Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 12
Diversity In Precision Medicine And Pharmacogenetics: Methodological And Conceptual Considerations For Broadening Participation
Authors Popejoy AB
Received 31 March 2019
Accepted for publication 12 September 2019
Published 14 October 2019 Volume 2019:12 Pages 257—271
DOI https://doi.org/10.2147/PGPM.S179742
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Alice B Popejoy1–3
1Department of Biomedical Data Science, Stanford University School of Medicine, Stanford, CA, USA; 2Center for Computational, Evolutionary and Human Genomics (CEHG), Stanford University, Stanford, CA, USA; 3Center for Integration of Research on Genetics and Ethics (CIRGE), Stanford University, Stanford, CA, USA
Correspondence: Alice B Popejoy
Medical School Office Building (MSOB), Stanford University School of Medicine, Third Floor, 1265 Welch Road, Stanford, CA 94305, USA
Tel +1 916 837 2951
Email [email protected]
Abstract: Genome-wide association studies (GWAS) have revealed important links between genetic markers across the human genome and phenotypic traits, including risk factors for disease. Studies have shown that GWAS continue to be overwhelmingly conducted on people of primarily European descent, despite the fact that the vast majority of human genomic variation is present in non-European populations such as those in Africa. To enhance our understanding of diversity in the pharmacogenomics and precision medicine literature, this review provides a window into the representation of biogeographical populations that have been studied for pharmacogenetic traits, such as enzyme metabolism and adverse drug response. Using the Medical Subject Headings (MeSH) ontology search terms in PubMed, studies were identified that are either population-based, or include a description of the study population on the basis of biological or environmental diversity. The results of this scoping review indicate that the majority of relevant papers (>95% of studies tagged in PubMed with MeSH terms “precision medicine” or “pharmacogenetics”, N=23,701) are not annotated with the “population group” MeSH term, suggesting that the majority of studies in this literature are not population-based, or the authors chose not to describe the study population. Among those studies related to pharmacogenetics or precision medicine that are specific to human population groups (N=1006) and were included in the analysis after filtering and screening on eligibility criteria (N=192), the majority of single-population studies included individuals of African, Asian, and European origins, or genetic ancestry. Combining studies of single and multiple populations, 33% involve participants of Asian origin or ancestry; 30% European; 24% African; 10% Hispanic or Latino; and < 3% American Indian or Alaska Native. These data provide a baseline for future comparison, indicating which biogeographic groups have informed the pharmacogenomic knowledgebase specific to diverse human populations. Challenges and potential solutions to improve diversity in the field and in genetics research more broadly are discussed.
Keywords: GWAS, population groups, biogeographic populations, scoping review, genomic variation, representation
Introduction
Genome-wide association studies (GWAS) investigate statistical links between genetic markers and phenotypic traits, revealing which regions of the genome are likely involved in disease pathways and other clinically relevant biological mechanisms. Results from these studies point to significant associations between single-nucleotide polymorphisms (SNPs) representing a region of the genome, and phenotypic measurements or classifications. Because of its widespread utility and effectiveness at identifying common genetic variants that are associated with disease risk, GWAS results shape the primary basis of scientific knowledge about genetic contributions to etiological pathways of health and disease.1 Related to the growing field of pharmacogenomics, which investigates the genetic basis of variation in response to pharmacologic exposures, GWAS results have revealed genomic regions that are involved in variable drug response, including metabolism, toxicity, and efficacy.2 These pharmacogenetic findings inform clinical decision-making about pharmaceutical selection and drug dosages on the basis of genetic information about a patient, thus establishing the foundation for precision, or personalized, medicine.3
Precision medicine is the translation of basic research on human variation at the individual and population level to increase the accuracy and precision of disease prevention, diagnosis, and treatment. However, the majority of GWAS (~80%) have been conducted in populations of European descent, meaning the genomic evidence-base for personalized medicine may be more informative for individuals sharing common ancestry with European populations.4,5 Therefore, the promise of precision medicine will only be realized when the genomic evidence-base is more representative of diverse ancestral groups. This article describes reasons diversity in genomics research is important, from both a scientific perspective and in terms of the ethical, legal, social implications (ELSI) that accompany the under-representation of marginalized groups. A scoping review of diversity in the research landscape of pharmacogenomics is presented in order to elucidate which ancestral groups have been most frequently studied and thus are most likely to benefit from personalized medicine. First, an overview of diversity measures is provided to contextualize the analysis, including a discussion of a biogeographic grouping system developed by Huddart and colleagues on the basis of continental (global) genetic ancestry.6 This framework for characterizing ancestral diversity was developed in order to classify study populations in pharmacogenetics research, and is used here to synthesize and describe populations in a scoping review of literature in the field. Following the results of this scoping review is a discussion of persistent challenges to broadening participation in genomics, as well as potential solutions for increasing the representation of diverse populations.
Defining Diversity In Human Populations
Before diversity (or a lack thereof) in GWAS and pharmacogenomics can be fully understood, it is necessary to formulate a clear concept of what “diversity” actually means in human populations. Modern humans have a single shared evolutionary origin, with estimates indicating that the most recent common ancestor (MRCA) of all humans lived in southern Africa around 200K years ago.7,8 Subsequent to the existence of modern humans’ MRCA, groups of its descendants migrated off the African continent to populate vast regions of the world. Each serially migrating population brought with them subsets of the genomic variation from the original, or founder population, such that increasing geographic distance is correlated with increasing genetic distance.9,10 But how is a human population defined? Given that all humans have a shared ancestral origin past a certain point in time, it is important to contextualize any further consideration of the ancestral origins of human populations as relative to demographic history, time, and place, while acknowledging that all humans are much more genetically similar than we are different. There is no objective or universal definition of a human “population” – whenever this term is used, it is always relative to some delineation on the basis of location, geo-political structure, socio-cultural factors, or a point in time. Nevertheless, both genetic and environmental differences do exist within and between groups of people across the world, and these differences must be accounted for in genomics research to avoid confounding by shared genetic and environmental factors that are both associated with a trait of interest. As such, genomic and environmental diversity are discussed in the sub-sections below, highlighting the importance of each in maximizing the power of discovery in GWAS.
Genomic Diversity
While the majority of the ~3 billion sites in the human genome are invariant, roughly 0.01% of the genome is polymorphic, meaning there is variation within and between populations at these sites. On average, people of African descent have the highest number of variable, or polymorphic, sites per genome compared to those of predominantly European or Asian descent,11 and because it is the birthplace of modern humans, the African continent is home to the majority of human genomic diversity.12 From a mathematical perspective, genomic diversity may be considered in terms of genetic distance, which can be calculated using various methodologies based on different evolutionary models. The most notable examples of these measures are Nei’s standard genetic distance,13 Cavalli-Sforza and Edwards’ chord distance,14 and Reynolds, Weir, and Cockerham’s measure of genetic divergence.15 Each of these measures is based on underlying assumptions about the cause(s) of genetic differences, including mutation, genetic drift, or a combination of the two. While there are still other examples,16 perhaps the most widely used measure of genetic distance is the fixation index, FST, derived from Wright’s F-statistics,17 which estimates the amount of variance in allele frequencies that is due to structure between populations, relative to the degree of genetic similarity within populations.18 That is, estimates of FST compare average genetic distances within and between populations, and this can be interpreted as the average amount of overall genetic diversity of individuals within a population, compared to genetic diversity between individuals from different populations. In the case of pharmacogenomic diversity across human populations, F-statistics have been used to demonstrate a high relative amount of variability in the genes encoding a superfamily of cytochrome P450 (CYP450) metabolizing enzymes, indicating functional differences in disease risk and drug response across populations.19
While the approaches described provide a useful quantitative framework for estimating genomic diversity, the models on which they are based have inherent biases and limitations. For one, the landscape of human genomic variation is impacted not only by mutation and drift, but also by genetic recombination and evolutionary forces such as migration (gene flow), bottlenecks, founder effects, and natural selection, which are not typically considered in classical models of genetic distance. As a result of these and other factors, the lines between “populations” are blurry, so the use of mathematical models that assume a priori classification of individuals into definitive populations are thus inherently limited and may even reinforce misleading notions about the existence of distinct and meaningful biological groups. At the same time, the very factors that contribute to fluidity in definitions of human populations also confer distinct genetic differences that do tend to cluster in groups of individuals with shared origins. It is for this reason that sampling individuals with diverse genetic backgrounds from across the globe is essential to represent the human genomic variation that has arisen in different geographic regions over long periods of time.
Ancestral Diversity
Variations in DNA continuously arise through random mutations and recombination, subsequently either rising to high frequency in the population, maintaining an intermediate prevalence, or falling out of existence, as a result of population-level effects such as gene flow, genetic drift and natural selection.20 The resulting differences in allele frequencies and linkage disequilibrium (LD), or the co-inheritance of alleles in close physical proximity on a chromosome, cluster in groups of individuals with shared ancestral origins. For example, ancestry informative markers (AIMs) are genetic variants that happen to exhibit this property of clustering within individuals of similar origins, and so are specifically selected for the purpose of assigning individuals into ancestral population groups.21 The most widely used methodology for stratifying individuals on the basis of ancestral background has been principal component analysis (PCA), since it was shown by Novembre and colleagues in 2008 to cluster individuals from European countries on the basis of genetic relatedness in concordance with their sampling location on a geographic map of Europe.22 While PCA provides a visual representation of how individuals in a sample cluster on the basis of genetic relatedness, other methods provide more fine-grained ancestry estimates including percentages of the genome that are derived from different ancestral populations, by comparison to global reference panels.23,24
The rise of direct-to-consumer (DTC) genetic ancestry testing has led to a demand for this type of analysis, but its utility for clinical genomics and personalized medicine is unclear. What is relevant about genetic ancestry for the purposes of assessing genomic diversity is the fact that certain SNPs at polymorphic sites in humans are more common in some ancestral populations than others, and that membership in a group or population with shared ancestral origins can thus serve as a proxy for the probability that an individual might have a population-specific allele, or as a proxy for genomic background. When study populations for GWAS are defined on the basis of ancestral background, then, GWAS findings from one population may not replicate in others due to ancestry-specific differences in allele frequencies and LD structure.25,26 It is for this reason that genetic ancestry, or ancestral background, needs to be taken into account both in conducting GWAS on samples with population structure, and as an essential element of assessing diversity.
Environmental Diversity
In contrast to the need for genomic diversity in GWAS and pharmacogenomics research, environmental diversity refers to the myriad ways in which people experience the world and interact with, or are affected by, their environments. Whereas genetic diversity is most relevant for research on Mendelian traits, which are typically caused by a single gene or variant and highly penetrant, environmental diversity is crucial for the investigation of underlying causes of complex traits. In addition to genomics, several other “-omics” play a role in disease biology, including epigenomics, transcriptomics, proteomics, metabolomics, and microbiomics.27 Many of these integrative assessments of non-genetic contributors to disease are highly influenced by the environment, including lifestyle factors such as stress, diet, and exercise, as well as institutionalized and structural factors such as building material and chemical exposures, air particulate matter, and others. As a result of these environmental influences, it is essential for research on complex disease to include and integrate multi-omics approaches to capture the variety of non-genetic factors that play a role in disease etiology. However, in order for variation in environmental exposures to be informative for research, study samples must involve participants with a variety of different environmental backgrounds, in addition to diversity in genomic background.
Socio-Cultural Factors
Due to systemic and institutionalized inequities on the basis of socio-economic, cultural, and historical factors, a person’s lived experience and environmental exposures may be correlated with those of other individuals with the same “socio-cultural background”, which is used here to refer to all of the factors listed above. For example, the United States has large disparities in age of onset and severity of illnesses, access to and quality of health care, and mortality rates on the basis of racial or ethnic identity.28 As a result of these inequities, members of racial and ethnic minority populations have worse health outcomes and higher risk for diseases with strong environmental components than others. In order to avoid confounding by this underlying structure of environmental variation along racial and ethnic lines, self-reported measures of “race” and “ethnicity” are often used in epidemiological models and association tests. However, it is important to note that these measures control for potentially confounding factors based on environmental and socio-cultural background, not genomic background. This is a mistake often made by researchers and non-scientists alike – conflating the self-reported, socio-cultural indicators of identity such as “race” and “ethnicity” with ancestral or genetic background.
While there is a correlation between genetic ancestry and self-reported race or ethnicity,29 (for example, most self-identified African Americans have at least some African ancestry) there is more genomic diversity within “racial” groups than there is between them, rejecting the idea that there is a biological or genetic basis for race.30,31 Even early research that would today be considered racist by its very framing (that the three “races of man: the Negroids, Mongoloids, and Caucasoids” are somehow biologically meaningful), found no evidence that genetic variation is higher between these groups than within them.32 As they are fluid and subject to change over time and across norms, social and cultural measures of identity should not be used as a proxy for ancestral background because they are not well aligned, especially for admixed individuals and communities, and each measure provides unique information about particular phenomena.33,34
Whereas genetic ancestry provides information about genomic background and demographic history, “race” refers to physical attributes such as skin and hair color, while “ethnicity” refers to cultural factors such as heritage, traditions, language, religion, and social practices including diet and exercise.35 Both of these concepts are culture- and context-specific, and are intricately tied to historical, political and hierarchical power dynamics, which reinforce institutional and structural inequalities, racism, and discrimination.36 Therefore, when socio-cultural diversity is considered as an important element of diversity in biomedical research, it should be because of the valuable insights that may be gained from investigating variable environmental exposures, not because it is a convenient proxy for genetic ancestry.
Genetic And Environmental Interactions
Complex disease etiology can be both multifactorial and polygenic, meaning both genetic and environmental factors can play a role, as well as multiple genetic pathways.37 When traits are polygenic, genes may interact with one another to cause variation in the phenotype, and when they are multifactorial, there are often gene-environment interactions (GxE) that modify the trait. When there are gene-gene interactions (GxG), genomic background plays a role in disease etiology, and thus ancestral (genetic) diversity in study samples is important, in order to gain a complete global picture of genetic contributions to the disease. When there are GxE, both genetic and environmental diversity must be present and accounted for in order to tease apart the contributions of each element individually, as well as the contribution of their interaction(s) to variation in a complex trait of interest. For example, asthma treatment failure has been found to be significantly more prevalent in African Americans than whites using long-acting β-agonists, despite fewer asthma symptoms in African Americans.38 However, genetic ancestry was not associated with variation in treatment response among African Americans, so researchers have hypothesized that socio-cultural factors may play a role in asthma treatment efficacy.39 Correlation between genetic, or ancestral, background and socio-cultural background poses methodological challenges for teasing apart and accounting for the contributions of each, especially for traits with GxE, because all of the above may be associated with shared environmental factors. As in the case of asthma, there may be genomic variants impacting variability in the efficacy of treatment, as well as socio-cultural and environmental factors that confound these associations. Inclusion of correlated variables as confounding factors may over-correct signals of association, and methods are underway to navigate these challenges.
Review Of Diversity In Pharmacogenomics Literature
One objective of personalized, or precision, medicine is to tailor clinical care to patients’ genetic traits and other biological pre-conditions. In particular, researchers and clinicians are interested in uncovering the unique combination of genetic variants that may impact diagnosis and drive treatment decisions. For example, mutations that influence the function of genes encoding drug-metabolizing enzymes (pharmacogenetic variants) may have implications for how well a patient is able to clear chemical compounds associated with various treatments. In order to gain a broad perspective on the kind of research that has been conducted in precision medicine and pharmacogenomics, and specifically which populations have been studied, this section describes a scoping review of the literature that was conducted in PubMed (Figure 1). The MeSH term search formula used for this scoping review was: “pharmacogenetics” OR “precision medicine” AND “population groups”, such that articles tagged with curated annotations for either pharmacogenetics or precision medicine were identified, in addition to annotations about study population. This initial search identified 1,006 articles in PubMed, which were then filtered by the presence of an abstract, as well as having the full free text available (as all NIH-funded research should), leading to a total of 326 abstracts and titles that were manually reviewed. Inclusion and exclusion criteria were developed with the goal of identifying articles describing original research that explicitly focuses on diverse study populations. Specifically, study populations need to be relevant for genetic and environmental diversity; therefore, groups or populations that are studied on the basis of age, disease status, or other categories that are not relevant to socio-cultural factors such as race, ethnicity, or ancestral background were excluded from the analysis. While study samples defined on the basis of other characteristics, such as disease status, are important for the pharmacogenomics knowledge-base, they are not informative for the purpose of this review, which is to sample the diverse populations that have been included in pharmacogenomics and precision medicine research.
Reviews and commentaries of existing literature were excluded, as well as descriptions of research databases or other resources. Articles that were included described a number of study designs, including genetic association (GWAS and candidate gene studies), population genetics, novel methods development to enable research on multi-ethnic or diverse cohorts, or technological innovations that enable or advance the ability to investigate biological mechanisms of disease across populations. In total, 192 articles were selected for inclusion, and all subsequently described analyses were conducted on this subset of the literature. Although this is a small number of studies relative to the entire scope of literature in pharmacogenomics and precision medicine, these articles were carefully selected using inclusion and exclusion criteria to identify studies most likely to be informative about the genomic and environmental diversity of study subjects that have been included in pharmacogenomics research to inform personalized medicine.
As seen in Figure 1, the studies included in this scoping review were selected through a structured process of identification, screening, eligibility, and inclusion. The study populations described in the included articles were then analyzed to determine their biogeographical origins or ancestry, and the type of study conducted was also noted for each article. Among the studies that were included, roughly half involved a single population, while the remainder included two or more study populations. In order to categorize each study population into groups that are informative about the genetic diversity of research participants, the classification system proposed by Huddart and colleagues as a standardized framework of biogeographic groupings for annotating populations in pharmacogenetics research was adopted and utilized.6 In this proposed grouping system, seven major continental regions are identified on the basis of global genetic ancestry estimates using PCA with 1000 Genomes (1KG) and Human Genome Diversity (HGDP) reference panels.40–42 Since individuals with ancestry from multiple populations do not easily fit into a single ancestral cluster, two additional categories of “admixed” populations are included for a total of nine broad biogeographical groups. Because it is primarily based on ancestral origins that can be detected using PCA, this grouping system is appropriate for classifying study populations that are informative for the scope of genetic diversity in the literature. As such, the biogeographical population groups utilized for this review are not meant to establish a standard for cataloging all human genomic and environmental diversity. Discussion of additional frameworks that are intended to provide more extensive classification of human populations, such as the PhenX Toolkit from the US National Institutes of Health (NIH)43 and Ancestro from curators of the GWAS Catalog at the European Biomedical Informatics Institute (EBI-EMBL),44 are outside the scope of this review.
Biogeographical Population Groups For Pharmacogenomics
The seven major continental groups as outlined by Huddart and colleagues6 in their framework for biogeographic groupings to annotate populations in pharmacogenetics research are as follows: American (AME), East Asian (EAS), European (EUR), Central/South Asian (SAS), Near Eastern (NEA), Oceanian (OCE), and Sub-Saharan African (SSA). The two admixed groups are African American/Afro-Caribbean (AAC) and Latino (LAT). In the AME group, individuals of American Indian or Alaska Native ancestry are included, as well as indigenous and native peoples of South America, and First Nations, Inuit, and Métis of Canada; such that the “America” referenced in the name of the category is that of pre-colonization by European settlers. EAS, EUR, SAS, and OCE are descriptive of geographical/continental regions, including individuals with ancestral origins from those regions. EUR is split from EAS along the Ural Mountains, such that Russia east of that border is included in EAS along with China and other east Asian countries, while west of the border is considered part of Europe. The SSA group includes most countries on the African continent, plus Madagascar, excluding the northern-most countries between Morocco (west) and Egypt (east), which are included in the NEA group along with the Arabian Peninsula and surrounding countries to the north.
While these boundaries may seem counter-intuitive from a geo-political standpoint, they are based on broad, global population structure indicated by detectable patterns of genetic variation via ancestral background, relative to reference panels. Two additional populations, AAC and LAT, are based on genetic ancestry reflecting the recent mixing of more ancient populations as a result of migration and colonization. The African and Afro-Caribbean group involves the key feature of African ancestral origins, which has become distinct from current African populations as a result of substantial ancestry components from Europe and other populations. Similarly, the admixed Latino group includes people from Latin America and self-identified Latinx from the United States.
Due to the heterogeneous and often vague language used to describe study populations in the literature, it was not possible to classify every article that was included in this review into one of the biogeographical groups outlined above. For instance, “African ancestry individuals” is not specific enough to distinguish between African American or Afro-Caribbean (AAC) and Sub-Saharan African (SSA) or even Near-Eastern (NEA), since this region also includes African countries from the north of the continent. Similarly, many study populations are described simply as “Asian” or “black”, which more closely resemble racial classifications than ancestral lineages. Therefore, in order to deal with the lack of specificity and consistency in the way populations are described in the literature, AAC and SSA are combined into a single category of African origin or ancestry. Similarly, EAS and SAS are combined into a single category for Asian origin or ancestry (“African” and “Asian”, respectively). The remaining categories into which pharmacogenomics and precision medicine study populations were grouped correspond to the biogeographic groupings described above: “American Indian or Alaska Native” (AME), “European” (EUR), “Hispanic/Latino” (LAT), and “Near Eastern” (NEA). Using these six broad categories outlined in Figure 1 (African, American Indian or Alaska Native, Asian, European, Hispanic/Latino, and Near Eastern), each article abstract was evaluated to determine the ancestral or socio-cultural background of the populations described and classified into one of the six groups, representing both genomic and environmental diversity.
 |
Figure 1 Scoping Review Flow Chart. The stages of a scoping review are highlighted, with the number of items evaluated at each step: 1) Identification of articles matching specified Mesh terms in PubMed (N=1,006); 2) Screening of articles using filters for availability and relevance (N=202); 3) Determination of eligibility by additional inclusion/exclusion criteria (N=192); and 4) Characteristics of articles included after the first three steps of identification, screening, and eligibility. These characteristics include whether studies were conducted on a single or multiple population(s), and the number of studies conducted in different biogeographical groups, as measured by ancestral origin or socio-cultural label of the population(s) studied. |
Half of the articles reviewed included analysis of a single study population, and the other half included two or more populations, most of which involved a comparison between one population of interest and individuals of European descent. Since many abstracts described their study populations on the basis of what they were not (for example, “non-whites” or “non-Asians”), it was not possible to ascertain which groups were being compared to the primary population in question. In these cases, the study was either excluded from the analysis because there was no specific description of any of the populations involved ("Other criteria" in Fig.1), or further investigation revealed that only a single population had actually been studied. The largest number of study populations fall into the African and European categories, and the majority of these (N=62, 32% of all articles reviewed) were analyzed together in the same study, often presented as a comparison of “white” and “black” study participants or “European Americans” and “African Americans”.
The next most prominently studied group is Asians, with 73 articles involving groups described as being from Asian countries, having Asian descent, or being otherwise referred to as “Asian”. In contrast to the large proportion of African origin study populations that are included in multi-population studies, 39 of the articles that describe research on Asian populations do not involve any other group. This is nearly twice as many single-population studies than for any other group. Perhaps most striking about the representation of populations in the literature is the near absence of research on American Indian and Alaska Natives, Hispanic/Latinx, and Near Eastern groups, relative to the other three described. As a result, the clinical knowledgebase for personalized, or precision, medicine (including pharmacogenomics) is based on an incomplete subset of the variation that is present in the human genome and is particularly sparse for some of the most marginalized global minority populations.
Geographic Distribution Of Pharmacogenomics Investigators
As shown by Popejoy and Fullerton, the diversity of GWAS participants is related to the country in which research investigators are located.4 In order to determine whether the same is true of the pharmacogenomics and precision medicine literature included in this review, country of first author affiliation was extracted using a custom python script to parse MEDLINE reference files downloaded from PubMed. Before 2014, only the first author affiliations are included in PubMed, so it was not possible to analyze country affiliations for other authors, which may have been informative about the geographic region driving the research, such as last author. However, in most cases of articles reviewed after 2014, the country of first and last author affiliations are concordant, so the use of first author affiliation is likely representative of where the research actually took place.
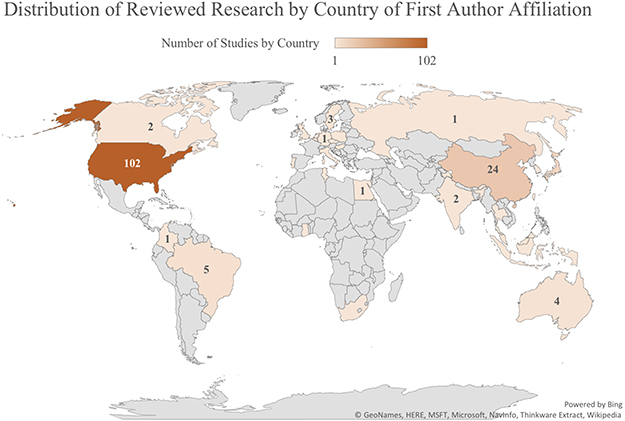 |
Figure 2 Global Concentrations of Studies by Country of First Author Affiliation. Heat map shows the number of studies included in this scoping review that were conducted in each country, as determined by the location of the first author’s affiliated institution. |
In addition to the number of studies conducted in each geographic region by country of first author affiliation (Figure 2), countries in which research was conducted were collapsed into major continental groups: Africa, Asia, Australia, Europe, Latin America, and North America in order to determine which study populations were investigated in each broad global region. Figure 3 illustrates the representation of study populations in research conducted in each global region. As predicted, the majority of studies involving individuals of Asian origin or ancestry were conducted in Asia, and the largest percentage of studies conducted in every other global region (Africa, Europe, Latin America) involved their corresponding study populations (Africa, European, and Hispanic or Latin American origins).
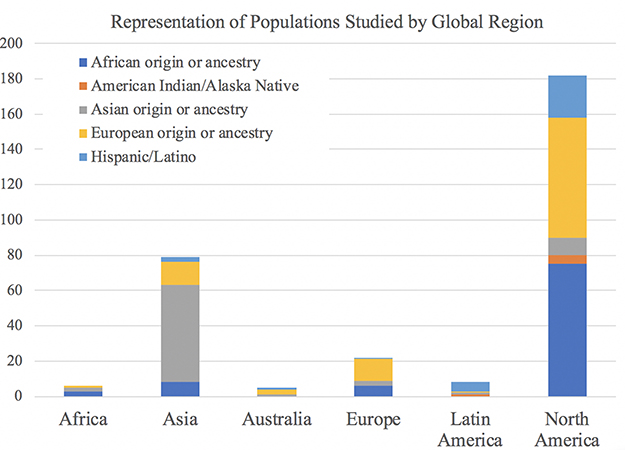 |
Figure 3 Ancestral Populations Included in Reviewed Research by Geographical Region of First Author Affiliation. For each study, geographical region was determined by pooling countries of first author affiliations into broad continental categories. Each study received a binary indicator of whether or not it included at least one population from each broad ancestral category, so studies with more than one study population received more than one indicator. Therefore, the total number on the Y-axis does not reflect the number of studies, but rather the distribution of populations studied within each geographic region. This graph thus illustrates the representation of ancestral populations in the body of research produced in each of the continental regions of the world. |
Because the majority of studies were conducted in North America alone (N=102, 53%), further detail is provided in Figure 4 about the combination of study populations that were investigated in these articles. The largest percentage of studies conducted in North America involved a combination of European and African ancestry participants (N=31, 30%), followed by a large percentage of studies on African participants only (N=24, 24%) and then European participants only (N=15, 15%). Only 5 studies were conducted on American Indian or Alaska Native populations, and 6 were in Hispanic/Latino groups; while 10 involved Africans, Europeans, and Hispanic/Latinos.
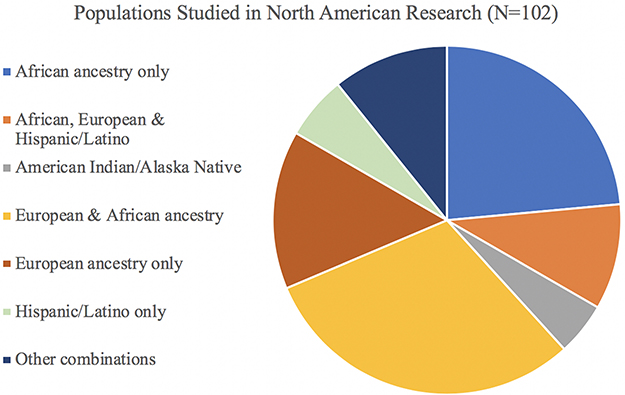 |
Figure 4 Populations Studied in Research Conducted in North America. Among articles included in the scoping review that had first author affiliations in North America (N=102), this figure illustrates which study populations were involved, both as single-population studies and combined in multiple-population studies. In contrast to Figure 3, which shows the total number of populations studied, this pie chart shows the distribution and combination(s) of those populations included across the total number of studies conducted in North America. |
Discussion
The results of this scoping review provide a window of insight into the biogeographic diversity of human populations that are reported and accessible from the pharmacogenomics and precision medicine literature. While the analysis demonstrates the diversity of study populations included in pharmacogenomics and precision medicine literature from broad continental regions, and specifically in North America (Figures 2–4), the representation of non-European populations is likely inflated due to the study design and review methodology. The fact that articles were screened on the basis of having a “population group” MeSH term associated with them in PubMed automatically eliminates any article that was not specific to any population(s). However, it is widely accepted that the majority of datasets analyzed in genomics research are based on European ancestry participants, so articles that do not make a point of differentiating research findings as specific to a European population would not likely meet the annotation screening criterion. Therefore, it is likely that the results of this scoping review underestimate the number of studies in pharmacogenomics and precision medicine that are specific to individuals and populations of European descent. As such, the results of this review should be interpreted with the intentional caveat of over-sampling non-European populations.
Out of 23,720 studies matching MeSH terms “pharmacogenetics” or “precision medicine” in PubMed, only 1,006 of them also match “population groups”, meaning fewer than 5% of studies in pharmacogenomics and precision medicine literature involve any characterization of the human population under investigation. It is likely that the majority of pharmacogenetics and precision medicine research on human populations is simply not tagged with the MeSH term “population groups” because they do not explicitly describe the race, ethnicity, or ancestral origins of the individuals studied. Among those cases, it is impossible to determine to which biogeographical population the study subjects belong; however, it may be the case that these are predominantly white individuals of European descent, since the overwhelming majority of biomedical research has historically been conducted in these populations and it may be considered the “default” for researchers. Alternatively, it may be that the race, ethnicity, or ancestral origins of participants is irrelevant or unknown to researchers, and thus they did not include a description of this characteristic of their study population. In any case, it bears noting that this review covers only 5% of the pharmacogenomics and precision medicine literature, as it is focused specifically on articles that explicitly describe the study populations included in their research. Additionally, while it is standard protocol for scoping reviews to exclude studies that do not include a free full text in PubMed in order to promote reproducibility of the review, the exclusion of 644 such articles may have influenced the results by exclusion of population-specific studies. Therefore, the results of this review should be interpreted as a sampling of the diversity that exists in the pharmacogenomics and precision medicine literature, rather than a comprehensive account of all studies in the field.
Relative to the genomic and environmental diversity that is present in African, American Indian/Alaska Native, and Hispanic/Latino populations worldwide, the pharmacogenomics and precision medicine literature is vastly under-representative. Considering that these populations already experience health disparities compared with people of European and Asian descent, the implementation of personalized medicine designed with diagnosis, treatment, and interventions based primarily on these two groups threatens to further increase health disparities. It is therefore critical that future research efforts in pharmacogenomics and precision medicine specifically target the most under-represented groups, in order to ensure that individuals from diverse genomic and environmental backgrounds benefit equitably from investments in research.
Diversity Requirements In Research
In 1990, the NIH Office of Minority Health Research was established to focus on health disparities,45 and in 1993, the United States Congress directed NIH to develop guidelines requiring that biomedical research funded by the federal government include women and minorities (PL 103–43). Subsequently, guidelines were developed for the inclusion of women and minorities in clinical research, stating that women and members of minority groups must be included in all research funded by the NIH, “unless a clear and compelling rationale and justification” are provided that such inclusion is inappropriate with respect to the health of the subjects, the purpose of the research, or under “other circumstances” (NOT-OD-18-014). However, despite this well-intentioned effort to increase representation in biomedical research, there is no provision in either the law passed by Congress or the regulation developed by the NIH that outlines consequences or ramifications for researchers who choose not to abide by them. As a result, a vast majority of research still does not include members of minority populations, and it is not common practice to justify the lack of representation in study samples. Recent efforts by US-funded research consortia have begun to focus on diverse populations, and the National Human Genome Research Institute (NHGRI) has funded several projects with the explicit goal of increasing diversity in genetics.46 It remains to be seen how influential these projects will be in the long run, but at the very least it sends a message to researchers that obtaining diverse research cohorts is a current priority for funding.
No such regulations on diversity and inclusion currently exist in the United Kingdom, and recent trends such as the development of the highly utilized UK Biobank47 indicate the exact opposite. Hundreds of GWAS conducted on data made publicly available by the UK Biobank continue to contribute to the European-biased knowledgebase of genetic associations with traits and disease, despite the fact that data are available on thousands of individuals of South Asian descent in this resource. This is likely due to a combination of factors, including the larger sample size of Europeans relative to other groups, perceived difficulty of conducting GWAS in multiple or non-European populations, and the historical view that white Europeans are more “homogeneous” with fewer variables that need to be controlled, thus leading to crisper statistical findings.
More powerful and authoritative regulations requiring diversity in biomedical research, and in particular the basic science leading to clinical applications such as pharmacogenomics and personalized medicine, are necessary in order to make a substantive impact. Both genomic and environmental diversity should be considered in the process of study design, such that factors including ancestral and socio-cultural background can be appropriately taken into account in the discovery and validation of underlying causes of Mendelian and complex disease.
Remaining Challenges And Solutions
Both challenges and solutions to the lack of diversity in genomics research involve ideological, analytic, cultural, demographic, and systemic elements. This is because deeply ingrained historical practices, structures and ideologies have shaped the way biomedical research is conducted, including who is prioritized as research subjects, who is conducting the research, and under what conditions and assumptions. From an ideological perspective, the assumption that people of European ancestry are the methodologically preferred study subjects is due to a perceived “homogeneity” in genomic and environmental factors, as well as sample sizes in existing datasets that far exceed other populations. A common practice in GWAS is to stratify study subjects by racial, ethnic, or ancestral categories to control for population structure. Often, only the largest sample is included in the analysis, which results in exclusion of non-European or admixed samples. However, the logic underlying these assumptions as a justification to focus on white or European study samples is flawed, both for scientific and values-based reasons.
Early genetic association analyses included very few genetic markers and relied on the LD structure inherent to European ancestry populations, which have longer stretches of LD than African genomes, meaning fewer tag-SNPs needed to be included in order to capture potentially disease-causing variation in larger regions of the genome. Now that the cost and technology of sampling more genomic loci that capture greater variation in non-European populations has become trivial, this methods-based justification for focusing on Europeans is becoming increasingly obsolete. Furthermore, the idea that anyone who is white, or appears to be of European descent, falls into a homogeneous group in terms of genomic and environmental diversity is based on outdated and harmful views that visual similarity (on the basis of typically “racial” characteristics such as skin and hair color) somehow confers biological meaning on its own – which it inherently does not, as described in detail in the introduction. Finally, the statistical argument for focusing on the largest sample size available in genetic association studies is based on privileging statistical significance over all other considerations, including the potential for novel discoveries of disease mechanisms. It is therefore important for researchers to challenge their own assumptions that chasing the lowest p-value in a larger sample size from Europeans will contribute more to the genomic knowledgebase than a larger (but still significant) p-value demonstrating previously unknown effects of variants that contribute to disease etiology in more diverse, traditionally under-represented populations.
Complex traits are influenced by a variety of genetic and environmental factors, including GxG and GxE interactions. In this case, a person’s self-reported race or ethnicity may influence their risk for disease or health outcomes because of tertiary confounding factors that are associated with both the phenotype and the identity measure, such as neighborhood, access to healthy food and health care, education level, and other community-level effects. In practice, self-reported measures such as race and ethnicity have been used in epidemiological models as a proxy for these environmental effects. However, it is critical to recognize that this is an imperfect measure, and it relies on the fact that there are historical, cultural, systemic and institutionalized inequalities that preserve the association between lower socio-economic status and race or ethnicity. It would be more rigorous to measure environmental factors directly and relegate the use of “race” as a variable to account for the phenomenon to which it directly applies: the experience of racism, which may impact a person’s health or risk for disease.
Unfortunately, barriers to integrating this nuanced awareness into research practice remain. From a practical standpoint, most datasets and even large cohorts simply do not contain detailed environmental data, and even when available, these data are often highly variable with regard to quality and consistency. However, from an ideological perspective, the continued use of self-reported identity measures as variables in research can only be justified so long as these measures are recognized for what they are: a proxy for environmental effects of systemic and institutionalized racism and inequality. Some researchers may choose to study white, European-ancestry populations to avoid the potential confusion or difficulty of accounting for this more detailed view of complex trait architecture. They may also believe that this approach is more likely to yield significant knowledge about the genetic components of disease. However, due to the fact that the majority of human genomic variation exists outside Europeans, the continued focus on these populations is unfounded. Conducting GWAS in non-European ancestry populations is likely to contribute substantially more to the knowledgebase of genetic mechanisms of disease than continuing to study people of primarily European ancestry.48 Continued development of more accurate means of accounting for genomic and environmental diversity will help streamline the selection of samples to prioritize in research, and measurement of variables that influence complex disease etiology. Finally, in addition to shifting historical, ideological and methodological practices that perpetuate a Euro-centric bias in genomics research, broadening participation of diverse groups in research, both as participants and as investigators, is essential to ensure equitable representation.
Broadening Participation In Research
Ancestral and socio-cultural backgrounds (or geographic location, in this review) of researchers influences the study populations on which they focus. Therefore, efforts to increase diversity in study participants must be accompanied by reciprocal efforts to increase the representation of people with diverse ancestral and socio-cultural backgrounds among leading research investigators. According to National Science Foundation (NSF) statistics, just 8.6% of all science and engineering doctorate holders employed as faculty in US academic institutions in 2015 were from under-represented minority groups, including African Americans, Hispanics or Latinos, and American Indian or Alaska Natives, combined.49 Evidence from earlier studies suggests that this percentage decreases when narrowing the analysis to tenured and tenure-track faculty in basic research departments.50 Institutional commitment to diversity efforts in hiring are important, but equally critical are illuminating and eradicating systemic and cultural factors that contribute to higher attrition rates for minority faculty researchers once they are hired.51
In theory, researchers with more diverse backgrounds will have a more nuanced understanding of the barriers to increased representation in genomics research and may be inherently committed to involving diverse community perspectives in the design and implementation of their research studies.52 In practice, however, institutional leadership must be careful to avoid creating additional administrative or service-related burdens for minority faculty members. These individuals should not be expected to carry a torch for diversity efforts, nor should they be treated as token members of certain groups and expected to speak on behalf of entire communities. As such, universities and funding agencies need to invest in systemic programs and training for all faculty members, including the development of community partnerships and databases, research methods for investigating multi-population cohorts, and incentives for conducting service-related activities that support the mentorship and promotion of minority students and faculty to leadership positions.
Historical abuses of power and harms against minority populations have contributed to a lack of trust in biomedical research institutions,53 and this is still an important challenge for researchers to overcome when seeking to establish community relations with diverse populations. Fortunately, resources are becoming available that instruct investigators on how to be effective allies and partners in research, engaging in meaningful community-based participatory research (CBPR), especially in some of the most under-represented populations such as Native American and other indigenous communities.54,55 A recent decision by the NIH to fund administrative supplements for bioethical components of existing research projects demonstrates how funding agencies can support efforts to enable ethical engagement and research in diverse populations. Ideally, this trend would expand to include funding mechanisms for community outreach and engagement for study design, in advance of research that is to be conducted in human populations.
Conclusion
This scoping review interrogates the precision medicine and pharmacogenomics literature to determine what proportion of studies focus on global human populations, thus providing a baseline of diversity in the knowledgebase for future comparison. The persistent under-representation of genomic and environmental diversity in research from individuals of African descent, as well as Hispanic/Latinx, American Indian/Alaska Natives and other indigenous communities worldwide may continue to widen health disparities. Until funding agencies, journals, and research institutions develop incentives for researchers to investigate diverse populations and provide systemic programs and training to enable this work, it is unlikely that there will be a huge shift in the diversity profile of genetics research. In order for pharmacogenomics and personalized medicine to equitably benefit people from all ancestral and socio-cultural backgrounds, there needs to be vastly greater representation of global populations and communities. In order for this to happen, researchers must commit to broader inclusion of diverse study participants and mentorship of under-represented trainees from the bottom-up, and institutions must facilitate the development of a diverse knowledgebase and workforce from the top-down.
Abbreviations
GWAS, Genome-wide association studies; SNP, single-nucleotide polymorphism; ELSI, ethical, legal, social implications; MRCA, most recent common ancestor; LD, linkage disequilibrium; AIMs, ancestry informative markers; PCA, principal components analysis; GxG, gene-gene interactions; GxE, gene-environment interactions; 1KG, 1000 Genomes Project; HGDP, Human Genome Diversity Project; AME, American; EAS, East Asian; EUR, European; SAS, Central/South Asian; NEA, Near Eastern; OCE, Oceanian; SSA, Sub-Saharan African; AAC, African American/Afro-Caribbean, LAT, Latino; NIH, National Institutes of Health; EBI-EMBL, European Biomedical Informatics Institute; NSF, National Science Foundation.
Acknowledgments
The author would like to thank Arturo Lopez Pineda for discussions and advice regarding the methods and visualization of a scoping review, and Genevieve Wojcik, Ragan Hart, and Carlos Bustamante for comments and feedback on early stages of review methods, findings, and figures.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24. doi:10.1016/j.ajhg.2011.11.029
2. Motsinger-Reif AA, Jorgenson E, Relling MV, et al. Genome-Wide Association Studies in Pharmacogenomics: successes and Lessons. Pharmacogenet Genomics. 2013;23(8):383–394. doi:10.1097/FPC.0b013e32833d7b45
3. Low SK, Takahashi A, Mushiroda T, et al. Genome-wide association study: a useful tool to identify common genetic variants associated with drug toxicity and efficacy in cancer pharmacogenomics. Clin Cancer Res. 2014;20(10):2541–2552. doi:10.1158/1078-0432.CCR-13-3045
4. Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. 2016;538(7624):161–164. doi:10.1038/538161a
5. Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell. 2019;177(1):26–31. doi:10.1016/j.cell.2019.02.048
6. Huddart R, Fohner AE, Whirl-Carillo M, et al. Standardized biogeographic grouping system for annotating populations in pharmacogenetic research. Clin Pharmacol Ther. 2018. Epub ahead of print. doi:10.1002/cpt.1322
7. Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325:31–36. doi:10.1038/325723a0
8. Henn BM, Gignoux CR, Jobin M. Hunter-gatherer genomic diversity suggests a southern African origin for modern humans. Proc Natl Acad Sci. 2011;108(13):5154–5162. doi:10.1073/pnas.1017511108
9. Ramachandran S, Deshpande O, Roseman CC. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci. 2005;102(44):15942–15947. doi:10.1073/pnas.0507611102
10. Nielsen R, Akey JM, Jakobsson M. Tracing the peopling of the world through genomics. Nature. 2017;541(7637):302–310. doi:10.1038/nature21347
11. Auton A, Brooks LD, Durbin RM; for the 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi:10.1038/nature15393
12. Rosenberg NA, Pritchard JK, Weber JL. Genetic structure of human populations. Science. 2002;298(5602):2381–2385. doi:10.1126/science.1078311
13. Nei M. Genetic distance between populations. Am Nat. 1972;106(949):283–292. doi:10.1086/282771
14. Cavalli-Sforza LL, Edwards AWF. Phylogenetic analysis, models and estimation procedures. Am J Hum Genet. 1967;19(3 Pt 1):233–257.
15. Reynolds J, Weir BS, Cockerham CC. Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics. 1983;105(3):767–779.
16. Nei M. Molecular Evolutionary Genetics. New York: Columbia University Press; 1987. Chapter 9.
17. Wright S. Genetical structure of populations. Nature. 1950;166(4215):247–249. doi:10.1038/166247a0
18. Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38(6):1358–1370.
19. Polimanti R, Piacentini S, Manfellotto D, Fuciarelli M. Human genetic variation of CYP450 superfamily: analysis of functional diversity in worldwide populations. Pharmacogenomics. 2013;13(16):
20. Lewontin RC. The Genetic Basis of Evolutionary Change. New York: Columbia University Press; 1974.
21. Paschou P, Lewis J, Javed A, et al. Ancestry informative markers for fine-scale individual assignment to worldwide populations. J Med Genet. 2010;47(12):835–847. doi:10.1136/jmg.2010.078212
22. Novembre J, Johnson T, Bryc K, et al. Genes mirror geography within Europe. Nature. 2008;456(7218):98–101. doi:10.1038/nature07331
23. Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi:10.1101/gr.094052.109
24. Lawson DJ, Hellenthal G, Myers S, Falush D, Copenhaver GP. Inference of population structure using dense haplotype data. PLoS Genet. 2012;8(1):e1002453. doi:10.1371/journal.pgen.1002453
25. Marigorta UM, Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 2013;9(6):e1003566. doi:10.1371/journal.pgen.1003566
26. Martin AR, Gignoux CR, Walters RK, et al. Human demographic history impacts genetic risk prediction across diverse populations. Am J Hum Genet. 2017;100(4):635–649. doi:10.1016/j.ajhg.2017.03.004
27. Hasin Y, Seldin M, Lusiscorresponding A. Multi-omics approaches to disease. Genome Biol. 2017;18:83. doi:10.1186/s13059-017-1215-1
28. Williams DR, Priest N, Anderson N. Understanding associations between race, socioeconomic status and health: patterns and prospects. Health Psychol. 2016;35(4):407–411. doi:10.1037/hea0000242
29. Banda Y, Kvale MN, Hoffman TJ. Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics. 2015;200:1285–1295. doi:10.1534/genetics.115.178459
30. Lewontin RC. The apportionment of human diversity. In: Dobzhansky T, Hecht MK, Steere WC, editors. Evolutionary Biology. New York: Springer; 1972. 381–397.
31. Hunley KL, Cabana GS, Long JC. The apportionment of human diversity revisited. Am J Phys Anthropol. 2016;160(4):561–569. doi:10.1002/ajpa.v160.4
32. Nei M, Roychoudhury AK. Genic variation within and between the three major races of man, caucasoids, negroids, and mongoloids. Am J Hum Genet. 1974;26:421–443.
33. Bonham VL, Green ED, Pérez-Stable EJ. Examining how race, ethnicity, and ancestry data are used in biomedical research. Jama. 2018;320(15):1533–1534. doi:10.1001/jama.2018.13609
34. Race, Ethnicity, & Genetics Working Group. The use of racial, ethnic, and ancestral categories in human genetics research. Am J Hum Genet. 2005;77(4):519–532. doi:10.1086/491747
35. Mersha TB, Abebe T. Self-reported race/ethnicity in the age of genomic research: its potential impact on understanding health disparities. Hum Genomics. 2015;9:1. doi:10.1186/s40246-014-0023-x
36. Ford CL, Collins O. Critical race theory, race equity, and public health: toward antiracism praxis. Am J Public Health. 2010;100(Suppl.1):S30–S35. doi:10.2105/AJPH.2009.171058
37. McCarthy MI, Abecasis GR, Cardon LR. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356–369. doi:10.1038/nrg2344
38. Wechsler ME, Castro M, Lehman E, et al.; The NHLBI Asthma Clinical Research Network. Impact of race on asthma treatment failures in the asthma clinical research network. Am J Respir Crit Care Med. 2011;184(11):1247–1253. doi:10.1164/rccm.201103-0514OC
39. Gould W, Peterson EL, Karungi G, et al. Factors predicting inhaled corticosteroid responsiveness in African American patients with asthma. J Allergy Clin Immunol. 2010;126(6):1131–1138. doi:10.1016/j.jaci.2010.08.002
40. 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi:10.1038/nature11632
41. Cann HM, De Toma C, Cazes L, et al. A human genome diversity cell line panel. Science. 2002;296(5566):261–262. doi:10.1126/science.296.5566.261b
42. Li JZ, Absher DM, Tang H, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319(5866):1100–1104. doi:10.1126/science.1153717
43. Hamilton CM, Strader LC, Pratt JG, et al. The PhenX Toolkit: get the most from your measures. Am J Epidemiol. 2011;174(3):253–260. doi:10.1093/aje/kwr193
44. Morales J, Welter D, Bowler EH, et al. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 2018;19:21. doi:10.1186/s13059-018-1396-2
45. Pérez-Stable EJ, Collins FS. Science visioning in minority health and health disparities. Am J Pub Health. 2019;109(S1):55. doi:10.2105/AJPH.2019.304962
46. Hindorff LA, Bonham VL, Brody LC, et al. Prioritizing diversity in human genomics research. Nat Rev Genet. 2018;19:175–185. doi:10.1038/nrg.2017.89
47. Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi:10.1038/s41586-018-0369-7
48. Hindorff LA, Bonham VL, Ohno-Machado L. Enhancing diversity to reduce health information disparities and build an evidence base for genomic medicine. Per Med. 2018;15(5):403–412. doi:10.2217/pme-2018-0037
49. National Science Foundation, National Center for Science and Engineering Statistics. 2017. Women, Minorities, and Persons with Disabilities in Science and Engineering: 2017. Special Report NSF 17-310. Arlington, VA. Available from: www.nsf.gov/statistics/wmpd/. Accessed October 02, 2019.
50. Leboy PS, Madden JF. Limitations on diversity in basic science departments. DNA Cell Biol. 2012;31:1365–1371. doi:10.1089/dna.2012.1756
51. Whittaker JA, Montgomery BL, Martinez Acosta VG. Retention of underrepresented minority faculty: strategic initiatives for institutional value proposition based on perspectives from a range of academic institutions. Undergrad Neurosci Educ. 2015;13(3):A136–A145.
52. Sierra-Mercado D, Lázaro-Muñoz G. Enhance diversity among researchers to promote participant trust in precision medicine research. Am J Bioeth. 2018;18(4):44–46. doi:10.1080/15265161.2018.1431323
53. Garrison NA, Cho MK. Awareness and acceptable practices: IRB and researcher reflections on the Havasupai Lawsuit. AJOB Prim Res. 2013;4(4):55–63. doi:10.1080/21507716.2013.770104
54. Claw KG, Anderson MZ, Begay RL, et al.; for the Summer internship for INdigenous peoples in Genomics (SING) Consortium. A framework for enhancing ethical genomic research with Indigenous communities. Nat Commun. 2018;9:2957. doi:10.1038/s41467-018-05188-3
55. Hudson M, Beaton A, Milne M, et al. Te Mata Ira: Guidelines for Genomic Research with Māori. Hamilton, New Zealand: Te Mata Hautū Taketake-Māori & Indigenous Governance Centre, University of Waikato; 2016.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.