Back to Journals » Clinical Interventions in Aging » Volume 12
Does eating less make you live longer and better? An update on calorie restriction
Authors Picca A, Pesce V, Lezza AMS
Received 11 July 2017
Accepted for publication 6 October 2017
Published 8 November 2017 Volume 2017:12 Pages 1887—1902
DOI https://doi.org/10.2147/CIA.S126458
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Richard Walker
Anna Picca,1 Vito Pesce,2 Angela Maria Serena Lezza2
1Department of Geriatrics, Neuroscience and Orthopedics, Catholic University of the Sacred Heart School of Medicine, Rome, 2Department of Biosciences, Biotechnologies and Biopharmaceutics, University of Bari, Bari, Italy
Abstract: The complexity of aging is hard to be captured. However, apart from its tissue-specific features, a structural and functional progressive decline of the whole organism that leads to death, often preceded by a phase of chronic morbidity, characterizes the common process of aging. Therefore, the research goal of scientists in the field moved from the search for strategies able to extend longevity to those ensuring healthy aging associated with a longer lifespan referred to as “healthspan”. The aging process is plastic and can be tuned by multiple mechanisms including dietary and genetic interventions. To date, the most robust approach, efficient in warding off the cellular markers of aging, is calorie restriction (CR). Here, after a preliminary presentation of the major debate originated by CR, we concisely overviewed the recent results of CR treatment on humans. We also provided an update on the molecular mechanisms involved by CR and the effects on some of the age-associated cellular markers. We finally reviewed a number of tested CR mimetics and concluded with an evaluation of future applications of such dietary approach.
Keywords: aging, calorie restriction, studies on humans, CR molecular mechanisms, CR mimetics
Introduction
The natural and multifactorial process of aging affects all organisms. Although presenting tissue-specific features, the trait of a progressive structural and functional decline with advancing age, ultimately leading to death, is unanimously shared. Often the lifelong experiences of aged people make them an invaluable reservoir of wisdom for younger people. However, the negative correlates of aging (eg, physical and cognitive decline) make such older persons progressively less independent in self-sustaining and more needy of support from society.
The constantly “graying” of Western countries has already led to a heavy economic social burden because the lifespan extension reached in the past decades has often been accompanied by the chronicity of age-related diseases. This indicated the need for reframing the goal of extended longevity to that of healthy aging associated with a longer lifespan, referred to as “healthspan”.1 A great deal of research demonstrated that aging has not a unique cause, whereas multiple mechanisms tune up the whole aging process.2 Thanks to the pioneering work carried out on the Caenorhabditis elegans model by Kenyon et al, it became clear that the aging process is plastic and capable of being accelerated or attenuated by a range of dietary and genetic interventions.3 This paved the way to the search of lifestyle interventions aimed at promoting healthy aging through prevention or delay of age-associated dysfunctions.
According to several authors,2,4–6 a successful approach to healthy aging should be able to counteract the following nine cellular markers of aging: 1) telomere erosion, 2) epigenetic alterations, 3) stem cells depletion, 4) cellular senescence, 5) mitochondrial dysfunction, 6) genomic instability, 7) proteostasis imbalance, 8) impaired nutrient sensing, and 9) abnormal intercellular communication. To date, the most robust intervention efficient in warding off the aforementioned cellular markers of aging is calorie restriction (CR) that involves the administration of a well-balanced, nutrient-dense diet that reduces calorie intake by 20%–40% without malnutrition.7 CR has a dramatic effect (two- to threefold) in extending both median and maximal lifespan in rodents, and it prevents or delays the onset of various age-related diseases such as obesity, type-2 diabetes, neurodegeneration, cardiomyopathy, and cancer.8
Here, after a preliminary presentation of the major debate originated by CR, we concisely overviewed the recent results of CR treatment on humans. We also provided an update on the molecular mechanisms involved by CR and the effects on some of the age-associated cellular markers. We finally reviewed a number of tested CR mimetics and concluded with an evaluation of future applications of such dietary approach.
CR: the issue of the real cause
The unified hypothesis about CR and longevity by Sinclair proposed that CR might increase the survival capability of the organism by evoking a highly conserved stress response.9 The “Hormesis hypothesis of CR” provided further support to such a model suggesting that the adaptive responses of cell and organs, induced by a moderate stress, prevent worse damage caused by a stronger similar stress.10–13 Since the initial experimental reports on CR, a major debate was arisen about the nature of the cause of the extended longevity namely whether it was due to the reduction of protein intake14 or to that of calories.15 For a long period, the identification of reduced calorie intake as a responsible factor for increased longevity prevailed.16 In the 2000 decade, works on insects (Drosophila melanogaster) by Partridge et al17,18 have reopened the question about the relevance of amino acids and proteins for the CR impact on longevity. It has been argued that the apparent effect of the decreased energy intake was really due to an altered ratio between protein and non-protein components of the diets.19,20 A novel approach, the “geometric framework” or “nutritional geometry”,20–23 provided a helpful tool for dissecting the consequences of composite nutritional manipulations of the diet on different phenotypic parameters including lifespan. Recent massive work has been performed on ad libitum-fed mice changing the protein to carbohydrate ratio (25 different diets) and then reporting the animals’ responses, including lifespan, in a two-dimensional plot.24 Evidence suggested that a decline in protein to carbohydrate ratio is the factor mostly affecting longevity. The clear indication drawn by Solon-Biet et al24 derived, however, from an intake restriction originated by additional indigestible cellulose that diluted the diet. Such diet “dilution” might have deeply affected the neuropeptide pathways linking restriction to lifespan benefits by inducing responses markedly different in comparison with those elicited by the simple reduction of food mass.25 Reanalysis of the insect results with the “geometric framework” tool led to suggest that something substantially different might be happening in these animals through protein restriction (PR) with respect to what a reduced calorie intake alone might induce in rodents.26 A recent application by Speakman et al27 of conventional statistical analyses as well as of the nutritional geometric framework approach to the literature data, through a very accurate and thoughtful survey, has prompted again to the conclusion that CR impact on rodents longevity is exerted independently of PR occurring or not and that it is due to calorie deficit. The evident contrast with previous experimental work by Solon-Biet et al24 has been reconciled, confirming the completely different effect of CR plus PR on rodents lifespan versus that of PR alone and demonstrating that reduced calories and not reduced protein intake cause the food restriction effect on longevity.27 A more recent contribution to the longstanding debate on the nature of the real cause of the benefits of CR has been made by Simpson et al28 by applying a nutritional geometry approach to the meta-analysis of previous reports on rodents and insects. In particular, this last study has questioned the conclusion drawn by Speakman et al,27 about the unique relevance of the reduced intake of calories versus that of protein to increase lifespan, indicating a series of factors (analysis of various rodents strains with different ages of diet onset, absence of the exact composition of all included diets, and impossible evaluation of the “protein leverage” effect occurring in ad libitum-fed animals exposed to a low-protein diet) that might have been underestimated, thus leading to a not-straightforward final evaluation. It has been also marked that Speakman et al,27 however, acknowledged a positive impact on longevity of the protein amount reduced below that of the reference diets and that the opposite conclusions might have derived from the difficult interpretation of the nutritional interventions by conventional instruments.28 Therefore, the application of the nutritional geometry approach might be very useful and might allow to modify the original question about the optimum diet for lifespan in order to include multiple variations in the intake of macronutrients and calories. Overall, experimental results of geometric framework studies24,29 are consistent with that of extensive meta-analyses.27,30 In fact, they all agree about the occurrence of the lowest risk of mortality with a protein content between 10 and 30% of total calories, while mortality increases together with protein content. This has shed a new light on CR, suggesting that it does not deal simply with a quantitative reduction in energy, but also with a qualitative change in macronutrients. In agreement with this, it might be helpful to introduce here the distinction between CR, implying a decreased total calorie intake, namely glucose dilution for yeast, dilution of bacterial density for worms, simple food dilution for fruit fly or food restriction for mammals, and dietary restriction (DR), involving the reduced intake of specific nutrients, each characterized by its respective caloric value. Such latter intervention causes a change in the total calorie intake as well as in the ratio between macronutrients, often in the proteins to carbohydrates ratio, which appears to be crucial for the impact on longevity.31,32 In particular, the application of DR regimens has allowed a more detailed definition of the assayed diet making more straightforward the interpretation of the experimental results and clarifying the relevance of specific nutrients.31
Results of CR treatment in humans
Results from three ongoing long-term studies on primates indicate an increased healthspan in animals treated with CR.33–36 The CR-mediated influences on disease prevention and longevity have yet to be clarified, but the beneficial consequences of CR on human metabolic and molecular adaptations have been already extensively analyzed. The most thorough and extended review of the results obtained from various kinds of CR regimens applied to humans is the recent analysis by Most et al.8
Short-term CR interventions in humans
The first reported CR study on humans involved a 10-week 20% CR that led to a reduced resting metabolic rate per kilogram of fat-free mass (FFM),37 thus showing that the energy expended per FFM could be changed through a metabolic adaptation. A reduction in both systolic and diastolic blood pressure values38 and in glucose concentrations was demonstrated in the CR group.39 Then the first controlled clinical trials of CR with adequate nutrient provision in healthy, non-obese humans were started by the US National Institute of Aging through the CALERIE-1 (Comprehensive Assessment of Long-term Effects of Reducing Intake of Energy) study. The project included three pilot studies carried out, respectively, at Pennington Biomedical Center in Louisiana, at Washington University in St Louis, and at Tufts University in Boston, and designed to evaluate feasibility and effects of CR on metabolic health after 6 months40,41 or 12 months.42 In these pilot studies, CR obtained a negative energy balance through different situations: 1) reduced CR, 2) increased exercise energy expenditure, or 3) combination of CR and exercise. Enrolled subjects’ fasting insulin concentrations decreased after CR and insulin sensitivity improved by 40% (p=0.08).43 In the CALERIE-1 trial, different markers related to cardiovascular diseases (CVDs) (eg, blood pressure, low-density lipoprotein [LDL], high-density lipoprotein [HDL], fibrinogen, and others) were not affected by CR, but it was calculated that the 25% CR diet for 6 months reduced by 29% the 10-year risk for CVD.44 In addition to this, a reduction in markers of oxidative stress (DNA damage and superoxide dismutase activity) was described45 for the first time. The following Phase II multi-center trial (CALERIE-2 study) was performed to analyze whether 2-year 25% CR in leaner and younger men and women was efficient and safe. Therefore, 220 healthy, young, and middle-aged (21–51 years old), non-obese men and women were examined for the efficacy of CR on energy metabolism, metabolic adaptations, immune function, chronic disease risk factors, and quality of life.46 Since the major contributor of metabolic rate is FFM, biopsies studies from CR-treated skeletal muscle could help to shed light on the molecular mechanisms of CR-induced metabolic adaptation. Mitochondrial biogenesis was promoted by CR in skeletal muscle. After 1-year of exposition to 20% CR, or to 20% increased energy expenditure because of endurance exercise, or to a healthy lifestyle,42 the CR-induced weight loss improved insulin sensitivity, increased adiponectin, and reduced the serum concentrations of leptin, insulin, LDL cholesterol, and C-reactive protein.47,48 Moreover, 30% CR sustained for 2 years, reducing energy intake to 75% of baseline (25% CR), resulted in adaptive changes identical to those reported in rodents subjected to CR.49 The 2-year of CR intervention was adopted also because the duration 6 months and 1 year in CALERIE-1 pilot trials was evaluated as not sufficient to induce in humans several of the metabolic and hormonal changes thought to favor rodents longevity. It has been demonstrated by the results of this large trial that mild CR can improve cardiometabolic risk factors, well below conventional clinical thresholds, even when healthy lean or slightly overweight young and middle-aged men and women are exposed. Total cholesterol, LDL cholesterol, triglycerides, C-reactive protein, tumor necrosis factor (TNF)-α, and blood pressure decreased significantly and HDL cholesterol increased in the CR group, even in people with normal risk factors.49
Long-term CR interventions in humans
At present, only the collection of data recorded from the members of the Calorie Restriction Society, who have imposed on themselves a regimen of severe CR with optimal nutrition (CRON), believing to extend in this way their healthy lifespan, provides direct evidence that CR may affect the aging process in humans. These very lean men and women (body mass index 19.7±1.8 kg/m2) voluntarily restricted their caloric intake (1,800 kcal/d) for an average of 15 years and consumed ~30% less energy than a counterpart (matched for age, sex, and socioeconomic status) fed with a regular Western diet. It is important that the CRON diet meets all recommendations for essential nutrients and is very rich in vegetable fiber and low glycemic foods including a wide variety of phytochemicals, which may modulate metabolic health.50 CRON data on longevity and mortality are not yet available, but the collected findings indicate that moderate/severe CR in humans induces the metabolic and molecular changes described in long-lived CR animals.8 Furthermore, such CR with optimal intake of nutrients has decreased metabolic and hormonal risk factors for type-2 diabetes, CVD, stroke, cancer, and vascular dementia in the participants.50 The total cholesterol–HDL cholesterol ratio was 2.6 and the range of triglycerides was 50 mg/dL. Systolic and diastolic blood pressure was 110/70 mmHg, even in people in their late 70s, and C-reactive protein was almost undetectable.50,51 Serum TNF-α, interleukin (IL) 6, fasting glucose, and insulin showed low values, and insulin sensitivity (HOMA-IR) improved.52 The variability of heart rate in the CRON practitioners was comparable with normal values of healthy men and women 20 years younger.53 Differently from the CALERIE trials, most hormonal adaptations described in long-lived CR rodents were found in subjects exposed to severe CR. It is relevant that studies about CR with adequate nutrition and behavioral support to participants have not reported an influence of CR on psychological and mental capacity both as a 20% CR for 10 weeks38 and 25% CR for 6 months in CALERIE-1.54 In the longer CALERIE-2 trial, bone mass significantly was reduced at sites of osteoporotic fractures as the hip and femoral neck and the lumbar spine.55 Such data might limit the application of CR for older persons, eventually affected by accelerated bone loss. However, results from CRON practitioners are consistent with those from animal studies indicating that CR reduces bone mineral density but improves quality and strength of bones through their reduced turnover and prevention of secondary hyperparathyroidism.56 Furthermore, absolute maximal aerobic capacity per kilogram of body mass was maintained or increased during CR, suggesting a positive effect of the body weight loss on physical functioning. Effectively, also quality of life improved according to scores from survey on physical component, depression, and physical functioning.49
CR: effects and mechanisms
CR has pleiotropic effects, and by improving multiple metabolic pathways, it generates benefits for the whole organism. In particular, some indications deriving from a thorough investigation of the CR counteracting action, with respects to the aforementioned cellular markers of aging, will be concisely outlined in the present paragraph, whereas a more detailed discussion of the specific mechanisms involved will be developed in the following paragraphs.
Genomic instability and epigenetic alterations
Telomere shortening and high levels of DNA damage (both at nuclear and mitochondrial level), including mutations, DNA breaks, and chromosomal rearrangements, are typical age-related alterations. With aging, the DNA repair capacity decreases causing genome instability.57 CR has a positive effect on the DNA repair and telomere machinery, thereby promoting genomic stability and healthy longevity.58 However, the genome is not unique in shaping cellular homeostasis, health, and aging, as “epigenetics” has unveiled mechanisms that adjust gene expression and directly affect disease/phenotypes, without changing DNA sequences. Epigenetic marks can be added/removed on histones or the DNA itself, modulating chromatin remodeling and gene expression according to various environmental signals.59 There exist some cellular pathways that are able to sense nutrients and/or energy levels and also to influence processes like epigenome remodeling, gene expression, protein activity, and organelle integrity.2 Such pathways are deeply involved in modulating aging and age-related diseases and in mediating the CR beneficial effects promoting proteostasis balance, genome stability, and stem cell survival.60 However, all CR mechanisms have not been yet clarified.
Proteostasis imbalance
Another mark of cellular aging is related to the crucial functional role performed by proteins inside cells where these molecules integrate all physiological pathways. Therefore, the stability of proteins (proteostasis) indicates the protection of protein structure and function, against environmental and internal stressors, operated by the cell. Vulnerability in proteostasis correlates with age-related changes and longevity rates among species.61 An intricate proteostasis network including synthesis of proteins, activity of chaperones, autophagy, unfolded-protein response, and ubiquitin-proteasome pathway, counteracts, inside cells and mitochondria, protein misfolding and unfolding, thus allowing a regular protein turnover.62 Effectively, CR appears associated with an increase in relevant chaperones and autophagic mediators active in protein quality control and removal of dysfunctional proteins and organelles.
Mitochondrial dysfunction, oxidative damage, and impaired nutrient sensing
Other two hallmarks among CR effects are reduction of oxidative damage and modulation of mitochondrial activity. With dietary treatment, endogenous antioxidants are induced and cell membranes peroxidability index is modulated, thereby decreasing oxidative damage.6 Pathways able to sense nutrients and/or energy levels – namely AMP-dependent kinase (AMPK) and sirtuins, the nicotinamide adenine dinucleotide (NAD)+-dependent deacetylases that regulate the activity of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), and forkhead box (FoxO) proteins, involved in mitochondrial biogenesis, turnover, and oxidative metabolism – are activated to induce mitochondrial activity with CR. Interestingly, two different pathways, target of rapamycin (TOR) inhibition and AMPK activation, which are both sensitive to energy/nutrient status, can induce FoxO activation. In general, CR decreases nutrient uptake that unbalances metabolism and requires the interaction of several regulatory pathways to reach a new equilibrium. A new balance is obtained via the coordination of growth-associated pathways, such as the insulin/insulin-like growth factor-1 (IGF-1) signaling (IIS) pathway and TOR, which are downregulated, and those activating a more efficient respiratory metabolism such as AMPK and sirtuins that are induced. This means that the diet-reduced calorie intake increases metabolic efficiency and protects against cellular damage and remodeling mechanisms, thereby reducing less efficient metabolism and synthetic pathways. It seems that an evolutionarily conserved response aiming to avoid useless expenditure of energy and to recycle structures for the organisms’ survival is activated by CR. Therefore, CR-induced block of IGF-1 receptor-dependent pathways and TOR-dependent activities inhibits processes involved in cell proliferation and glycolysis. Recently, it has been shown that CR elicits also an anti-inflammatory effect, by inhibiting nuclear factor-kB (NF-kB) activity and by reducing pro-inflammatory profile of aging. Therefore, CR-related extension in healthspan and longevity derives from the interrelated contributions of all the mechanisms described here (Figure 1).
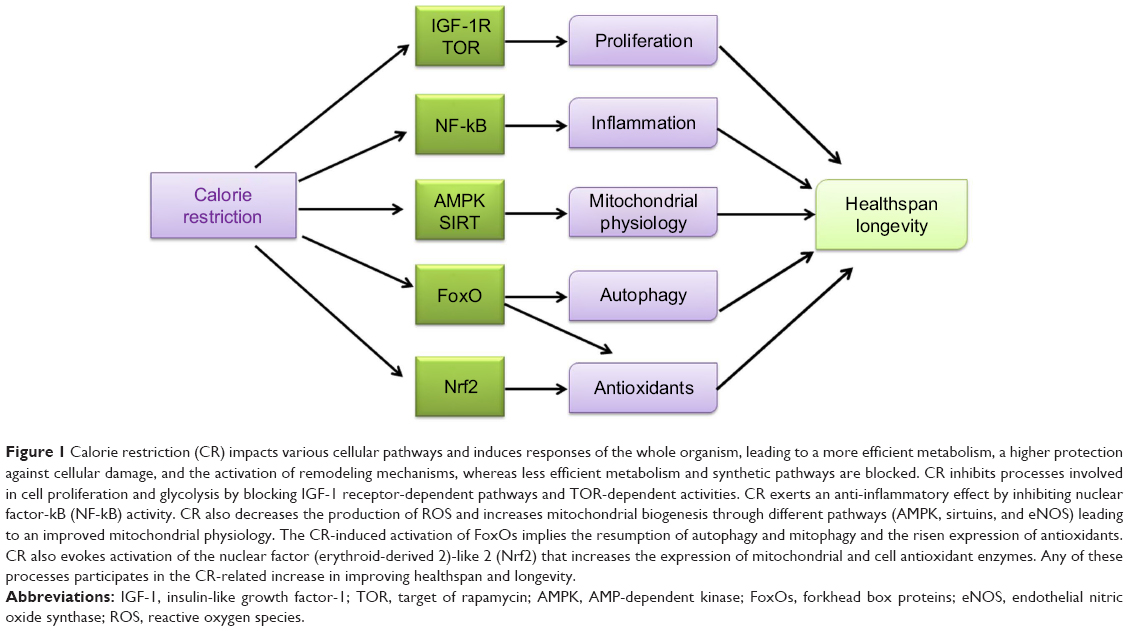 | Figure 1 Calorie restriction (CR) impacts various cellular pathways and induces responses of the whole organism, leading to a more efficient metabolism, a higher protection against cellular damage, and the activation of remodeling mechanisms, whereas less efficient metabolism and synthetic pathways are blocked. CR inhibits processes involved in cell proliferation and glycolysis by blocking IGF-1 receptor-dependent pathways and TOR-dependent activities. CR exerts an anti-inflammatory effect by inhibiting nuclear factor-kB (NF-kB) activity. CR also decreases the production of ROS and increases mitochondrial biogenesis through different pathways (AMPK, sirtuins, and eNOS) leading to an improved mitochondrial physiology. The CR-induced activation of FoxOs implies the resumption of autophagy and mitophagy and the risen expression of antioxidants. CR also evokes activation of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) that increases the expression of mitochondrial and cell antioxidant enzymes. Any of these processes participates in the CR-related increase in improving healthspan and longevity. |
Management of reactive oxygen species (ROS), lipid peroxidation, and mitochondrial fitness
A relevant increase in ROS level, reduction in antioxidant defenses, and appearance of mitochondrial dysfunction, implying accumulation of oxidative damage to mitochondrial DNA, proteins, and lipids, are usually associated with aging.63–65 However, age-related mitochondrial dysfunction exceeds just the increased oxidative damage because it affects other key mitochondrial functions such as biogenesis, turnover, dynamics, and protein quality control.66–70 On the other side, the “Hormesis hypothesis” has highlighted the relevance of ROS as signaling molecules in several cellular processes among which lifespan extension is included, which is attained via the regulation of nuclear genes expression by retrograde signaling pathways.71 Aging correlates with increased mitochondrial and cell ROS production and with increased degree of fatty acid unsaturation in membranes, due to a major fraction of more oxidizable polyunsaturated fatty acids.72 Such two factors, together with a reduction in membrane-linked antioxidant enzymes, trigger age-increased lipid peroxidation73 that impacts on various cell processes. CR in mitochondria reduces the membrane potential and the production of ROS, as well as it counteracts the age-related membranes deterioration by changing the saturation/unsaturation index of fatty acids with positive effects on oxidative damage and membrane fluidity. In particular, it has been recently demonstrated that the fatty acid types introduced with the diet may influence the CR effect.74 As for the age-related increase of ROS, these reactive species, prevalently produced inside the cells by the mitochondrial electron transport chain, are released in higher amounts by the damaged mitochondria, which become also inefficient for energy production. In particular, age-related mitochondrial dysfunction affects the assembling of respiratory complexes in supercomplexes, involved in ROS regulation, thus increasing ROS production.75 Furthermore, it has been demonstrated that the relevance of a cascade that, through the activation of nuclear factor (erythroid-derived 2)-like 2 (Nrf2), increases the expression of NAD(P)H dehydrogenase and quinone 1 (Nqo1) and ensures the maintenance of mitochondrial membrane potential compromised during aging in mice.76 CR at the plasma membrane also induces the expression of coenzyme Q-dependent enzymes in old rodents but not in young animals,77,78 suggesting that the age of onset of CR might be important for organisms. Age-related mitochondrial dysfunction impairs mitochondrial biogenesis especially in those tissues largely dependent on oxidative energy for their metabolism such as muscle, brain, or heart.79 CR induces mitochondrial biogenesis in human heart and skeletal muscle and in rodents liver and skeletal muscle66,78,80–82 by means of at least three different pathways that have been analyzed in muscle: induction of endothelial nitric oxide synthase (eNOS), activation of PGC-1α, and adiponectin-dependent activation of the SIRT1/AMPK axis. The CR-related induction of NOS in mice and human muscle45,83 is very relevant for the sequential activation of mitochondrial biogenesis. SIRT1 is also essential since it activates eNOS by deacetylation,84 thus favoring mitochondrial biogenesis. However, mitochondrial biogenesis, dynamics, and genome copy number as well as oxidative phosphorylation are all controlled by the master regulator PGC-1α85 that adapts mitochondrial population to energy needs. In fact, more recently, it has been suggested that the beneficial impact of CR on aging might be reached through the attenuation of age-related mitochondrial decline obtained by a complex crosstalk among energy-sensing pathways.86 In fact, PGC-1α activity is directly modulated at transcriptional and posttranslational level87 by two major cell metabolic sensors namely AMPK and SIRT1, through phosphorylation and deacetylation.88,89 Also a factor important in age-related mitochondrial dysfunction is the accumulation of damaged organelles due to reduced mitochondrial turnover by inhibition of mitophagy, specifically dedicated to the removal of damaged mitochondria.65 Effectively, the renovation of mitochondrial network is very relevant in the CR-dependent healthspan increase90 and in counteracting the aging process.91
CR: the contribution of genetics and epigenetics of longevity
The studies about genes influencing lifespan led to the conclusion that human longevity can be influenced by about 25% by genetic factors, whereas environmental factors strongly affect lifespan by targeting relevant pathways in which alterations of the expression pattern are induced,1 also by changes in chromatin structure.92,93 The object of epigenetics is “chromatin” and the mechanisms through which it modulates gene expression. Such mechanisms operate by means of changes in the chromatin structure that may involve DNA methylation profile, addition/removal of methyl groups to/from cytosines, involvement of histones via posttranslational modifications (methylation, acetylation, or phosphorylation), and nucleosomes positioning via ATP-dependent chromatin remodelers. The presence of a marked DNA methylation and specific histone modifications can render chromatin so tight and compact (closed) to become poorly accessible to the transcription machinery. Vice versa, unmethylated DNA and a different array of histone modifications can favor a looser or “open” form of DNA that might be actively transcribed. With aging, the epigenome undergoes remodeling at both the histone and DNA levels and the hope is that, by ensuring epigenetic stability, a disease-free lifespan might be obtained.94 It appears likely the existence of a crosstalk between the chromatin and metabolome making of chromatin the nexus where the balance between fecundity and longevity is decided.95 Effectively, chromatin and metabolome are very closely linked through metabolic cofactors. Products of the tricarboxylic acid (TCA) cycle, glycolysis, and β-oxidation feed into chromatin-modifying enzymes and adjust their activity, adapting epigenetic responses to the metabolic phenotype of the cell.95 CR implies large changes in metabolic cofactors likely altering the cellular availability of enzyme co-factors and substrates, such as s-adenosyl-methionine, acetyl-CoA, and NAD+, so much to affect all enzymes using these factors, among which many control methylation and acetylation reactions. It appears that age-related decline may be causally related to age-dependent loss of cell-specific transcriptional regulation and genomic integrity due to changes in the genome methylation profile.96 Such changes might be attenuated by CR, as short-term CR has reduced age-dependent alterations of methylation in the promoters of genes involved in age-associated degenerative phenotypes in 25-month-old rats.97 CR also works by inhibiting anabolic pathways, allowing cells to invest resources for maintenance and stress resistance, which improve health and longevity, via alteration of programs of gene expression. CR, in fact, increases the activity of histone deacetylases, particularly SIRT1, which is localized to age-associated genes such as p16 in CR, thus contributing to their repression through histone H3 hypoacetylation. This effect increases the replicative lifespan of human fibroblasts in vitro.98 Histone acetylation should favor gene activation through cumulative neutralization of histones’ positive charges, leading to disruption of nucleosomes and finally to generation of a more “open” chromatin structure.99 Modified histone acetylation profiles, particularly hyperacetylation of histones, have been linked to aging phenotypes. However, we now begin to understand that histone acetylation plays a more nuanced role in gene regulation, with site-specific changes in histone acetylation being as important as more global alterations.100 Few recent reports have demonstrated that longevity interventions can attenuate age-related chromatin decline, but the specific crucial alterations that impact on healthspan await to be highlighted by further studies.94
Sirtuins
The role of sirtuins pathway in regulating lifespan was discovered in yeast,101 but the family of these NAD+-dependent protein deacetylases is present in all organisms where intervenes favoring longevity102 and, by removing acetyl residues, it regulates the activity of many enzymes. In humans, this family of ubiquitously expressed proteins comprises seven members, each featuring a specific localization. The most thoroughly studied SIRT1 regulates gene expression and the function of various proteins active in different molecular pathways. Furthermore, SIRT1 and the nuclear SIRT6 and 7 act as DNA repairing enzymes,103 whereas SIRT3, SIRT4, and SIRT5 regulate mitochondrial functions. Sirtuins are strongly involved by CR as reported in mammals, where the diet induces in many organs the expression and the activity of such enzymes, and several CR-related metabolic changes are implied by sirtuins’ activities.104 In particular, the deacetylating activity of sirtuins is coupled to NAD+ hydrolysis, thus originating the deacetylated substrate, O-acetyl-ADP-ribose and nicotinamide, a sirtuin inhibitor. Sirtuins depend on NAD+ for their activity, which link them to the availability of this co-substrate and to the cell energy balance; therefore, shifts in the NAD+/NADH ratio are directly detected by sirtuins acting as nutrient and metabolic sensors that influence enzymes to rapidly recuperate cell homeostasis.105,106 When NAD+ accumulates, such as during scarcity of nutrients, especially glucose, sirtuins are activated. These deacetylases affect both epigenome and acetylome since they remove acetyl groups from the lysine residues of histones and non-histone proteins. In particular, acetylome modulation has recently gained a great consideration because of the constantly growing number of proteins regulated by acetylation/deacetylation, involved in various crucial cellular processes like cell cycle, metabolism regulation, antioxidant protection, autophagy, and more. Therefore, the relevance of sirtuins for CR impact on longevity and healthspan has been thoroughly analyzed.107 In particular, the CR-activated SIRT1 senses NAD+ abundance and deacetylates specific histone-acetylated residues, acting as an epigenetic regulator, but at the same time, by deacetylating transcription factors such as NF-kB, PGC-1α and FOXO3a regulate cell expression.108
For example, CR induces in mice and human muscle the SIRT1-mediated activation of eNOS45,83 that is essential for promoting mitochondrial biogenesis. This is also favored through the activation of PGC-1α obtained by means of the SIRT1-mediated deacetylation.88,89 Thus, regulators of metabolism such as sirtuins are involved in the multiple effects of CR also through the complex network between their activities modulating mitochondrial biogenesis, dynamics, and turnover. However, after the initial general consensus, the need of sirtuins’ activation for CR efficacy has been objected109 and the overall anti-aging activity of sirtuins has been questioned because pro-aging actions of specific sirtuins (Sir2 in S. cerevisiae and SirT1 in murine neurons) have been demonstrated.110,111 It has been reported that the ablation of the SirT1 gene reduced age-related oxidative stress markers in the neurons of old mice, thus supporting its neuronal pro-aging role. Such ablation, although, induced very serious consequences for the whole organism namely severe developmental defects and a shorter lifespan in both ad libitum-fed and restricted animals that confirmed the SirT1 general protective role.111,112 More recently, the debate about the relevance of sirtuins activity for lifespan has continued with negative113,114 and positive102,115 reports that led to the final assessment of sirtuins as evolutionarily conserved regulators of aging/longevity.116 In particular, the control over longevity exerted by sirtuins appears very tightly linked to cell NAD+ levels and this has led to include NAD+ among the CR mimetics (see the following specific section). Another pathway responsible for the involvement of sirtuins in the modulation of aging and related conditions acts through the age-dependent nuclear genome instability driving the activity of SIRT1, SIRT6, and SIRT7 that participate in nuclear DNA repair and regulate mitochondrial homeostasis and mitochondrial unfolded protein response,117 a signaling pathway contributing to longevity.115 DNA damage and repair are also relevant because their balance addresses the cell destiny toward apoptosis and death or autophagy/mitophagy and survival. Sir2 in yeast and SIRT1 in mammals have been reported to activate, respectively, mitophagy118 and autophagy.119 It has also been demonstrated that SIRT1 interacts cooperatively with the energy sensor pathway of AMPK (see the following section) to promote mitophagy, likely through a reciprocal feedback loop.120 This suggests that SIRT1 may protect from aging by inhibiting apoptosis and maintaining mitochondrial integrity at the expenses of NAD+ cellular content. The regulation of NAD+ content depends, however, also on the competition between two NAD+-consuming enzymes namely SIRT1 and poly(ADP-ribose) polymerase 1 (PARP1), with the last activated to repair DNA damages. Therefore, when the damage level is so high to induce PARP1, this uses NAD+ up, which is not anymore available for SIRT1121 and causes inhibition of the sirtuin and the subsequent reduction of autophagy and mitophagy. The finding that in some premature aging diseases, where defective mitophagy should trigger pathology progression, the restoration of the NAD+–SIRT1 (SIR2.1) pathway has improved healthspan122,123 strongly supports the link between sirtuins and autophagy/mitophagy.
IGF/TOR/FoxO and APMK pathways
The target of IGF-1 signaling (IIS) pathway is sensitive to nutrients and integrates growth/survival signals with glucose abundance. Aging appears modulated by changes in the insulin/IGF-1 receptor signaling system,124–126 as longevity is enhanced by a decrease in IIS signaling in worms, flies, and mice. The IIS pathway includes, among the main constituents, the gene expression regulatory factors FoxOs, whose localization either in the cytoplasm or in the nucleus depends on nutrient conditions. The IGF-1 signaling pathway is activated by nutrients abundance and induces activation of TOR and ribosomal protein 6 kinase (S6K) with sequential protein synthesis, while inhibiting FoxO-mediated transcription. These factors are relevant for CR impact on lifespan, as longevity in both C. elegans and D. melanogaster is reduced by the inhibition of the FoxO orthologue daf-16 by IGF-1-dependent signaling.3 The relevance of FoxO for the induction of various CR protective mechanisms is supported by its involvement in the activation of several pathways induced by stress response genes.127 CR decreases IGF-1, insulin, and glucose plasma levels in rodents128 and also in humans.48 The bacteria Streptomyces hygroscopicus produces the antibiotic rapamycin, largely used for immunosuppression and cancer therapy. Later on, in mammals, the material target of rapamycin was pointed out as the mammalian target of rapamycin (mTOR) gene. The mTOR is a major metabolic switch that controls cell resources assignment so that growth factors and amino acid availability activate it to promote cell anabolism, while conditions of nutrient deprivation or rapamycin treatment repress it to favor cellular catabolism, through derepression of autophagic programs and drastic reduction of protein synthesis to free up cytosolic resources. Insulin, amino acids, and hormones trigger the product of mTOR that is a serine/threonine protein kinase involved in the regulation of relevant cell processes such as growth, proliferation, motility, survival, protein and lipid synthesis, autophagy, inflammation, mitochondrial function, and glucose metabolism.129,130 TOR kinase functions through the formation of two distinct complexes: TORC1 and TORC2, with only TORC1 being rapamycin sensitive in yeast and mammals, although prolonged rapamycin treatment can also inhibit TORC2.131 Thus TORC1 activation after glucose input or insulin/IGF signaling or abundant amino acids availability has an opposite effect compared to that of sirtuins that are supposed to reduce TORC1 signaling130 as stress signals or energy deficiencies do.132 The activation of TORC1 induces protein translation and cell growth, whereas its inhibition stops growth and elicits stress responses as autophagy.129 Reduced mTOR signaling has been demonstrated to extend lifespan in different organisms.133 The mTOR inhibition has been largely identified as a longevity assurance mechanism,133 and the availability of rapamycin and other mTOR inhibitors makes this pathway a valuable target for interventions to extend healthspan.134 An increase in the AMP/ATP ratio, such as in conditions when cells are scarce of glucose, activates the very sensitive energy sensor AMPK, leading to reduced ATP utilization and raised energy production, whereas its activity is reduced when the cell energy load is high, such as in the conditions of low AMP/ATP ratio.135 CR activates the AMPK pathway in heart, liver, and skeletal muscle of rodents,136 and the AMPK impact on lifespan, described in several organisms from yeast to mammals, can be obtained through the activation of FoxO factors, the main constituents of the IIS pathway137 (Figure 2). A longer lifespan is linked to an increased AMPK activity, while a reduced longevity is linked with its inhibition.138,139 However, in mammals, AMPK activity is not induced by CR, thereby raising doubts about its importance in these organisms.140
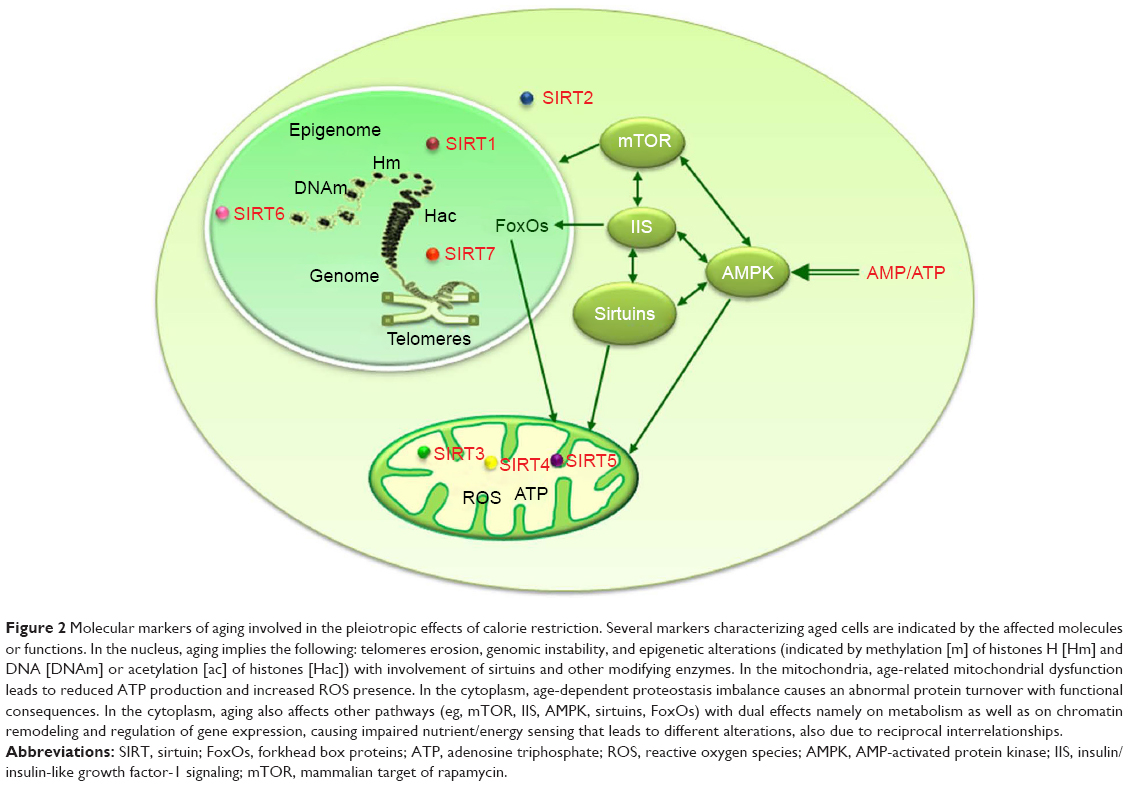 | Figure 2 Molecular markers of aging involved in the pleiotropic effects of calorie restriction. Several markers characterizing aged cells are indicated by the affected molecules or functions. In the nucleus, aging implies the following: telomeres erosion, genomic instability, and epigenetic alterations (indicated by methylation [m] of histones H [Hm] and DNA [DNAm] or acetylation [ac] of histones [Hac]) with involvement of sirtuins and other modifying enzymes. In the mitochondria, age-related mitochondrial dysfunction leads to reduced ATP production and increased ROS presence. In the cytoplasm, age-dependent proteostasis imbalance causes an abnormal protein turnover with functional consequences. In the cytoplasm, aging also affects other pathways (eg, mTOR, IIS, AMPK, sirtuins, FoxOs) with dual effects namely on metabolism as well as on chromatin remodeling and regulation of gene expression, causing impaired nutrient/energy sensing that leads to different alterations, also due to reciprocal interrelationships. |
Inflammation and CR
Aging is also characterized by a condition of relevant inflammation, demonstrated by the age-associated rise of several pro-inflammatory factors including TNF-α, interferon-γ, IL-1β, and IL-18.141–143 Furthermore, induction of such chronic inflammation during aging can be favored by age-related elevation of oxidative stress that also appears to raise the incidence of age-related diseases.144,145 Another feature of aging with pathological consequences is the increased production of danger-associated molecular patterns that, originated by damage accumulated with age, are recognized by immune receptors and induce the activation of the inflammasome,146,147 leading to chronic inflammation often accompanying different age-associated diseases.148 Such chronic inflammation associated with aging has been identified by the new idea of “inflammaging”.149 The CR-positive effects on inflammation have been demonstrated by the reduction of inflammation and insulin resistance obtained in a rat model of age-associated inflammation150 as well as by the regulation of GSH redox status and expression of NF-κB, SIRT1, peroxisome proliferator-activated receptors, and FoxOs.151
CR mimetics
A workshop entitled “Interventions to Slow Aging in Humans: Are We Ready?” was held in Erice, Italy, on October 2013, in order to define the more comprehensive set of pathways involved in aging and of safe counteracting strategies to slow the whole process and extend healthspan. Multiple interventions and final indications were very thoroughly exposed in the review,130 to which it is referred for deeper analyses and that was a major source for this section. In fact, a relevant concern about the feasibility of CR for most people, especially in the developed countries, deals with the severity of such regimen that has prompted the search for less drastic diets and safe drugs that may obtain the CR benefits with a more practical treatment. In recent years, growing attention has been dedicated to compounds, of different chemical natures, able to induce some of the effects evoked by CR in various organisms and thus grouped as CR mimetics. Such molecules share the ability to activate one or more pathways normally induced by CR.6 Studies in animals demonstrated that a decrease in IGF-1 levels or IGF-1 action can extend lifespan. The obese, long-living mice without the growth hormone receptor (GHR−/−) have reduced levels of IGF-1 and decreased risk for cancer and diabetes while are sensitive to insulin.152,153 In patients suffering from the GHR-deficient Laron syndrome, similar results were obtained. In order to pharmacologically decrease IGF-1 action, designed drugs can target cells/tissues that produce or respond to GH and/or IGF-1. Among such drugs, somatostatin analogues are not only able to decrease serum GH levels and IGF-1 levels, but they also abolish secretion of other endocrine hormones, including insulin, which makes their use inappropriate to prolong healthspan.130 Another approved drug for treating acromegaly is the GHR antagonist pegvisomant154,155 that inhibits GH action by binding to and blocking the GHR.156 Thus, pegvisomant might be assayed for longevity and healthy aging effects. Sirtuins are involved in pathways implying health benefits and are a relevant drug target. Efficient sirtuin-activating compounds (STACs) are plant-derived metabolites, such as flavones, stilbenes, chalcones, and anthocyanidins, that activate SIRT1 in vitro.157 Resveratrol (3,5,40-trihydroxystilbene) is still the strongest natural STAC158 and the best CR mimetic13 since it decreases insulin secretion and insulin level while enhancing sensitivity to insulin, reduces fat mass, promotes mitochondrial biogenesis and oxidative phosphorylation, raises the NAD+/NADH ratio thus inducing sirtuins, elevates AMPK activity, and supports mitophagy and autophagy.13 It has been recently reported that obese humans treated with resveratrol present effects similar to those induced by CR.159 In mice kept on a high fat diet, resveratrol increases lifespan160 as the synthetic activators SRT1720 and SRT2104 also prolong the lifespan of mice fed either a high-calorie or a low-calorie diet and both protect from tissue age-related changes.161 In preclinical trials, STACs have also shown to be very promising to cope with age-associated diseases and complications.158 To activate sirtuins in an alternate way, their need for NAD+ can be satisfied by providing NAD+ precursors or by inducing NAD+ biosynthetic enzymes162 or by inhibiting NAD+ hydrolase CD38.163 However, due to its multiple connections with energy/nutrient sensing pathways, mTOR signaling is a major candidate for targeted intervention to prolong healthspan. Many lifespan-extending interventions in model organisms, such as protein and CR/DR, decreased insulin/IGF signaling, activation of AMP kinase and possibly of sirtuins, reduce mTORC1 signaling,133,164 thereby raising the question of whether their positive effects depend on mTORC1 inactivation. Also rapamycin (sirolimus), probably the best characterized inhibitor of mTOR, has gained consideration because it increases lifespan and improves healthspan in several organisms.165 It has been demonstrated that stress, nutrient, and growth factors elicit responses in the conserved family of kinases of TOR proteins. The mimetic action of rapamycin is justified by the extended longevity attained by CR through downregulation of TOR in yeast and other organisms, as well as in mice.166–168 Furthermore, inhibition of TOR increases FoxO-dependent autophagy that is crucial to extend lifespan in yeast169 and in C. elegans.170 However, rapamycin has serious side effects (eg, hyperglycemia, hyperinsulinemia, insulin resistance, and proliferative defects in hematopoietic lineages),171 which limit its use as an anti-aging drug.
In 1998, 2-deoxy-D-glucose (2DG), an inhibitor of glycolysis, was indicated by Lane et al172 as another CR mimetic since it increased C. elegans lifespan173 and produced in rats various CR-like consequences such as reduced insulin level174 and increased glucocorticoids concentrations.175 But long-term administration of 2DG can induce cardiotoxicity and increased tumorigenesis in adrenal medulla.176 Another target of CR mimetics is AMPK activation that induces insulin sensitivity resulting in increased glucose uptake in skeletal muscles, reduced hepatic glucose production, and generalized raised fatty acid oxidation.177 Activators of AMPK as biguanides and thiazolidinediones have been developed. Metformin, a biguanine drug used for type-2 diabetes therapy, likely through AMPK activation, increases the lifespan of C. elegans and D. melanogaster.178 CR-like effects such as enhanced insulin sensitivity, reduced plasma levels of LDL and cholesterol, increased antioxidant protection, and reduced chronic inflammation are induced by low doses of metformin in mice.6 The consideration of metformin has grown also because of its association with the induction of SIRT1-mediated autophagy independently of AMPK.179 However, consideration of metformin for anti-aging clinical trials requires a deeper understanding of its mechanisms of action.180 This presentation of the pharmacological CR mimetics is not absolutely exhaustive, as approaches directed to mitigate telomere erosion, counteract epigenetic alterations, reduce stem cells depletion, and ward off cellular senescence have also been discussed in this paper reporting about the aforementioned meeting,130 but they exceed the limits of this review. Furthermore, recently, accumulating data have suggested that severe CR is not necessary, but different dietary interventions such as intermittent fasting, prolonged fasting, restricted feeding, and protein or selective amino acid restriction may recapture some of the positive results of severe CR in modulating certain anti-aging pathways.60 In fact, revitalization of mice endocrine, immune, and nervous systems and improvements in biomarkers of human diseases (diabetes, CVD, and cancer) have been obtained with intermittent CR, as a result of subjecting mice and humans to fasting for few days per week, without major negative consequences.181 Although all the effects involved by intermittent or prolonged fasting are not yet known, preliminary clinical trials in humans support these approaches as safe and efficacious to be tested in anti-aging studies.182 However, the fasting interventions have to be carefully applied because they can be dangerous for old people or patients receiving insulin or insulin-like drugs. Therefore, the alternate group of dietary interventions, namely the restriction of specific nutrients, has recently gained growing consideration. In fact, studies in humans, analyzing the composition of the diet, have reported very interesting findings. The evaluation performed by Pedersen et al183 led to the indication that an increase in all-cause mortality, type-2 diabetes mellitus, and CVD is possibly associated with low carbohydrate, high protein diets. The worst consequences were linked with animal-based proteins and other animal product–based diets, while vegetable-based proteins and other vegetable product–based diets were able to reverse the tendency. A later NHANES study published by Levine et al184 has showed, in agreement with previous data, that the problems implicated by a higher protein diet in people under 65 years of age are eliminated by the consumption of vegetable- rather than animal-based protein. In participants aged 50–65 years with the highest protein intake (more than 20% of calories from protein), all-cause mortality increased by 75% and cancer and diabetes mortality increased four times. However, in participants over 65 years, a reverse association occurred as subjects consuming the highest protein intake had a 28% reduction in mortality. Therefore, the source of protein may be as crucial as the amount of protein in the diet. However, it seems that the ratio of dietary protein to other macronutrient calorie sources (eg, carbohydrates and fats), likely affecting the regulation of mTORC1 signaling,24 is mainly responsible for the longevity benefits of PR.130 Other studies have focused on the DR of specific essential amino acids as that in which the reduction of branched chain amino acids to the levels found in a low protein diet caused an improvement of mouse metabolic health similar to that obtained with a low protein diet.185 Restriction of another essential amino acid, methionine, extends the longevity of flies, rats, and mice186–188 and reverses the metabolic dysfunction in aged mice.186 The mechanisms involved in the sensing of amino acid levels are not fully understood, but they certainly include the activation of GCN2 (general control non-derepressible 2) and mTOR. In mammals, while mTOR is induced by abundance of amino acids, particularly leucine, GCN2 responds to the absence of several distinct amino acids. In conclusion, although much research remains to be done, it appears that restriction of dietary protein contributes to the metabolic benefits of a CR diet and that alterations in protein quality may mimic some of the CR beneficial effects.189 This type of intervention, which does not reduce calorie intake, may extend the benefits of CR to a much broader population. However, CR or PR in the elderly has to be applied cautiously, as the preservation of muscle and bone mass is a priority in the aged to delay or prevent sarcopenia that is a major driver of frailty and loss of independence.190,191 Indeed, an increasing amount of data suggests that in the elderly protein intake may need to be increased to preserve muscle mass.192–196 Effectively, high protein diets, particularly in association with exercise, can improve muscle mass, if not function, in old age, by promoting anabolic responses in muscles.197,198 Therefore, recent findings suggest that there are valid alternates to conventional CR and that diets including altered protein contents demonstrate proteins’ activity in the regulation of longevity through the positive impact on metabolic health.199
Future applications
Overall, the results from multiple studies show that both reduced calories intake (CR) and the ratio between macronutrients, namely the protein to carbohydrates ratio, positively impact lifespan. In particular, it seems that dietary restriction of proteins or other individual nutrients (DR), with respects to carbohydrates, produces an effect on longevity independent on that of CR. DR appears to be efficacious at a much lower degree and is induced in a more severe range of restriction (50%–85%: relative to a reference intake of 18%–26% protein in the diet) than that affected by CR (10%–65% relative to ad libitum intake).27 Available data from the CRON practitioners confirm the positive effects of a reduced calorie intake on various parameters usually affected by age-related decline. However, it needs to be stressed that the CRON regimen is very rich in essential nutrients, vegetable fiber, low glycemic foods, and phytochemicals, which may modulate metabolic health. Furthermore, a dietetic indication, shared by all recent studies,27,28,30 suggests the opportunity of a reduced protein intake (between 10 and 30% of total calories) to lower the risk of mortality. However, Nakagawa et al30 also showed that very low level of protein was associated with reduced viability. It has been demonstrated that CR and DR have a large impact on healthspan and longevity through the involvement of multiple mechanisms affecting several metabolic pathways in tissues and organs, which have only begun to be dissected in details. It needs also to be considered that although DR can be a more feasible dietary regimen than severe CR, the pharmacological search for CR mimetics has developed a number of drugs that are already or next to be tested in clinical studies on humans and that will open a new, more walkable avenue toward increased healthspan. But in a nutshell, a non-severe reduction of the calorie with particular attention to a reduced protein intake versus a high carbohydrate one in not-aged subjects and a high protein diet in old persons might help people to better cope with a large number of age-related dysfunctions and diseases.
Acknowledgments
This research was supported by grants to AMSL (University of Bari-Progetti di Ateneo, 2012) and grant to AMSL (Istituto Banco di Napoli-Fondazione, 2015). We thank Flavio Fracasso, MS, for assisting with the figures.
Disclosure
The authors report no conflicts of interest in this work.
References
Passarino G, De Rango F, Montesanto A. Human longevity: genetics or lifestyle? It takes two to tango. Immun Ageing. 2016;13:12–17. | ||
Carmona JJ, Michan S. Biology of healthy aging and longevity. Rev Invest Clin. 2016;68(1):7–16. | ||
Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. | ||
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. | ||
Kennedy BK, Berger SL, Brunet A, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159:709–713. | ||
Lopez-Lluch G, Navas P. Calorie restriction as an intervention in aging. J Physiol. 2016;594(8):2043–2060. | ||
Fontana L. Modulating human aging and age-associated diseases. Biochim Biophys Acta. 2009;1790(10):1133–1138. | ||
Most J, Tosti V, Redman LM, Fontana L. Calorie restriction in humans: an update. Ageing Res Rev. 2017;39:36–45. | ||
Sinclair DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005;126:987–1002. | ||
Mattson MP. Dietary factors, hormesis and health. Ageing Res Rev. 2008;7:43–48. | ||
Rattan SI. Hormesis in aging. Ageing Res Rev. 2008;7:63–78. | ||
Martins I, Galluzzi L, Kroemer G. Hormesis, cell death and aging. Aging (Albany NY). 2011;3:821–828. | ||
Testa G, Biasi F, Poli G, Chiarpotto E. Calorie restriction and dietary restriction mimetics: a strategy for improving healthy aging and longevity. Curr Pharm Des. 2014;20:2950–2977. | ||
Slonaker JR. The effect of different per cents of protein in the diet VII. Life span and cause of death. Am J Physiol. 1931;2:266–275. | ||
McCay CM, Crowell MF, Maynard LA. The effect of retarded growth upon the length of life and upon the ultimate body size. J Nutr. 1935;10:63–79. | ||
Masoro E. Subfield history: caloric restriction, slowing aging, and extending life. Sci Aging Knowledge Environ. 2003;2003(8):RE2. | ||
Mair W, Piper MD, Partridge L. Calories do not explain extension of life span by dietary restriction in Drosophila. PLoS Biol. 2005;3:e223. | ||
Piper MD, Partridge L. Dietary restriction in Drosophila: delayed aging or experimental artefact? PLoS Genet. 2007;27(3):e57. | ||
Raubenheimer D, Simpson SJ, Mayntz D. Nutrition, ecology and nutritional ecology: toward an integrated framework. Funct Ecol. 2009;23:4–16. | ||
Piper MD, Partridge L, Raubenheimer D, Simpson SJ. Dietary restriction and aging: a unifying perspective. Cell Metab. 2011;14:154–160. | ||
Simpson SJ, Raubenheimer D. A multilevel analysis of feeding-behavior – the geometry of nutritional decisions. Philos Trans R Soc Lond Ser B Biol Sci. 1993;342:381–402. | ||
Simpson SJ, Raubenheimer D. Caloric restriction and aging revisited: the need for a geometric analysis of the nutritional bases of aging. J Gerontol A Biol Sci Med Sci. 2007;62:707–713. | ||
Raubenheimer D, Simpson SJ, Tait AH. Match and mismatch: conservation, physiology, nutritional ecology and the timescales of biological adaptation. Philos Trans R Soc Lond Ser B Biol Sci. 2012;367:1628–1646. | ||
Solon-Biet SM, McMahon AC, Ballard JW, et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014;19:418–430. | ||
Lusseau D, Mitchell SE, Barros C, et al. The effects of graded levels of calorie restriction: IV. Non-linear change in behavioural phenotype of mice in response to short-term calorie restriction. Sci Rep. 2015;5:13198. | ||
Taormina G, Mirisola MG. Calorie restriction in mammals and simple model organisms. Biomed Res Int. 2014;2014:308690. | ||
Speakman JR, Mitchell SE, Mazidi M. Calories or protein? The effect of dietary restriction on lifespan in rodents is explained by calories alone. Exp Gerontol. 2016;86:28–38. | ||
Simpson SJ, Le Couteur DG, Raubenheimer D, et al. Dietary protein, aging and nutritional geometry. Ageing Res Rev. 2017;39:78–86. | ||
Le Couteur DG, Solon-Biet S, Cogger VC, et al. The impact of low protein, high carbohydrate diets on aging and lifespan. Cell Mol Life Sci. 2016;73:1237–1252. | ||
Nakagawa S, Lagisz M, Hector KL, Spencer HG. Comparative and meta-analytic insights into life-extension via dietary restriction. Aging Cell. 2012;11:401–409. | ||
Kapahi P, Kaeberlein M, Hansen M. Dietary restriction and lifespan: lessons from invertebrate models. Ageing Res Rev. 2017;39:3–14. | ||
Lushchak O, Strilbytska O, Piskovatska V, Storey KB, Koliada A, Vaiserman A. The role of the TOR pathway in mediating the link between nutrition and longevity. Mech Ageing Dev. 2017;164:127–138. | ||
Zainal TA, Oberley TD, Allison DB, Szweda LI, Weindruch R. Caloric restriction of rhesus monkeys lowers oxidative damage in skeletal muscle. FASEB J. 2000;14(12):1825–1836. | ||
Black A, Allison DB, Shapses SA, et al. Calorie restriction and skeletal mass in rhesus monkeys (Macaca mulatta): evidence for an effect mediated through changes in body size. J Gerontol A Biol Sci Med Sci. 2001;56(3):B98–B107. | ||
Mattison JA, Roth GS, Beasley TM, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–321. | ||
Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun. 2014;5:3557. | ||
Velthuis-te Wierik EJ, Westerterp KR, van den Berg H. Impact of a moderately energy-restricted diet on energy metabolism and body composition in non-obese men. Int J Obes Relat Metab Disord. 1995;19:318–324. | ||
Velthuis-te Wierik EJ, van den Berg H, Schaafsma G, Hendriks HF, Brouwer A. Energy restriction, a useful intervention to retard human ageing? Results of a feasibility study. Eur J Clin Nutr. 1994;48:138–148. | ||
Velthuis-te Wierik EJ, van Leeuwen RE, Hendriks HF, et al. Short-term moderate energy restriction does not affect indicators of oxidative stress and genotoxicity in humans. J Nutr. 1995;125:2631–2639. | ||
Das SK, Gilhooly CH, Golden JK, et al. Long-term effects of 2 energy-restricted diets differing in glycemic load on dietary adherence, body composition, and metabolism in CALERIE: a 1-y randomized controlled trial. Am J Clin Nutr. 2007;85:1023–1030. | ||
Heilbronn LK, de Jonge L, Frisard MI, et al. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: a randomized controlled trial. JAMA. 2006;295:1539–1548. | ||
Racette SB, Weiss EP, Villareal DT, et al. One year of caloric restriction in humans: feasibility and effects on body composition and abdominal adipose tissue. J Gerontol Ser A Biol Sci Med Sci. 2006;61:943–950. | ||
Larson-Meyer DE, Heilbronn LK, Redman LM, et al. Effect of calorie restriction with or without exercise on insulin sensitivity, beta-cell function, fat cell size, and ectopic lipid in overweight subjects. Diabetes Care. 2006;29:1337–1344. | ||
Lefevre M, Redman LM, Heilbronn LK, et al. Caloric restriction alone and with exercise improves CVD risk in healthy non-obese individuals. Atherosclerosis. 2009;203:206–213. | ||
Civitarese AE, Carling S, Heilbronn LK, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. | ||
Rochon J, Bales CW, Ravussin E, et al. Design and conduct of the CALERIE study: comprehensive assessment of the long-term effects of reducing intake of energy. J Gerontol Ser A Biol Sci Med Sci. 2011;66:97–108. | ||
Fontana L, Villareal DT, Weiss EP, et al. Calorie restriction or exercise: effects on coronary heart disease risk factors. A randomized: controlled trial. Am J Physiol Endocrinol Metab. 2007;293:E197–E202. | ||
Weiss EP, Racette SB, Villareal DT, et al. Improvements in glucose tolerance and insulin action induced by increasing energy expenditure or decreasing energy intake: a randomized controlled trial. Am J Clin Nutr. 2006;84:1033–1042. | ||
Ravussin E, Redman LM, Rochon J, et al. A 2-year randomized controlled trial of human caloric restriction: feasibility and effects on predictors of health span and longevity. J Gerontol Ser A Biol Sci Med Sci. 2015;70:1097–1104. | ||
Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci U S A. 2004;101:6659–6663. | ||
Meyer TE, Kovacs SJ, Ehsani AA, Klein S, Holloszy JO, Fontana L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J Am Coll Cardiol. 2006;47:398–402. | ||
Fontana L, Klein S, Holloszy JO. Effects of long-term calorie restriction and endurance exercise on glucose tolerance, insulin action, and adipokine production. Age. 2010;32:97–108. | ||
Stein PK, Soare A, Meyer TE, Cangemi R, Holloszy JO, Fontana L. Caloric restriction may reverse age-related autonomic decline in humans. Aging Cell. 2012;11:644–650. | ||
Martin CK, Anton SD, Han H, et al. Examination of cognitive function during six months of calorie restriction: results of a randomized controlled trial. Rejuvenation Res. 2007;10:179–190. | ||
Villareal DT, Fontana L, Das SK, et al. Effect of two-year caloric restriction on bone metabolism and bone mineral density in non-obese younger adults: a randomized clinical trial. J Bone Miner Res. 2016;31:40–51. | ||
Kalu DN, Hardin RR, Cockerham R, Yu BP, Norling BK, Egan JW. Lifelong food restriction prevents senile osteopenia and hyperparathyroidism in F344 rats. Mech Ageing Dev. 1984;26:103–112. | ||
Lu T, Pan Y, Kao S-Y, et al. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. | ||
Vera E, Bernardes de Jesus B, Foronda M, Flores JM, Blasco MA. Telomerase reverse transcriptase synergizes with calorie restriction to increase health span and extend mouse longevity. PLoS One. 2013;8:e53760. | ||
Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015;16:593–610. | ||
Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell. 2015;161:106–118. | ||
Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011;3:a00444. | ||
Dokladny K, Zuhl MN, Mandell M, et al. Regulatory coordination between two major intracellular homeostatic systems: heat shock response and autophagy. J Biol Chem. 2013;288:14959–14972. | ||
Pesce V, Cormio A, Fracasso F, et al. Age-related mitochondrial genotypic and phenotypic alterations in human skeletal muscle. Free Radic Biol Med. 2001;30(11):1223–1233. | ||
Lezza AM, Fallacara FP, Pesce V, Leeuwenburgh C, Cantatore P, Gadaleta MN. Localization of abasic sites and single-strand breaks in mitochondrial DNA from brain of aged rat, treated or not with caloric restriction diet. Neurochem Res. 2008;33(12):2609–2614. | ||
Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int. 2014;2014:238463. | ||
Picca A, Pesce V, Fracasso F, Joseph AM, Leeuwenburgh C, Lezza AM. Aging and calorie restriction oppositely affect mitochondrial biogenesis through TFAM binding at both origins of mitochondrial DNA replication in rat liver. PLoS One. 2013;8(9):e74644. | ||
Picca A, Fracasso F, Pesce V, et al. Age- and calorie restriction-related changes in rat brain mitochondrial DNA and TFAM binding. Age (Dordr). 2013;35(5):1607–1620. | ||
Picca A, Lezza AM. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion. 2015;25(11):67–75. | ||
Rugarli E, Trifunovic A. Is mitochondrial free radical theory of aging getting old? Biochim Biophys Acta. 2015;1847(11):1345–1346. | ||
Picca A, Pesce V, Sirago G, Fracasso F, Leeuwenburgh C, Lezza AM. “What makes some rats live so long?” The mitochondrial contribution to longevity through balance of mitochondrial dynamics and mtDNA content. Exp Gerontol. 2016;85(12):33–40. | ||
Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011;51:327–336. | ||
Barja G. The mitochondrial free radical theory of aging. Prog Mol Biol Transl Sci. 2014;127:1–27. | ||
Yu BP. Membrane alteration as a basis of aging and the protective effects of calorie restriction. Mech Ageing Dev. 2005;126:1003–1010. | ||
Lopez-Dominguez JA, Ramsey JJ, Tran D, et al. The influence of dietary fat source on life span in calorie restricted mice. J Gerontol A Biol Sci Med Sci. 2015;70:1181–1188. | ||
Genova ML, Lenaz G. The interplay between respiratory supercomplexes and ROS in aging. Antioxid Redox Signal. 2015;23:208–238. | ||
Kwon J, Han E, Bui CB, et al. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 2012;13:150–156. | ||
Hyun DH, Emerson SS, Jo DG, Mattson MP, de Cabo R. Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc Natl Acad Sci U S A. 2006;103:19908–19912. | ||
Rodriguez-Bies E, Navas P, Lopez-Lluch G. Age-dependent effect of every-other-day feeding and aerobic exercise in ubiquinone levels and related antioxidant activities in mice muscle. J Gerontol A Biol Sci Med Sci. 2015;70:33–43. | ||
Short KR, Bigelow ML, Kahl J, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. | ||
Feige JN, Lagouge M, Auwerx J. Dietary manipulation of mouse metabolism. Curr Protoc Mol Biol. 2008;66:Chapter 29:Unit 29B.5. | ||
Baur JA, Chen D, Chini EN, et al. Dietary restriction: standing up for sirtuins. Science. 2010;329:1012–1013. | ||
Picca A, Pesce V, Fracasso F, Joseph A-M, Leeuwenburgh C, Lezza AMS. A comparison among the tissue-specific effects of aging and calorie restriction on TFAM amount and TFAM-binding activity to mtDNA in rat. Biochim Biophys Acta. 2014;1840:2184–2191. | ||
Nisoli E, Tonello C, Cardile A, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. | ||
Mattagajasingh I, Kim CS, Naqvi A, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–14860. | ||
Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. | ||
Ruetenik A, Barrientos A. Dietary restriction, mitochondrial function and aging: from yeast to humans. Biochim Biophys Acta. 2015;1847:1434–1447. | ||
Martin-Montalvo A, de Cabo R. Mitochondrial metabolic reprogramming induced by calorie restriction. Antioxid Redox Signal. 2013;19:310–320. | ||
Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J Biol Chem. 2005;280:16456–16460. | ||
Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. | ||
Lopez-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Exp Gerontol. 2008;43:813–819. | ||
Weber TA, Reichert AS. Impaired quality control of mitochondria: aging from a new perspective. Exp Gerontol. 2010;45:503–511. | ||
Prolla TA, Mattson MP. Molecular mechanisms of brain aging and neurodegenerative disorders: lessons from dietary restriction. Trends Neurosci. 2001;24(11 Suppl):S21–S31. | ||
Ghosh S, Wanders D, Stone KP, Van NT, Cortez CC, Gettys TW. A systems biology analysis of the unique and overlapping transcriptional responses to caloric restriction and dietary methionine restriction in rats. FASEB J. 2014;28(6):2577–2590. | ||
Field AE, Adams PD. Targeting chromatin aging – the epigenetic impact of longevity-associated interventions. Exp Gerontol. 2017;94(8):29–33. | ||
Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. | ||
Rakyan VK, Down TA, Maslau S, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. | ||
Kim CH, Lee EK, Choi YJ, et al. Short-term calorie restriction ameliorates genome wide, age-related alterations in DNA methylation. Aging Cell. 2016;15:1074–1081. | ||
Li Y, Tollefsbol TO. P16INK4A suppression by glucose restriction contributes to human cellular lifespan extension through SIRT1-mediated epigenetic and genetic mechanisms. PLoS One. 2011;6(2):e17421. | ||
Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–266. | ||
Peleg S, Feller C, Ladurner AG, Imhof A. The metabolic impact on histone acetylation and transcription in ageing. Trends Biochem Sci. 2016;41:700–711. | ||
Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. | ||
Satoh A, Brace CS, Rensing N, et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013;18:416–430. | ||
Vazquez BN, Thackray JK, Serrano L. Sirtuins and DNA damage repair: SIRT7 comes to play. Nucleus. 2017;8:107–115. | ||
Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. 2010;31:212–220. | ||
Yang H, Yang T, Baur JA, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. | ||
Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. | ||
Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013;27:2072–2085. | ||
Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. 2011;93:884S–890S. | ||
Kaeberlein M, Kirkland KT, Fields S, Kennedy BK. Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol. 2004;2(9):E296. | ||
Fabrizio P, Gattazzo C, Battistella L, et al. Sir2 blocks extreme life-span extension. Cell. 2005;123:655–667. | ||
Li Y, Xu W, McBurney MW, Longo VD. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008;8:38–48. | ||
Longo VD. Linking sirtuins, IGF-I signaling, and starvation. Exp Gerontol. 2009;44:70–74. | ||
Herranz D, Munoz-Martin M, Canamero M, et al. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:3. | ||
Burnett C, Valentini S, Cabreiro F, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011;477:482–485. | ||
Mouchiroud L, Houtkooper RH, Moullan N, et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. | ||
Imai SI, Guarente L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. NPJ Aging Mech Dis. 2016;2:16017. | ||
Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol. 2016;17:308–321. | ||
Sampaio-Marques B, Felgueiras C, Silva A, et al. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy. 2012;8:1494–1509. | ||
Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57:456–466. | ||
Price NL, Gomes AP, Ling AJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. | ||
Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411–424. | ||
Fang EF, Scheibye-Knudsen M, Brace LE, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD+/SIRT1 reduction. Cell. 2014;157(4):882–896. | ||
Scheibye-Knudsen M, Mitchell SJ, Fang EF, et al. A high fat diet and NAD+ rescue premature aging in Cockayne syndrome. Cell Metab. 2014;20(5):840–855. | ||
Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–1351. | ||
Selman C, Lingard S, Choudhury AI, et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008;22:807–818. | ||
Kenyon C. The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing. Philos Trans R Soc Lond B Biol Sci. 2011;366:9–16. | ||
Goto T, Takano M. Transcriptional role of FOXO1 in drug resistance through antioxidant defense systems. Adv Exp Med Biol. 2009;665:171–179. | ||
Argentino DP, Dominici FP, Al-Regaiey K, Bonkowski MS, Bartke A, Turyn D. Effects of long-term caloric restriction on early steps of the insulin-signaling system in mouse skeletal muscle. J Gerontol A Biol Sci Med Sci. 2005;60:28–34. | ||
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. | ||
Longo VD, Antebi A, Bartke A, et al. Interventions to slow aging in humans: are we ready? Aging Cell. 2015;14(4):497–510. | ||
Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. | ||
Kim YM, Kim DH. dRAGging amino acid-mTORC1 signaling by SH3BP4. Mol Cell. 2013;35:1–6. | ||
Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. | ||
Heiss C, Spyridopoulos I, Haendeler J. Interventions to slow cardiovascular aging: dietary restriction, drugs and novel molecules. Exp Gerontol. Epub 2017 Jun 27. | ||
Hardie DG. Sensing of energy and nutrients by AMP-activated protein kinase. Am J Clin Nutr. 2011;93:891S–896S. | ||
Cantò C, Auwerx J. Calorie Restriction: is AMPK a key sensor and effector? Physiology. 2011;26:214–224. | ||
Greer EL, Dowlatshahi D, Banko MR, et al. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17:1646–1656. | ||
Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. | ||
Tohyama D, Yamaguchi A. A critical role of SNF1A/dAMPKalpha (Drosophila AMP-activated protein kinase alpha) in muscle on longevity and stress resistance in Drosophila melanogaster. Biochem Biophys Res Commun. 2010;394:112–118. | ||
Gonzalez AA, Kumar R, Mulligan JD, Davis AJ, Weindruch R, Saupe KW. Metabolic adaptations to fasting and chronic caloric restriction in heart, muscle, and liver do not include changes in AMPK activity. Am J Physiol Endocrinol Metab. 2004;287:E1032–E1037. | ||
Chung HY, Kim HJ, Kim KW, Choi JS, Yu BP. Molecular inflammation hypothesis of aging based on the anti-aging mechanism of calorie restriction. Microsc Res Tech. 2002;59:264–272. | ||
Salvioli S, Capri M, Valensin S, Tieri P, Monti D, Ottaviani E, Franceschi C. Inflamm-aging, cytokines and aging: state of the art, new hypotheses on the role of mitochondria and new perspectives from systems biology. Curr Pharm Des. 2006;12:3161–3171. | ||
Goto M. Inflammaging (inflammation + aging): a driving force for human aging based on an evolutionarily antagonistic pleiotropy theory? Biosci Trends. 2008;2:218–230. | ||
Solana R, Pawelec G, Tarazona R. Aging and innate immunity. Immunity. 2006;24:491–494. | ||
Candore G, Caruso C, Jirillo E, Magrone T, Vasto S. Low grade inflammation as a common pathogenetic denominator in age-related diseases: novel drug targets for anti-ageing strategies and successful ageing achievement. Curr Pharm Des. 2010;16:584–596. | ||
Feldman N, Rotter-Maskowitz A, Okun E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res Rev. 2015;24:29–39. | ||
Picca A, Lezza AMS, Leeuwenburgh C, et al. Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int J Mol Sci. 2017;18(5): pii: E933. | ||
Pawelec G, Goldeck D, Derhovanessian E. Inflammation, ageing and chronic disease. Curr Opin Immunol. 2014;29:23–28. | ||
Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;(69 Suppl1):S4–S9. | ||
Horrillo D, Sierra J, Arribas C, et al. Age-associated development of inflammation in Wistar rats: effects of caloric restriction. Arch Physiol Biochem. 2011;117:140–150. | ||
Chung HY, Lee EK, Choi YJ, et al. Molecular inflammation as an underlying mechanism of the aging process and age-related diseases. J Dent Res. 2011;90:830–840. | ||
Zhou Y, Xu BC, Maheshwari HG, et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc Natl Acad Sci U S A. 1997;94:13215–13220. | ||
Ikeno Y, Hubbard GB, Lee S, et al. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J Gerontol A Biol Sci Med Sci. 2009;64:522–529. | ||
Trainer PJ, Drake WM, Katznelson L, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med. 2000;342:1171–1177. | ||
Van der Lely AJ, Biller BM, Brue T, et al. Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab. 2012;97:1589–1597. | ||
Kopchick JJ, Parkinson C, Stevens EC, Trainer PJ. Growth hormone receptor antagonists: discovery, development, and use in patients with acromegaly. Endocr Rev. 2002;23:623–646. | ||
Howitz KT, Bitterman KJ, Cohen HY, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. | ||
Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014;35:146–154. | ||
Timmers S, Konings E, Bilet L, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14:612–622. | ||
Pearson KJ, Baur JA, Lewis KN, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. | ||
Minor RK, Baur JA, Gomes AP, et al. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;1:70. | ||
Wang G, Han T, Nijhawan D, et al. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014;158:1324–1334. | ||
Canto C, Houtkooper RH, Pirinen E, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet induced obesity. Cell Metab. 2012;15:838–847. | ||
Kennedy BK, Pennypacker JK. Drugs that modulate aging: the promising yet difficult path ahead. Transl Res. 2014;163:456–465. | ||
Wilkinson JE, Burmeister L, Brooks SV, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11:675–682. | ||
Sharp ZD. Aging and TOR: interwoven in the fabric of life. Cell Mol Life Sci. 2011;68:587–597. | ||
Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. | ||
Selman C, Tullet JM, Wieser D, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. | ||
Kamada Y, Sekito T, Ohsumi Y. Autophagy in yeast: a TOR-mediated response to nutrient starvation. Curr Top Microbiol Immunol. 2004;279:73–84. | ||
Toth ML, Sigmond T, Borsos E, et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy. 2008;4:330–338. | ||
Soefje SA, Karnad A, Brenner AJ. Common toxicities of mammalian target of rapamycin inhibitors. Target Oncol. 2011;6:125–129. | ||
Lane MA, Ingram DK, Roth GS. 2-Deoxy-D-glucose feeding in rats mimics physiologic effects of calorie restriction. J Anti Aging Med. 1998;1:327–337. | ||
Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. | ||
Ingram DK, Roth GS. Glycolytic inhibition as a strategy for developing calorie restriction mimetics. Exp Gerontol. 2011;46:148–154. | ||
Wan R, Camandola S, Mattson MP. Dietary supplementation with 2-deoxy-D-glucose improves cardiovascular and neuroendocrine stress adaptation in rats. Am J Physiol Heart Circ Physiol. 2004;287:H1186–H1193. | ||
Minor RK, Smith DL Jr, Sossong AM, et al. Chronic ingestion of 2-deoxy-D-glucose induces cardiac vacuolization and increases mortality in rats. Toxicol Appl Pharmacol. 2010;243:332–339. | ||
Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Investig. 2013;123:2764–2772. | ||
Slack C, Foley A, Partridge L. Activation of AMPK by the putative dietary restriction mimetic metformin is insufficient to extend lifespan in Drosophila. PLoS One. 2012;7:e47699. | ||
Song YM, Lee YH, Kim JW, et al. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy. 2015;11:46–59. | ||
Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. | ||
Brandhorst S, Choi IY, Wei M, et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 2015;22(1):86–99. | ||
Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19:181–192. | ||
Pedersen AN, Kondrup J, Borsheim E. Health effects of protein intake in healthy adults: a systematic literature review. Food Nutr Res. 2013;57(1):21245. | ||
Levine ME, Suarez JA, Brandhorst S, et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014;19:407–417. | ||
Fontana L, Cummings NE, Arriola Apelo SI, et al. Decreased consumption of branched-chain amino acids improves metabolic health. Cell Rep. 2016;16:520–530. | ||
Lees EK, Krol E, Grant L, et al. Methionine restriction restores a younger metabolic phenotype in adult mice with alterations in fibroblast growth factor 21. Aging Cell. 2014;13:817–827. | ||
Miller RA, Buehner G, Chang Y, Harper JM, Sigler R, Smith-Wheelock M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell. 2005;4:119–125. | ||
Orentreich N, Matias JR, DeFelice A, Zimmerman JA. Low methionine ingestion by rats extends life span. J Nutr. 1993;123:269–274. | ||
Cummings NE, Lamming DW. Regulation of metabolic health and aging by nutrient-sensitive signaling pathways. Mol Cell Endocrinol. 2017;455:13–22. | ||
Jensen MD, Ryan DH, Apovian CM, et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American college of cardiology/American heart association task force on practice guidelines and the obesity society. J Am Coll Cardiol. 2014;63:2985–3023. | ||
Landi F, Calvani R, Cesari M, et al. Sarcopenia as the biological substrate of physical frailty. Clin Geriatr Med. 2015;31:367–374. | ||
Nowson C, O’Connell S. Protein requirements and recommendations for older people: a review. Nutrients. 2015;7:6874–6899. | ||
Paddon-Jones D, Rasmussen BB. Dietary protein recommendations and the prevention of sarcopenia. Curr Opin Clin Nutr Metab Care. 2009;12:86–90. | ||
Wolfe RR, Miller SL, Miller KB. Optimal protein intake in the elderly. Clin Nutr. 2008;27:675–684. | ||
Le Couteur DG, Solon-Biet S, Wahl D, et al. Dietary protein, ageing and the Okinawan ratio. Age Ageing. 2016;45:443–447. | ||
Volpi E, Campbell WW, Dwyer JT, et al. Is the optimal level of protein intake for older adults greater than the recommended dietary allowance? J Gerontol A Biol Sci Med Sci. 2013;68:677–681. | ||
Baum JI, Kim IY, Wolfe RR. Protein consumption and the elderly: what is the optimal level of intake? Nutrients. 2016;8:359–378. | ||
Moore DR. Keeping older muscle young through dietary protein and physical activity. Adv Nutr. 2014;5:599S–607S. | ||
Solon-Biet SM, Mitchell SJ, de Cabo R, Raubenheimer D, Le Couteur DG, Simpson SJ. Macronutrients and caloric intake in health and longevity. J Endocrinol. 2015;226(1):R17–R28. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.