Back to Journals » International Medical Case Reports Journal » Volume 19
Diffuse Maxillofacial Neurofibromas with Orbital and Cranial Extension Suggestive of Neurofibromatosis Type I: A Comparative Case Series from Uganda
Authors Mbuliro A, Mawanda A, Nakitto BM, Kamulegeya A ![]()
Received 10 January 2026
Accepted for publication 30 June 2026
Published 14 July 2026 Volume 2026:19 595092
DOI https://doi.org/10.2147/IMCRJ.S595092
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Aisha Mbuliro,1 Anatoli Mawanda,2– 4 Barbara Mukasa Nakitto,5 Adriane Kamulegeya1,5
1School of Dentistry, Makerere University, Kampala, Uganda; 2Department of Pathology, Uganda Cancer Institute, Kampala, Uganda; 3Department of Pathology, King Ceasor University, Kampala, Uganda; 4Department of Pathology, Mulago National Referral Hospital, Kampala, Uganda; 5Department of Oral and Maxillofacial Surgery, Mulago National Referral Hospital, Kampala, Uganda
Correspondence: Aisha Mbuliro, Email [email protected]
Background: Diffuse maxillofacial neurofibromas with orbital and cranial extension are rare manifestations of suspected Neurofibromatosis Type I (NF1). These tumors present diagnostic and therapeutic challenges, particularly in low resource settings.
Case Presentation: Two Ugandan patients with diffuse maxillofacial neurofibromas are described. A 13-year-old girl developed early-onset, rapidly progressive right facial swelling with bilateral orbital involvement, monocular blindness, and sphenoid bone destruction. Multiple café-au-lait macules were present with bilateral Lisch nodules but absent skeletal deformities. Histopathology and immunohistochemistry confirmed diffuse neurofibroma. Conservative debulking was performed with a stable but cosmetically disfiguring outcome. A middle-aged man presented with a 15-year history of right facial swelling following trauma, associated with proptosis, trismus, hearing loss, and intraoral masses. Imaging revealed tumor extension to the skull base. Initial biopsy was suggestive of a myofibroblastic tumor, but final excisional biopsy confirmed neurofibroma. Partial resection undertaken due to extensive involvement, left a residual tumor under surveillance. Both patients had café-au-lait macules and Lisch nodules but did not fully meet NIH criteria for NF1, and genetic testing was unavailable.
Conclusion: The cases highlight diagnostic uncertainty of NF1 in low-resource environments, were reliance on clinical and radiological features substitutes for genetic confirmation. Pediatric disease tends to be more aggressive with earlier morbidity, while adult disease shows slower but infiltrative growth. Conservative surgery and multidisciplinary care are key to preserving function and quality of life, underscoring the need for long-term follow-up and access to emerging targeted therapies.
Keywords: neurofibromatosis type I, diffuse neurofibroma, craniofacial tumors, orbital involvement, resource-limited settings, case series
Background
Neurofibromatosis was first formally described by the German pathologist Friedrich Daniel von Recklinghausen in 1882, who identified the neural origin of these tumors and characterized the condition that now bears his name.1 Neurofibromatosis Type I (NF1), also known as von Recklinghausen disease, is an autosomal dominant genetic disorder caused by mutations in the NF1 gene on chromosome 17q11.2, with a prevalence of 1 in 2500–3500 individuals worldwide.2,3 Clinical features include café-au-lait macules, neurofibromas, Lisch nodules, optic gliomas, and skeletal abnormalities.3,4 Diagnosis is based on fulfilling at least two of the National Institutes of Health (NIH) criteria, which were revised in 2021 to improve sensitivity, particularly in younger patients.4,5
Diffuse maxillofacial neurofibromas with orbital and cranial extension are rare but clinically significant due to their potential for visual loss, cranial involvement, and severely disfiguring outcomes.6,7 Skull base involvement, as seen in plexiform neurofibromas extending intracranially, represents one of the most challenging surgical scenarios in NF1 management.7 In resource-limited settings, absence of advanced genetic testing, limited access to imaging, and shortage of multidisciplinary expertise further complicate diagnosis and management.8 Two cases; one pediatric and one adult are reported to illustrate the clinical presentation, diagnostic challenges, and treatment outcomes of diffuse maxillofacial neurofibromas in Uganda.
Case Presentation
Case 1: Pediatric Patient
A 13-year-old African girl presented with progressive right-sided facial swelling that had been present since she was 6 months of age (Figure 1). She had undergone debulking surgery at age 3, but recurrence led to orbital expansion and monocular blindness. Examination showed right preauricular, temporal, and orbital swelling, a phthisical right eye, pulsatile proptosis of the left eye, and multiple café-au-lait macules with bilateral Lisch nodules but absent skeletal deformities.
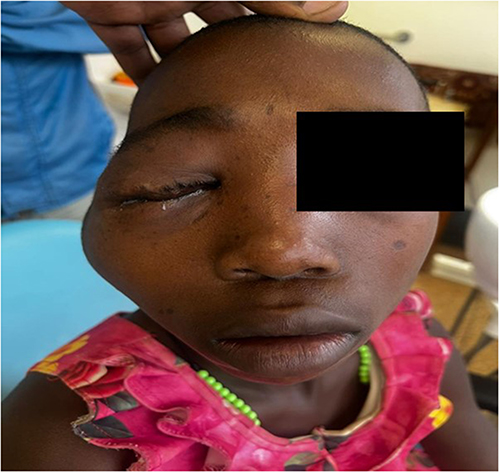 |
Figure 1 Pre-operative clinical photograph of the pediatric patient (Case 1) demonstrating right-sided preauricular, temporal, and orbital swelling. Photograph taken on 6 August 2025. |
Histopathology revealed spindle-shaped cells with hyperchromatic nuclei, pseudomeissnerian corpuscles, and collagenous stroma (Figures 2 and 3). Immunohistochemistry showed strong S-100 positivity (Figure 4), CD34 staining in a fingerprint pattern (Figure 5), EMA negativity (Figure 6), and a low Ki-67 proliferation index of less than 5% (Figure 7), confirming diffuse neurofibroma.9
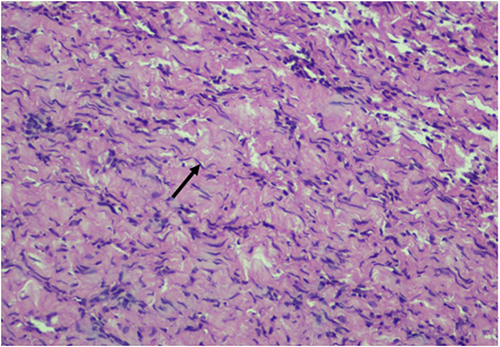 |
Figure 2 Hematoxylin and eosin (H&E) sections showing haphazardly arranged spindle-shaped cells with poorly defined borders and hyperchromatic wavy and comma-shaped nuclei demonstrating a “diving dolphin” resemblance, shown in black arrows (H&E x40). |
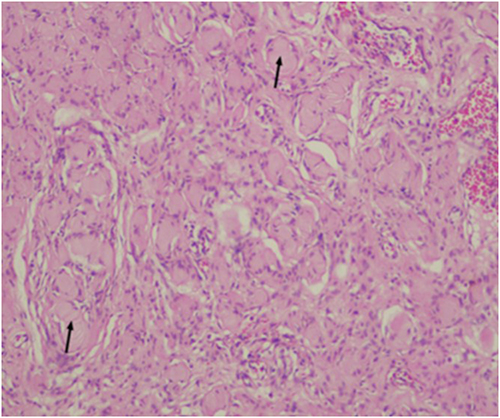 |
Figure 3 H&E sections at higher magnification showing Pseudomeissnerian corpuscles (Wagner–Meissner bodies) shown in black arrows, (H&E x100). |
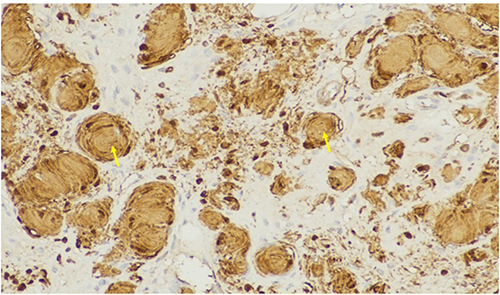 |
Figure 4 Immunohistochemical staining with S-100 protein demonstrating strong nuclear and cytoplasmic positivity (+) in Schwann cells, confirming neural differentiation, shown in yellow arrows. |
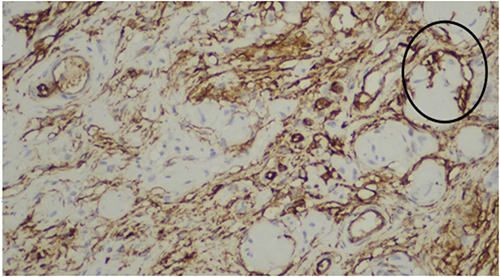 |
Figure 5 Immunohistochemical staining for CD34 demonstrating positivity between collagen bundles in a characteristic fingerprint pattern, supporting the diagnosis of diffuse neurofibroma. Shown in circles (black). |
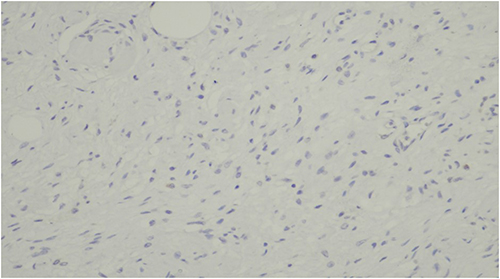 |
Figure 6 Negative immunohistochemical staining for Epithelial Membrane Antigen (EMA), helping to exclude schwannoma and perineurioma. |
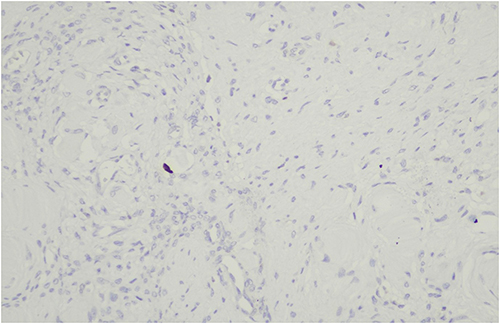 |
Figure 7 Immunohistochemical staining for Ki-67 demonstrating a low proliferation index of less than 5%, consistent with a benign neoplasm. |
Conservative debulking was performed by oral and maxillofacial surgeons. Intraoperatively, the tumor was found to adhere firmly to the overlying skin; attempts to separate the tumor from the skin were unsuccessful and were associated with skin perforation. Postoperative recovery was uneventful. At 4 weeks and at 8 weeks (Figure 8) postoperatively the patient remained monocular with persistent facial deformity but demonstrated no progression of tumor.
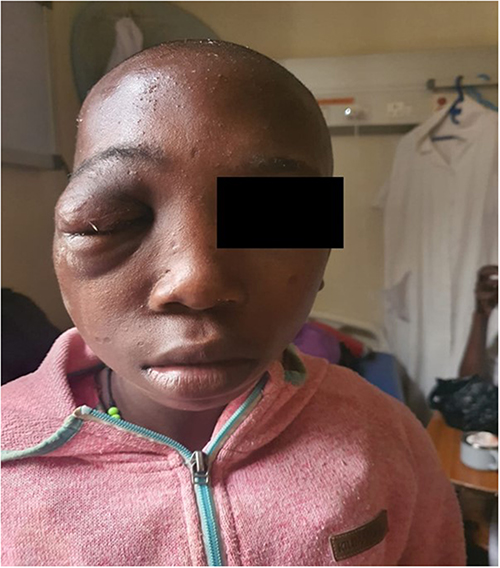 |
Figure 8 Post-operative clinical photograph of the pediatric patient (Case 1) at 8-week follow-up demonstrating residual facial deformity with no tumor progression. Photograph taken on 13 August 2025. |
Case 2: Adult Patient
A middle-aged African man presented with a 15-year history of progressive right facial swelling that began following trauma, associated with proptosis, hearing loss, trismus, and difficulty chewing (Figure 9). Multiple café-au-lait macules were noted on examination (Figure 10).10 Examination revealed interconnected soft tissue masses involving the right cheek, parotid region, infraorbital area, and intraoral mucosa. Bilateral Lisch nodules were present, but no skeletal abnormalities were observed.
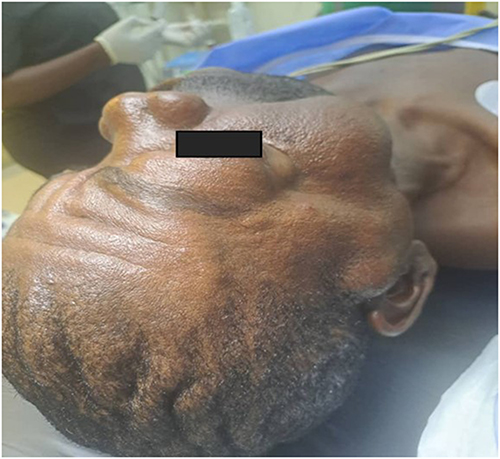 |
Figure 9 Pre-operative clinical photograph of the adult patient (Case 2) demonstrating right facial swelling involving the cheek, parotid, and infraorbital regions. Photograph taken intraoperatively on 30 June 2026. |
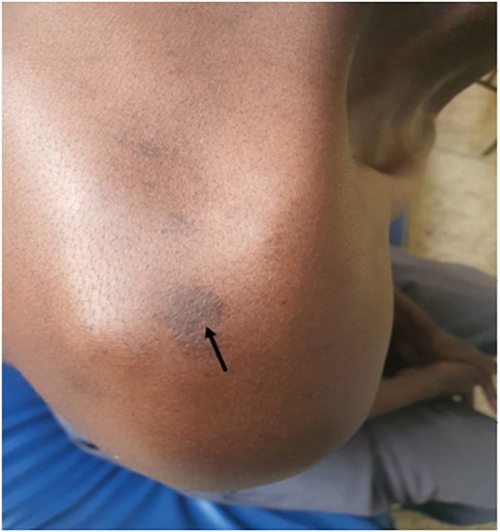 |
Figure 10 Café au lait spots on the posterior shoulders of the adult patient (Case 2). Multiple hyperpigmented macules are visible (black arrows). Photograph taken on 1 June 2025. |
CT showed isodense soft tissue masses with hyperdense rims involving the right maxillary sinus, ethmoid sinus, orbital floor, and anterior cranial fossa (Figure 11).7 Fine needle aspiration cytology was inconclusive, and initial biopsy suggested a myofibroblastic tumor. Surgical excision revealed fusiform tan-brown masses with tortuous nerve bundles consistent with a “bag of worms” appearance (Figures 12 and 13).11 Final histopathology confirmed neurofibroma, though S-100 staining was unavailable at the time of analysis.
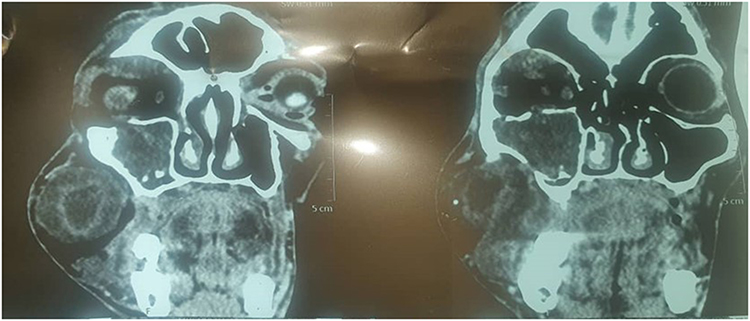 |
Figure 11 CT scan of the adult patient (Case 2) taken on 1 June 2025 demonstrating isodense soft tissue masses with hyperdense rims. Tumor extension into the anterior cranial fossa, involvement of the orbital floor, cranial extension into the ethmoid and maxillary sinuses is also demonstrated. |
 |
Figure 12 Gross morphology of the excised fusiform neurofibroma masses demonstrating the characteristic “bag of worms” appearance of tortuous nerve bundles. Photograph taken intraoperatively on 30 June 2026 (adult case). |
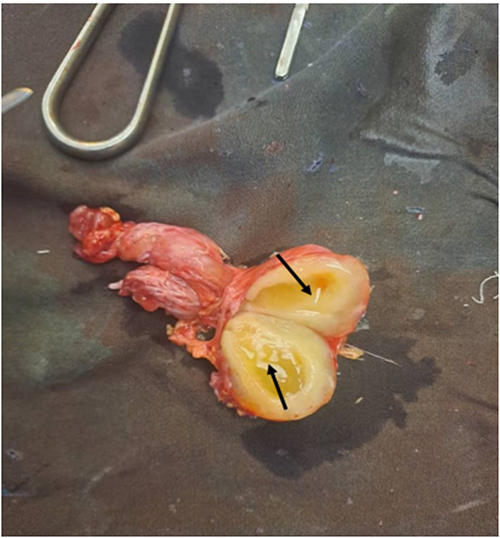 |
Figure 13 Gross internal morphology of one of the excised neurofibroma masses demonstrating the cut surface, shown in black arrows. Photograph taken intraoperatively on 30 June 2026 (adult case). |
Partial surgical excision was performed, leaving residual tumor due to extensive skull base involvement. At 2 months postoperatively (Figure 14), recovery was complicated by trismus, right eye obliteration, numbness, and facial asymmetry. The patient remains under active surveillance.
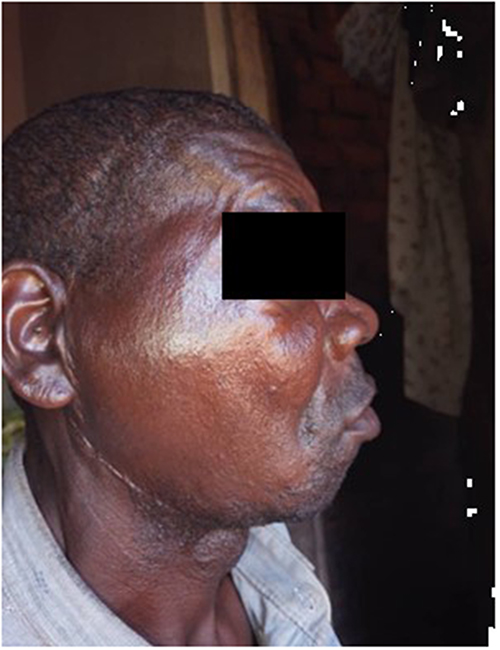 |
Figure 14 Follow-up clinical photograph of the adult patient (Case 2) at 2 months postoperatively demonstrating residual facial asymmetry and right eye obliteration. Photograph taken on 2 December 2025. |
Discussion
The cases demonstrate substantial difficulty of diagnosing NF1 in settings where genetic testing and advanced diagnostics are unavailable.6,8 Both patients exhibited multiple café-au-lait macules, diffuse neurofibromas, and bilateral Lisch nodules all strongly suggestive of NF1 but neither fully met the revised NIH diagnostic criteria.4,5 The child’s sphenoid bone destruction strongly supported a presumptive diagnosis of NF1.7
Diagnosis was particularly challenging in the adult case, where FNAC and initial biopsy yielded misleading results. This illustrates the well-recognized limitations of superficial biopsy in spindle cell tumors and underscores the importance of excisional sampling for definitive histopathological diagnosis.9,11 Immunohistochemical markers, including S-100, CD34, and EMA, are critical for distinguishing neurofibroma from other spindle cell neoplasms such as schwannoma and solitary fibrous tumor.9
The disease course differed markedly by age: the pediatric patient developed aggressive early-onset disease with rapid orbital destruction and blindness, whereas the adult experienced slow, infiltrative progression involving the skull base. Literature supports that NF1-associated tumors in children demonstrate higher proliferative activity, while adult tumors tend to be more diffuse and surgically unresectable.12,13 Cranial extension of neurofibromas, including rare associations with gliomatosis cerebri, further underscores the complexity of CNS involvement in NF1.14
Surgical management must balance functional preservation against tumor control. In children, conservative debulking helps preserve appearance and function despite incomplete removal.13,15 In adults, skull base involvement often necessitates partial resection, as demonstrated in this case16 Both cases underscore the critical importance of multidisciplinary input including neurosurgery and ophthalmology which was unfortunately unavailable in this setting.8 The management of optic nerve glioma in resource-limited environments has been described elsewhere, with reports of successful outcomes using intensity-modulated radiotherapy even under constrained conditions.17
The risk of malignant transformation of NF1-associated neurofibromas into malignant peripheral nerve sheath tumors is estimated at 2–3%, necessitating long-term surveillance.18 Targeted therapies, particularly MEK inhibitors such as selumetinib, have demonstrated efficacy in reducing the volume of inoperable plexiform neurofibromas in children.19 However, the practical challenges of delivering selumetinib therapy in resource-limited settings including drug availability, cost, and monitoring requirements remain significant barriers.20 These barriers highlight the need for context-appropriate treatment protocols and international support for NF1 management in sub-Saharan Africa.8
Emerging tools including artificial intelligence-assisted variant identification and liquid biopsy technologies may further improve diagnostic accuracy in the future.21,22 Long-term psychosocial support and structured transition programs for adolescents and young adults with NF1 are also increasingly recognized as essential components of comprehensive care.23
Conclusions
Diffuse maxillofacial neurofibromas with orbital and cranial extension are rare, debilitating manifestations of NF1. In resource-limited contexts such as Uganda, diagnosis relies primarily on clinical and radiological features, and treatment requires tailored surgical approaches. Early detection, multidisciplinary collaboration, and long-term surveillance are essential to improving functional outcomes and quality of life for affected patients. Improved access to targeted therapies and genetic testing remains an urgent priority.
Abbreviations
CT, Computed Tomography; EMA, Epithelial Membrane Antigen; FNAC, Fine Needle Aspiration Cytology; IHC, Immunohistochemistry; MEK, Mitogen-activated protein kinase/extracellular signal-regulated kinase; MPNST, Malignant Peripheral Nerve Sheath Tumor; NF1, Neurofibromatosis Type I; NIH, National Institutes of Health.
Data Sharing Statement
All data relevant to this study are included in this article. Additional details are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
This case series did not require formal institutional ethical approval for publication of case details in accordance with the policies of Makerere University, Kampala, Uganda, and Mulago National Referral Hospital, Kampala, Uganda. Written informed consent was obtained from the adult patient and from the parent/guardian of the pediatric patient for participation and publication.
Consent for Publication
Written informed consent was obtained from the adult patient and from the parent of the pediatric patient for publication of this case report and any accompanying images.
Acknowledgments
The authors thank the patients and their families for their cooperation and consent to publish.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No external funding was received for this study.
Disclosure
The authors declare no competing interests.
References
1. Ghalayani P, Saberi Z, Sardari F. Neurofibromatosis type I (von Recklinghausen’s disease): a family. Dent Res J. 2012;9(4):483.
2. Anderson J, Gutmann D. Neurofibromatosis type 1. In: Handbook of Clinical Neurology. Vol. 132. Elsevier;2015:75–10. doi:10.1016/B978-0-444-62702-5.00004-4
3. Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med. 2010;12(1):1–11. doi:10.1097/GIM.0b013e3181bf15e3
4. Legius E, Messiaen L, Wolkenstein P, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. 2021;23(8):1506–1513. doi:10.1038/s41436-021-01170-5
5. Angelova-Toshkina D, Holzapfel J, Huber S, et al. Neurofibromatosis type 1: a comparison of the 1997 NIH and the 2021 revised diagnostic criteria in 75 children and adolescents. Genet Med. 2022;24(9):1978–1985. doi:10.1016/j.gim.2022.05.013
6. Kehrer-Sawatzki H, Cooper DN. Challenges in the diagnosis of neurofibromatosis type 1 (NF1) in young children facilitated by means of revised diagnostic criteria including genetic testing for pathogenic NF1 gene variants. Hum Genet. 2022;141(2):177–191. doi:10.1007/s00439-021-02410-z
7. Maqsood H, Saim M, Anjum AS, Younus S. A rare case of diffuse neurofibroma of the scalp with destructive lesions involving the base of the skull in a patient with neurofibromatosis type 1. Cureus. 2021. doi:10.7759/cureus.13930
8. Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81–88. doi:10.1136/jmg.2006.045906
9. Park JY, Park H, Park NJ, Park JS, Sung HJ, Lee SS. Use of Calretinin, CD56, and CD34 for differential diagnosis of schwannoma and neurofibroma. Korean J Pathol. 2011;45(1):30. doi:10.4132/KoreanJPathol.2011.45.1.30
10. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61(1):1–14. doi:10.1016/j.jaad.2008.12.051
11. Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13(8):834–843. doi:10.1016/S1474-4422(14)70063-8
12. Dombi E, Solomon J, Gillespie AJ, et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: relationship to age and body weight. Neurology. 2007;68(9):643–647. doi:10.1212/01.wnl.0000250332.89420.e6
13. Wise JB, Cryer JE, Belasco JB, Jacobs I, Elden L. Management of head and neck plexiform neurofibromas in pediatric patients with neurofibromatosis type 1. Arch Otolaryngol Head Neck Surg. 2005;131(8):712. doi:10.1001/archotol.131.8.712
14. Onal C, Bayindir C. Gliomatosis cerebri with neurofibromatosis: an autopsy-proven case. Child’s Nerv Syst. 1999;15(5):219–221. doi:10.1007/s003810050376
15. Needle MN, Cnaan A, Dattilo J, et al. Prognostic signs in the surgical management of plexiform neurofibroma: the Children’s Hospital of Philadelphia experience, 1974–1994. J Pediatr. 1997;131(5):678–682. doi:10.1016/S0022-3476(97)70092-1
16. Parsons CM, Canter RJ, Khatri VP. Surgical management of neurofibromatosis. Surg Oncol Clin N Am. 2009;18(1):175–196. doi:10.1016/j.soc.2008.08.009
17. Tackie JNO, Schandorf E, Bankah P, Gbadamosi H, Daniels J, Dadzie MA. Optic nerve glioma successfully treated with definitive intensity-modulated radiotherapy in a limited-resource setting: a case report. Ecancer J. 2024;18. doi:10.3332/ecancer.2024.1800
18. Evans DGR, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311–314. doi:10.1136/jmg.39.5.311
19. Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382(15):1430–1442. doi:10.1056/NEJMoa1912735
20. Khalaf TM, Alqadhi AA. Challenges of Selumetinib therapy for neurofibromatosis in a resource-limited setting. Cureus. 2025. doi:10.7759/cureus.81071
21. Grech VS, Lotsaris K, Touma TE, Kefala V, Rallis E. The role of artificial intelligence in identifying NF1 gene variants and improving diagnosis. Genes. 2025;16(5):560. doi:10.3390/genes16050560
22. Rafanan J, Ghani N, Kazemeini S, Nadeem-Tariq A, Shih R, Vida TA. Modernizing neuro-oncology: the impact of imaging, liquid biopsies, and AI on diagnosis and treatment. IJMS. 2025;26(3):917. doi:10.3390/ijms26030917
23. Siegel A, Lockridge R, Struemph KL, et al. Perceived transition readiness among adolescents and young adults with neurofibromatosis type 1 and plexiform neurofibromas: a cross-sectional descriptive study. J Pediatr Psychol. 2024;49(6):383–391. doi:10.1093/jpepsy/jsae006
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.