivity, the expression of SPDEF has been detected in 86 clinical specimens and then analyzed by receiver–operator curves (ROCs). The result showed that ER positivity with 98.3% sensitivity (58/59) and 76.9% specificity (20/26) when SPDEF expression is at or above the MCF7 level, with the area under the curve (AUC) 0.902.40 In another word, predicting ER+BC by SPDEF markers seems to be reliable. Moreover, numerous studies have demonstrated that the loss of SPDEF protein expression and high expression of SPDEF mRNA may be a powerful indicator to estimate the migration promoting characteristics of advanced BC.
Prognosis of BC is also influenced by SPDEF. There is a correlation between higher SPDEF expression and shorter overall survival (OS) in 246 patients with ER−BC, but SPDEF expression is not associated with disease-free survival (DFS). Meanwhile, the multivariate analysis confirmed that SPDEF expression is a significant independent prognostic variable affecting OS.25,74 Moreover, three independent data sets were downloaded from GEO and ArrayExpress databases, and analyzed by Kaplan-Meier. The results showed that the high SPDEF expression is related to the poor OS in ER+BC patients. In the Cox regression model, SPDEF is an important predictor of survival when SPDEF is a continuous variable in ER+BC, which is consistent with the previous reports.27,30 In summary, SPDEF can be used as a valuable indicator to evaluate the prognosis of BC.
The Functional Duality of SPDEF Arouses New Considerations on BC Therapies
The targeted therapy model has opened up a new field of tumor chemotherapy, which has been widely used in the treatment of various molecular subtypes of BC. In Her2+ BC population, Her2-blocking therapies and the achievement of trastuzumab have considerably modified the prognosis of these patients.75 Further, the addition of pertuzumab to trastuzumab and docetaxel demonstrated an improved progression-free survival (PFS) and OS, as well as validating the dual Her2 blockade concept by CLEOPATRA trial.76,77 Endocrine therapy is still the main treatment for luminal BC and selective adjuvant chemotherapy. More recently, the addition of a targeted therapy to endocrine treatment has offered new therapeutic options for luminal BC patients. Indeed, the addition of everolimus to exemestane has improved DFS in this population.78 For TNBC, there is no target to easily block yet, and the anthracycline and paclitaxel schemes remain mainstream. Specifically, in the Phase 3 trial of IMPASSION-130, atezolizumab combined with nab-paclitaxel significantly improved PFS in patients with metastatic TNBC and is more pronounced in PDL1-positive tumors.79 Since tumor cells are interindividual heterogeneity and increase with the treatment, these lead to poor therapeutic effect or serious toxicity. And the dual-functional characteristics of genes are related to tumor heterogeneity. Hence, finding the most suitable tumor candidate antigen based on this dual nature will benefit the development of tumor-targeted immunotherapy.
At present, Her2 is the only antibody-mediated immunotherapy target for BC,80 which is critical for BC treatment. Current therapeutic applications have shown that targeting OCG and its related pathways are expected to develop new drugs, including antibodies and small synthetic molecules.81 Moreover, the overall limited expression of SPDEF in normal human tissues suggests that SPDEF-based anti-tumor therapies will have minimal toxicity against vital normal tissues.82 These observations support SPDEF as a highly desirable novel candidate antigen against luminal BC. Meanwhile, Sood found that SPDEF might be immunogenic and intolerant in female BC patients, and the SPDEF sequence appears to contain HLA-A2-binding peptides that are potentially capable of eliciting HLA-A2-restricted T cell responses.83 Additionally, SPDEF as TF can significantly affect the biological characteristics of tumors by inducing large-scale changes in gene expression. And these alter genes may encode the cell surface and/or secreted molecules capable of influencing the behavior of the neighboring tumor and/or stromal cells.31 This concept is supported by the characteristics of ER expression in BC and its implication for endocrine therapy. Similarly, the eliminating SPDEF expressed cells from BC by SPDEF-targeted vaccines/immunotherapy should not only elicit the killing of SPDEF expressing tumor cells but also modulate the tumor microenvironment and inhibit tumor progression.83 Furthermore, SPDEF is essential for luminal BC cell survival and models of endocrine resistance, which may have therapeutic value for BC patients treated with endocrine therapy.27 Therefore, SPDEF should be a useful novel target for co-targeting with endocrine therapy to minimize endocrine resistance in BC.82 Although there are no reports of clinical trials on SPDEF targeted therapy, these considerations provide a compelling rationale to evaluate the potential of SPDEF as a novel luminal breast tumor antigen. In future work, to redefine “SPDEF gene” by considering each mRNAs, regulatory RNA, protein isoform, and posttranslational modification from the same genomic locus6 instead one-sided thinking that SPDEF is an OCG or a TSG in BC, may be conducive to better understand tumor biology and to select targets for different cancer subtypes for individualized treatment.
Conclusions
SPDEF discussed in this review has attracted much attention because of its role in oncogenesis and progression. The definition of SPDEF in the dichotomy of cancer-regulatory genes has been controversial. It is more reasonable to consider SPDFE is a dual-functional gene by thorough summarizing the current knowledge, which will contribute to comprehend the heterogeneity of tumors and facilitate future research. The mechanism underlying the regulation of SPDEF must be investigated due to a better understanding of the dual-functional nature of SPDEF and is of great significance to the development of targeted therapy for each BC subgroup. Therefore, SPDEF has the potential of being a new diagnostic and therapeutic target in tumor biology in the near future.
Acknowledgments
This work was supported by the Sichuan Science and Technology Program for International Cooperation, China (Grant No. 2017HH0105), the Doctoral Research Initiation Fund of Affiliated Hospital of Southwest Medical University (19077), the Sichuan Science and Technology Program for Foundation Platform, China (Grant No. 2018TJPT0023), the Sichuan Science and Technology Program for Major Project, China (Grant No. 2019YFS0332; Grant No. 2019YFS0038).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Zepeda-Castilla EJ, Recinos-Money E, Cuellar-Hubbe M, Robles-Vidal CD, Maafs-Molina E. [Molecular classification of breast cancer]. Cir Cir. 2008;76(1):87–93. Spanish.
3. Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009;101(10):736–750. doi:10.1093/jnci/djp082
4. Provenzano E, Ulaner GA, Chin SF. Molecular classification of breast cancer. PET Clin. 2018;13(3):325–338. doi:10.1016/j.cpet.2018.02.004
5. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi:10.1200/JCO.2008.18.1370
6. Lou X, Zhang J, Liu S, Xu N, Liao DJ. The other side of the coin: the tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13(11):1677–1693. doi:10.4161/cc.29082
7. Isobe M, Emanuel BS, Givol D, Oren M, Croce CM. Localization of gene for human p53 tumour antigen to band 17p13. Nature. 1986;320(6057):84–85. doi:10.1038/320084a0
8. Soussi T, Wiman KG. TP53: an oncogene in disguise. Cell Death Differ. 2015;22(8):1239–1249. doi:10.1038/cdd.2015.53
9. Yang L, Han Y, Suarez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21(5):868–876. doi:10.1038/sj.leu.2404624
10. Yip SC, Saha S, Chernoff J. PTP1B: a double agent in metabolism and oncogenesis. Trends Biochem Sci. 2010;35(8):442–449. doi:10.1016/j.tibs.2010.03.004
11. Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci U S A. 1989;86(22):8763–8767. doi:10.1073/pnas.86.22.8763
12. Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249(4971):912–915. doi:10.1126/science.2144057
13. Diller L, Kassel J, Nelson CE, et al. p53 functions as a cell cycle control protein in osteosarcomas. Mol Cell Biol. 1990;10(11):5772–5781. doi:10.1128/MCB.10.11.5772
14. Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell. 1990;62(4):671–680. doi:10.1016/0092-8674(90)90113-S
15. Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352(6333):345–347. doi:10.1038/352345a0
16. Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–1238. doi:10.1126/science.1978757
17. Wang H, Bao W, Jiang F, et al. Mutant p53 (p53-R248Q) functions as an oncogene in promoting endometrial cancer by up-regulating REGgamma. Cancer Lett. 2015;360(2):269–279. doi:10.1016/j.canlet.2015.02.028
18. Ortega-Molina A, Boss IW, Canela A, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199–1208. doi:10.1038/nm.3943
19. Toska E, Osmanbeyoglu HU, Castel P, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017;355(6331):1324–1330. doi:10.1126/science.aah6893
20. Sun X, Wang SC, Wei Y, et al. Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell. 2017;32(5):574–589 e576. doi:10.1016/j.ccell.2017.10.007
21. Fang JZ, Li C, Liu XY, Hu TT, Fan ZS, Han ZG. Hepatocyte-specific Arid1a deficiency initiates mouse steatohepatitis and hepatocellular carcinoma. PLoS One. 2015;10(11):e0143042. doi:10.1371/journal.pone.0143042
22. Turner DP, Findlay VJ, Kirven AD, Moussa O, Watson DK. Global gene expression analysis identifies PDEF transcriptional networks regulating cell migration during cancer progression. Mol Biol Cell. 2008;19(9):3745–3757. doi:10.1091/mbc.e08-02-0154
23. Findlay VJ, Turner DP, Yordy JS, et al. Prostate-derived ETS factor regulates epithelial-to-mesenchymal transition through both SLUG-dependent and independent mechanisms. Genes Cancer. 2011;2(2):120–129. doi:10.1177/1947601911410424
24. Schaefer JS, Sabherwal Y, Shi HY, et al. Transcriptional regulation of p21/CIP1 cell cycle inhibitor by PDEF controls cell proliferation and mammary tumor progression. J Biol Chem. 2010;285(15):11258–11269. doi:10.1074/jbc.M109.073932
25. Cao L, Xu C, Xiang G, et al. AR-PDEF pathway promotes tumour proliferation and upregulates MYC-mediated gene transcription by promoting MAD1 degradation in ER-negative breast cancer. Mol Cancer. 2018;17(1):136. doi:10.1186/s12943-018-0883-0
26. Mukhopadhyay A, Khoury T, Stein L, Shrikant P, Sood AK. Prostate derived Ets transcription factor and Carcinoembryonic antigen related cell adhesion molecule 6 constitute a highly active oncogenic axis in breast cancer. Oncotarget. 2013;4(4):610–621. doi:10.18632/oncotarget.934
27. Buchwalter G, Hickey MM, Cromer A, et al. PDEF promotes luminal differentiation and acts as a survival factor for ER-positive breast cancer cells. Cancer Cell. 2013;23(6):753–767. doi:10.1016/j.ccr.2013.04.026
28. Ghadersohi A, Pan D, Fayazi Z, Hicks DG, Winston JS, Li F. Prostate-derived Ets transcription factor (PDEF) downregulates survivin expression and inhibits breast cancer cell growth in vitro and xenograft tumor formation in vivo. Breast Cancer Res Treat. 2007;102(1):19–30. doi:10.1007/s10549-006-9314-9
29. Findlay VJ, Turner DP, Moussa O, Watson DK. MicroRNA-mediated inhibition of prostate-derived Ets factor messenger RNA translation affects prostate-derived Ets factor regulatory networks in human breast cancer. Cancer Res. 2008;68(20):8499–8506. doi:10.1158/0008-5472.CAN-08-0907
30. Sood AK, Wang J, Mhawech-Fauceglia P, Jana B, Liang P, Geradts J. Sam-pointed domain containing Ets transcription factor in luminal breast cancer pathogenesis. Cancer Epidemiol Biomarkers Prev. 2009;18(6):1899–1903. doi:10.1158/1055-9965.EPI-09-0055
31. Sood AK, Saxena R, Groth J, et al. Expression characteristics of prostate-derived Ets factor support a role in breast and prostate cancer progression. Hum Pathol. 2007;38(11):1628–1638. doi:10.1016/j.humpath.2007.03.010
32. Feldman RJ, Sementchenko VI, Gayed M, Fraig MM, Watson DK. Pdef expression in human breast cancer is correlated with invasive potential and altered gene expression. Cancer Res. 2003;63(15):4626–4631.
33. Turner DP, Moussa O, Sauane M, Fisher PB, Watson DK. Prostate-derived ETS factor is a mediator of metastatic potential through the inhibition of migration and invasion in breast cancer. Cancer Res. 2007;67(4):1618–1625. doi:10.1158/0008-5472.CAN-06-2913
34. Oettgen P, Finger E, Sun Z, et al. PDEF, a novel prostate epithelium-specific ets transcription factor, interacts with the androgen receptor and activates prostate-specific antigen gene expression. J Biol Chem. 2000;275(2):1216–1225. doi:10.1074/jbc.275.2.1216
35. Charlot C, Dubois-Pot H, Serchov T, Tourrette Y, Wasylyk B. A review of post-translational modifications and subcellular localization of Ets transcription factors: possible connection with cancer and involvement in the hypoxic response. Methods Mol Biol. 2010;647:3–30.
36. Yordy JS, Muise-Helmericks RC. Signal transduction and the Ets family of transcription factors. Oncogene. 2000;19(55):6503–6513. doi:10.1038/sj.onc.1204036
37. Gunawardane RN, Sgroi DC, Wrobel CN, Koh E, Daley GQ, Brugge JS. Novel role for PDEF in epithelial cell migration and invasion. Cancer Res. 2005;65(24):11572–11580. doi:10.1158/0008-5472.CAN-05-1196
38. Tamura RE, Paccez JD, Duncan KC, et al. GADD45alpha and gamma interaction with CDK11p58 regulates SPDEF protein stability and SPDEF-mediated effects on cancer cell migration. Oncotarget. 2016;7(12):13865–13879. doi:10.18632/oncotarget.7355
39. Ghadersohi A, Sood AK. Prostate epithelium-derived Ets transcription factor mRNA is overexpressed in human breast tumors and is a candidate breast tumor marker and a breast tumor antigen. Clin Cancer Res. 2001;7(9):2731–2738.
40. Turcotte S, Forget MA, Beauseigle D, Nassif E, Lapointe R. Prostate-derived Ets transcription factor overexpression is associated with nodal metastasis and hormone receptor positivity in invasive breast cancer. Neoplasia. 2007;9(10):788–796. doi:10.1593/neo.07460
41. He J, Pan Y, Hu J, Albarracin C, Wu Y, Dai JL. Profile of Ets gene expression in human breast carcinoma. Cancer Biol Ther. 2007;6(1):76–82. doi:10.4161/cbt.6.1.3551
42. Frietsch JJ, Grunewald TG, Jasper S, et al. Nuclear localisation of LASP-1 correlates with poor long-term survival in female breast cancer. Br J Cancer. 2010;102(11):1645–1653. doi:10.1038/sj.bjc.6605685
43. Tsujimoto Y, Nonomura N, Takayama H, et al. Utility of immunohistochemical detection of prostate-specific Ets for the diagnosis of benign and malignant prostatic epithelial lesions. Int J Urol. 2002;9(3):167–172. doi:10.1046/j.1442-2042.2002.00444.x
44. Findlay VJ, LaRue AC, Turner DP, Watson PM, Watson DK. Understanding the role of ETS-mediated gene regulation in complex biological processes. Adv Cancer Res. 2013;119:1–61.
45. Dittmer J. The biology of the Ets1 proto-oncogene. Mol Cancer. 2003;2:29. doi:10.1186/1476-4598-2-29
46. Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. doi:10.1016/S0378-1119(02)01156-3
47. Seth A, Watson DK. ETS transcription factors and their emerging roles in human cancer. Eur J Cancer. 2005;41(16):2462–2478. doi:10.1016/j.ejca.2005.08.013
48. Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47–54. doi:10.1038/nature17676
49. Thompson HG, Harris JW, Wold BJ, Lin F, Brody JP. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene. 2003;22(15):2322–2333. doi:10.1038/sj.onc.1206325
50. Li SS, Xu LZ, Zhou W, et al. p62/SQSTM1 interacts with vimentin to enhance breast cancer metastasis. Carcinogenesis. 2017;38(11):1092–1103. doi:10.1093/carcin/bgx099
51. Luo RZ, Yuan ZY, Li M, Xi SY, Fu J, He J. Accumulation of p62 is associated with poor prognosis in patients with triple-negative breast cancer. Onco Targets Ther. 2013;6:883–888. doi:10.2147/OTT.S46222
52. Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10(1):26–35. doi:10.1038/sj.cdd.4401186
53. Keane MM, Ettenberg SA, Lowrey GA, Russell EK, Lipkowitz S. Fas expression and function in normal and malignant breast cell lines. Cancer Res. 1996;56(20):4791–4798.
54. Turner DP, Findlay VJ, Moussa O, et al. Mechanisms and functional consequences of PDEF protein expression loss during prostate cancer progression. Prostate. 2011;71(16):1723–1735. doi:10.1002/pros.21389
55. Sabherwal Y, Mahajan N, Zhang M. Epigenetic modifications of prostate-derived Ets transcription factor in breast cancer cells. Oncol Rep. 2013;30(4):1985–1988. doi:10.3892/or.2013.2661
56. Choong PF, Nadesapillai AP. Urokinase plasminogen activator system: a multifunctional role in tumor progression and metastasis. Clin Orthop Relat Res. 2003;415 Suppl:S46–S58. doi:10.1097/01.blo0000093845.72468.bd
57. Han B, Nakamura M, Mori I, Nakamura Y, Kakudo K. Urokinase-type plasminogen activator system and breast cancer (Review). Oncol Rep. 2005;14(1):105–112.
58. Kruger A, Soeltl R, Lutz V, et al. Reduction of breast carcinoma tumor growth and lung colonization by overexpression of the soluble urokinase-type plasminogen activator receptor (CD87). Cancer Gene Ther. 2000;7(2):292–299. doi:10.1038/sj.cgt.7700144
59. Andronicos NM, Ranson M. The topology of plasminogen binding and activation on the surface of human breast cancer cells. Br J Cancer. 2001;85(6):909–916. doi:10.1054/bjoc.2001.2022
60. Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57(1):25–40. doi:10.1007/s000180050497
61. Ramos-DeSimone N, Hahn-Dantona E, Sipley J, Nagase H, French DL, Quigley JP. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem. 1999;274(19):13066–13076. doi:10.1074/jbc.274.19.13066
62. Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3(12):932–943. doi:10.1038/nrm977
63. Festuccia C, Angelucci A, Gravina GL, et al. Epidermal growth factor modulates prostate cancer cell invasiveness regulating urokinase-type plasminogen activator activity. EGF-receptor inhibition may prevent tumor cell dissemination. Thromb Haemost. 2005;93(5):964–975. doi:10.1160/TH04-09-0637
64. Chapman HA, Wei Y. Protease crosstalk with integrins: the urokinase receptor paradigm. Thromb Haemost. 2001;86(1):124–129. doi:10.1055/s-0037-1616208
65. Sager R, Sheng S, Pemberton P, Hendrix MJ. Maspin. A tumor suppressing serpin. Adv Exp Med Biol. 1997;425:77–88.
66. Hendrix MJ. De-mystifying the mechanism(s) of maspin. Nat Med. 2000;6(4):374–376. doi:10.1038/74624
67. Zhang M, Maass N, Magit D, Sager R. Transactivation through Ets and Ap1 transcription sites determines the expression of the tumor-suppressing gene maspin. Cell Growth Differ. 1997;8(2):179–186.
68. Zou Z, Anisowicz A, Hendrix MJ, et al. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263(5146):526–529. doi:10.1126/science.8290962
69. Bear JE, Svitkina TM, Krause M, et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109(4):509–521. doi:10.1016/S0092-8674(02)00731-6
70. Lin YH, Park ZY, Lin D, et al. Regulation of cell migration and survival by focal adhesion targeting of Lasp-1. J Cell Biol. 2004;165(3):421–432. doi:10.1083/jcb.200311045
71. Storci G, Sansone P, Trere D, et al. The basal-like breast carcinoma phenotype is regulated by SLUG gene expression. J Pathol. 2008;214(1):25–37. doi:10.1002/path.2254
72. Jethwa P, Naqvi M, Hardy RG, et al. Overexpression of Slug is associated with malignant progression of esophageal adenocarcinoma. World J Gastroenterol. 2008;14(7):1044–1052. doi:10.3748/wjg.14.1044
73. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi:10.3322/caac.21208
74. Cao L, Li C, Xu C, et al. Clinical significance of PDEF factor expression and its relation to androgen receptor in ER(-) breast cancer. Histopathology. 2018;73(5):819–831. doi:10.1111/his.13699
75. Le Du F, Perrin C, Brunot A, et al. Therapeutic innovations in breast cancer. Presse Med. 2019;48(10):1131–1137. doi:10.1016/j.lpm.2019.04.005
76. Baselga J, Swain SM. CLEOPATRA: a Phase III evaluation of pertuzumab and trastuzumab for HER2-positive metastatic breast cancer. Clin Breast Cancer. 2010;10(6):489–491. doi:10.3816/CBC.2010.n.065
77. Swain SM, Baselga J, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372(8):724–734. doi:10.1056/NEJMoa1413513
78. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–529. doi:10.1056/NEJMoa1109653
79. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121. doi:10.1056/NEJMoa1809615
80. Mittendorf EA, Holmes JP, Ponniah S, Peoples GE. The E75 HER2/neu peptide vaccine. Cancer Immunol Immunother. 2008;57(10):1511–1521. doi:10.1007/s00262-008-0540-3
81. Osborne C, Wilson P, Tripathy D. Oncogenes and tumor suppressor genes in breast cancer: potential diagnostic and therapeutic applications. Oncologist. 2004;9(4):361–377. doi:10.1634/theoncologist.9-4-361
82. Sood AK, Geradts J, Young J. Prostate-derived Ets factor, an oncogenic driver in breast cancer. Tumour Biol. 2017;39(5):1010428317691688. doi:10.1177/1010428317691688
83. Sood AK. PDEF and PDEF-induced proteins as candidate tumor antigens for T cell and antibody-mediated immunotherapy of breast cancer. Immunol Res. 2010;46(1–3):206–215. doi:10.1007/s12026-009-8129-2
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.