Back to Archived Journals » Research and Reviews in Parkinsonism » Volume 5
Diagnosis of Parkinson’s disease: progress and future prospects
Received 4 April 2015
Accepted for publication 16 June 2015
Published 29 July 2015 Volume 2015:5 Pages 19—32
DOI https://doi.org/10.2147/JPRLS.S62065
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Abdul Qayyum Rana
Roshni A Patel,1 Jennifer G Goldman2
1Rush Medical College, Rush University, Chicago, IL, USA; 2Section of Parkinson’s Disease and Movement Disorders, Department of Neurological Sciences, Rush University Medical Center, Chicago, IL, USA
Abstract: This review focuses on methods for diagnosing Parkinson’s disease (PD), highlighting clinical, pathological, and biomarker-driven approaches. For each of these diagnostic approaches, the past, present, and future prospects of diagnosing PD will be explored. Our understanding of PD – its pathogenesis and clinical spectrum – is ever changing. PD is now recognized as a complex multisystem disorder with motor and nonmotor features, including those preceding the onset of classic motor features as well as different phenotypes, often associated with different genetic mutations. Theories regarding the pathophysiology of PD and the role of α-synuclein, its deposition outside of the central nervous system, and possible progression or propagation are discussed. It has been almost 200 years since James Parkinson first described this disorder, and since then, our understanding of the disease process has evolved and become more sophisticated. However, the clinical diagnostic criteria used have remained fairly static for decades. Proposals for redefining PD’s diagnostic criteria incorporate clinical, pathological, and biomarker-driven elements, individually and/or collectively, and may need to consider whether individualized risk and personalized profiles could be ascertained.
Keywords: α-synuclein, biomarker, cerebrospinal fluid, imaging, neuropathology
Introduction
With a prevalence of 1.6% among Medicare beneficiaries over the age of 65, Parkinson’s disease (PD) is the second most common neurodegenerative disease in the United States after Alzheimer’s disease (AD).1 The economic burden of PD is projected to be $23 billion annually, and it will likely increase given the country’s rapidly aging population.2 Diagnostic techniques should be improved to identify PD patients, so they can benefit from early pharmacologic and nonpharmacologic treatments for symptomatic improvement at the onset of disease. Future disease-modifying and neuroprotective agents may be targeted toward those persons at risk of developing PD, though such interventions have not yet come to fruition.
The clinical diagnosis of PD has traditionally been made on the basis of clinical symptoms, patient history, and neurological examination. The clinical diagnosis can be confirmed on autopsy by “gold standard” neuropathological findings, though in many cases, autopsies are infrequently done or primarily used for research purposes. This review article presents new thoughts in the field regarding establishing the diagnosis of PD, including the incorporation of its premotor and nonmotor symptoms, genetic forms, and biomarkers with pathological, imaging, and biofluid modalities. Each diagnostic category (ie, clinical, pathologic, and biomarker driven) will be discussed in the context of its past, progress, and future prospects.
Clinical diagnosis of PD
Past
In his seminal 1817 “Essay on a Shaking Palsy”, James Parkinson described six patients with a progressive disease characterized by “involuntary tremulous motion, with lessened muscular power, in parts not in action”.3 Since James Parkinson’s first description of the disease that would subsequently carry his name, the clinical definition of PD has focused primarily on its cardinal motor features of bradykinesia, muscle rigidity, resting tremor, and postural instability. These features, along with supportive and exclusionary criteria, are incorporated in the United Kingdom PD Society Brain Bank criteria, which is one of the most widely used definitions for diagnosing PD in clinical and research settings.4
The accuracy of this clinical definition has been tested against the histopathological “gold standard” of PD. Conducting autopsy studies of 100 patients with clinical diagnosis of idiopathic PD, Hughes et al5 found that 76% of the patients had Lewy bodies in the substantia nigra at postmortem examination. Rajput et al6 reported similar accuracy of clinical diagnosis at 76%. A follow-up study by Hughes et al7 showed an increased diagnostic accuracy of PD among movement disorder specialists, reporting a positive predictive value (PPV) of 98.6% and sensitivity of 91.1%. This improvement in diagnostic accuracy was proposed to result from movement disorder specialists’ use of pattern recognition for diagnoses beyond that inherent in any formal set of diagnostic criteria.7
More recently, a clinicopathological study by Adler et al8 revealed more modest accuracy rates in diagnosing PD. Among medication-responsive patients, the accuracy of clinical diagnosis of PD was only 53%. Increased disease duration (>5 years) raised the diagnostic accuracy to >85%. On the other hand, among untreated or medication-unresponsive patients, accuracy of clinical diagnosis was merely 26%.8 This study, which was based on data from a longitudinal study of aging and neurodegenerative disorders, suggests a need for greater caution in the accuracy of clinical diagnosis of early PD, particularly in those patients who are unresponsive to dopaminergic medication or very early in their disease course. Furthermore, clinical features overlapping with progressive supranuclear palsy (PSP), multisystem atrophy (MSA), AD, and other degenerative conditions, especially early on, suggest that clinical criteria can be insufficient for differentiating PD from atypical parkinsonian and other neurodegenerative diseases and that other diagnostic markers are needed.8 Indeed, there is a broad differential for PD, including the atypical parkinsonian syndromes of MSA, PSP, and corticobasal degeneration (CBD); dementia with Lewy bodies (DLB), vascular parkinsonism, AD with parkinsonism, secondary parkinsonism, essential tremor (ET); and others. Although atypical parkinsonian disorders (MSA, DLB, PSP, and CBD) have unique diagnostic criteria and pathological features (Table 1), they account for the majority of misdiagnosed conditions of PD.9–15 The atypical symptoms may appear later in the parkinsonian disease course, which may partially explain the misdiagnosis. Generally speaking, MSA is characterized by early autonomic dysfunction, whereas PSP typically has early postural instability and falls accompanied by vertical gaze palsy or slowed vertical saccades.13–15 Some PSP patients, however, may have a more parkinsonian phenotype with asymmetry, tremor, and moderate initial response to levodopa.16 CBD is often associated with limb apraxia, myoclonus, frontal lobe signs, and speech, language and cognitive impairments, though some clinical features can also overlap with PSP.12
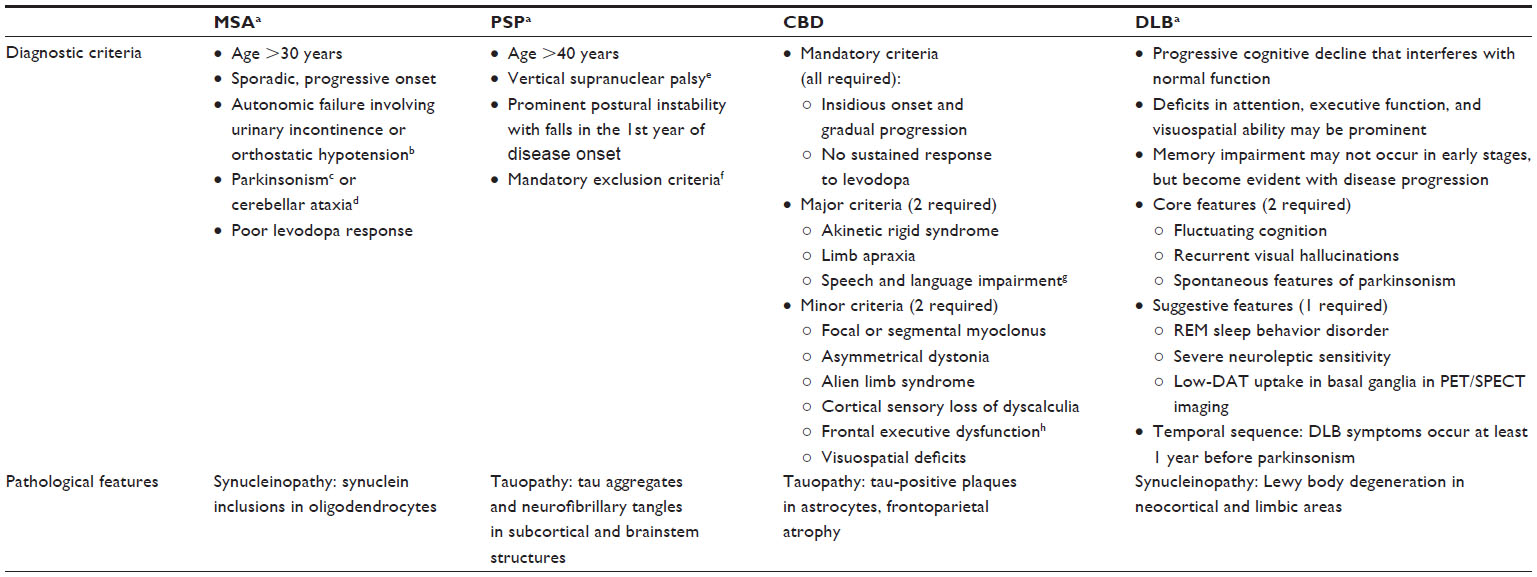 | Table 1 Features of atypical parkinsonian syndromes |
Present
PD is now thought of as a complex multisystem disorder with premotor and nonmotor features, in addition to its classic motor features. Indeed, motor parkinsonism only represents the “tip of the iceberg” of this diverse disease.10 Nonmotor symptoms include autonomic dysfunction, cognitive and behavioral changes, sleep problems, and sensory disturbances (eg, vision problems, pain). These symptoms are now recognized as part of the PD symptom spectrum. Some of these nonmotor symptoms manifest years before the typical motor symptoms, and as such, these premotor features have the potential to serve as early diagnostic markers of PD.17
Common premotor symptoms include hyposmia, constipation, mood changes, and sleep disturbance.17 In a questionnaire filled out by newly diagnosed PD patients in Spain and Austria regarding premotor symptoms, smell loss, taste loss, constipation, dream-enacting behavior, and frequent nightmares were reported significantly more among PD patients compared to controls (odds ratios with 95% confidence interval [95% CI] were 5.4 [2.6–10.9], 11.9 [1.5–93.9], 2.7 [1.4–5.2], 9.9 [3.0–26.4], and 4.1 [1.7–9.5], respectively).18 A large-scale UK study reported similar findings when they assessed premotor symptoms in a primary care database of newly diagnosed PD patients. In addition, they found a high incidence at 5 years before PD diagnosis of premotor hypotension, erectile dysfunction, urinary dysfunction, dizziness, fatigue, depression, and anxiety (relative risk [95% CI] 3.23 [1.85–5.52], 1.3 [1.11–1.51], 1.96 [1.34–2.80], 1.99 [1.67–2.37], 1.56 [1.27–1.91], 1.76 [1.41–2.17], 1.41 [1.09–1.79], respectively), as well as tremor and balance impairments (relative risk [95% CI] 13.7 [7.82–24.31], 2.19 [1.09–4.16], respectively), in PD patients compared to healthy controls. At 10 years prior to PD diagnosis, constipation, along with tremor (relative risk [95% CI] 2.01 [1.62–2.49], 7.59 [1.11–44.83], respectively), was higher in those who subsequently developed PD compared to controls.19 In a meta-analysis that included many early nonmotor features such as constipation and mood disorders and multiple risk factors of PD spanning genetic, environmental, lifestyle, comorbidities, and medication factors, the strongest associations with a later diagnosis of PD were genetic (ie, having a first degree or any relative with PD, or any relative with tremor), followed by constipation, and lack of smoking history; for a later diagnosis of PD, the relative risk of constipation was 2.34 (95% CI, 1.55–3.53) and mood disorders 1.86 (95% CI, 1.64–2.11).20 Some premotor symptoms (eg, hyposmia, constipation, and mood changes) have higher sensitivity for predicting PD development than others, but may have low specificity since they occur frequently in the general population and/or in other neurodegenerative diseases. In contrast, other symptoms (eg, rapid eye movement Behavior Disorder [RBD]) have high specificity, but are not necessarily common. For example, RBD is reported in <1% of newly diagnosed PD patients,19 though another study cites a higher rate of RBD at 25%,21 which may reflect differences in study methodologies and RBD ascertainment. However, among patients with idiopathic RBD, there is a high rate of developing PD or other synucleinopathies, with rates greater than 50%. Two separate prospective cohorts reported that 81% and 82% of RBD subjects developed parkinsonism or neurodegenerative disease at 16- and 14-year follow-up, respectively,22,23 while Postuma et al24 reported a frequent but slightly lower conversion rate at 52% in 12 years.
Combining premotor symptoms with other clinical signs and/or biomarkers can improve the sensitivity and specificity of PD detection. For example, in the aforementioned study, Pont-Sunyer et al18 found that the combination of three premotor symptoms, smell loss, constipation, and dream-enacting behavior, could discriminate between PD and controls with high specificity (97.2%) but sacrificing sensitivity (25.7%). The Parkinson Associated Risk Study (PARS) reported that combining hyposmia with two other risk factors, male sex and constipation, increased the likelihood of having an abnormal dopamine transporter (DAT) scan to over 40%, compared to hyposmia alone at 11%.25 Busse et al26 demonstrated that the combination of three signs, hyposmia, hyperechogenicity on transcranial sonography (TCS), and asymmetric motor signs, had a 49% sensitivity, 98% specificity, and 97% PPV in discriminating PD from other parkinsonian syndromes, including vascular PD, atypical parkinsonism, ET, and major depressive disorder with motor slowing.26 In another study, the combination of major depressive disorder, mild asymmetric motor slowing, hyposmia, and substantia nigra hyperechogenicity on TCS predicted subsequent PD in all their subjects at 10-year follow-up.27
In recent years, PD has been characterized by different genetic forms, which may lead to differences in clinical symptoms, age of onset, course, and thereby PD diagnosis. Genetic forms account for approximately 10% of PD cases. To date, genome-wide association studies have identified 20 PD-related loci, designated as PARK 1–20.28 The clinical and pathological features of each genetic form are unique and may deviate from those of idiopathic PD. For example, the most common autosomal dominant form of parkinsonism is a mutation in the LRRK2 gene (PARK 8).29,30 This mutation has high prevalence in populations with North African and Ashkenazi Jewish ancestry, and in some series, is associated with lower prevalence of hyposmia and cognitive dysfunction.31 Though the motor features are similar to presentations of idiopathic PD, the pathological changes associated with LRRK2 mutation are variable, and differences in synuclein deposition may be seen. The synuclein (SNCA) mutations (PARK 1, PARK 4) lead to α-synuclein misfolding and aggregation within Lewy bodies. These mutations are associated with atypical clinical features including myoclonus, dysautonomia, dementia, and poor levodopa response. The autosomal recessive forms of PD due to parkin (PARK 2), DJ-1 (PARK 7), and PINK1 (PARK 6) gene mutations are associated with early-onset PD.20,21 Autosomal recessive mutations that lead to juvenile onset parkinsonism include ATPase type 13A2 gene (PARK 9) and F-box protein 7 gene (PARK 15).29,30 New genetic mapping tools, such as whole exome sequencing, have led to the identification of additional genes associated with increased risk of PD or parkinsonism. For example, DNAJC6 and SYNJ1 were recently identified as rare autosomal recessive genes associated with juvenile parkinsonism in some family cohorts. Both genes encode proteins involved in postendocytic recycling of synaptic vesicles.32 Unique molecular markers have also been identified in PD cohorts, such as decreased gene expression of Nurr1, a transcription factor essential for development, survival and maintenance of nigral dopaminergic neurons, in PD patients compared to patients with non-PD neurological diseases and healthy controls.33
Future
The future of PD may bring forth new clinical definitions. The Movement Disorder Society (MDS) created a task force to update the definition of PD, and this group recently published an article challenging some of the traditional notions about PD and introducing new definitions and criteria. The MDS task force recommended that the “gold standard” of PD diagnosis remain a clinical motor syndrome accompanied by pathological evidence of substantia nigra neurodegeneration and synuclein deposition. However, they also suggested creating a “clinicogenetic” category of PD diagnosis that takes into account genetic mutations that result in PD symptoms, regardless of synuclein pathology. In addition, they recommended incorporation of the preclinical/prodromal stage into the definition of PD, elimination of the “1-year” rule that separates PD dementia and DLB, and further delineation of atypical syndromes (eg, MSA, PSP, CBD).34
Though the MDS task force paper provides only an introductory statement (the formal definition statement/diagnostic criteria is yet to be published), it has already incited some controversy. In an online feedback forum setup by the MDS task force, excerpted comments include that it may be futile to reclassify PD until robust, noninvasive biomarkers (eg, either advanced imaging or biochemical markers) can identify α-synuclein; urges that the new criteria need not stray far from the traditional one; requests to include unilateral onset and persistent asymmetry (from the UK Parkinson’s Disease Society Brain Bank criteria); and that if premotor symptoms are indeed included in the new criteria, they must be considered as second-line manifestations, with the more typical motor symptoms remaining first line.35
Regardless of whether premotor and/or nonmotor symptoms are incorporated into new diagnostic criteria for PD, the increased awareness and treatment of these symptoms will likely have positive effects on quality of life and symptom management for PD patients. Perhaps the greatest diagnostic challenge lies in combining premotor symptoms with other robust, reliable, and potentially noninvasive biomarkers to increase sensitivity and specificity of predicting PD development. These techniques could be used to make preclinical diagnoses (before the onset of classical motor symptoms) or identify at-risk populations who might be candidates for research studies and interventions for disease modification or neuroprotection.
Pathological diagnosis of PD
Past
The pathological hallmark of PD is neuronal loss and Lewy bodies in the substantia nigra. This definition evolved from 1912, when German pathologist Fredrick Lewy described the characteristic neuronal eosinophilic cytoplasmic concentric inclusion bodies in the brainstem, and from 1919, when Russian pathologist Konstantin Tretiakoff noted the presence of such inclusion bodies in substantia nigra neurons of parkinsonian patients and named them “corps de Lewy” in honor of Fredrick Lewy.36 Though it was suspected that Lewy body formation may cause neurodegeneration in PD, the mechanism was unknown. Initially ubiquitin and neurofilaments were implicated,37 but eventually α-synuclein staining was identified in histopathological studies of patients diagnosed with PD and DLB.38 α-Synuclein is a protein normally abundant in presynaptic terminals of the brain. Though its exact function is unknown, it is thought to be involved with vesicle trafficking given its interactions with microtubules and phospholipids. Usually it adopts a helical structure, but when misfolded, α-synuclein forms β-sheets that polymerize to form toxic oligomers and fibrils. Accumulation of misfolded α-synuclein is thought to contribute to dysfunction and death of neurons in the substantia nigra of PD patients.39
Present
Pathological stages of PD have been proposed by Braak et al,40,41 with a predictable topography of Lewy body deposition and a sequence that progresses with disease severity, seeming to spread along the nervous system. Specifically, α-synuclein pathology has been proposed to start in the peripheral nervous system (dorsal motor nucleus of cranial nerves [CN] 9 and 10 and the olfactory bulb), followed by deposition and ascent through the brainstem, and eventually to the cortical areas. Braak et al40–42 suggest that this pattern occurs because certain “vulnerable” neuronal cell types (specifically, those with long, thin projections and unmyelinated or poorly myelinated axons) are more susceptible to the formation and effects of synuclein aggregates. Thereby, PD affects such cells in a predictable sequence, moving from cell-to-cell via a consecutive series of projection neurons.40–42
PD-associated pathology has been described outside of the central nervous system, such as in the olfactory bulb and gastrointestinal (GI) system. The GI system is a region of particular interest since Lewy-related pathology and α-synuclein staining have been found in the gastric mucosa and enteric nervous system of PD patients. Braak et al42 identified α-synuclein in the submucosal (Meissner’s) plexus on autopsy of 5 PD or incidental Lewy body disease (iLBD) patients, but in contrast, did not find any in five control subjects. Beach et al43 identified synuclein pathology at autopsy in the GI tract of 16/26 (62%) patients with PD or DLB, but only in 2/26 (8%) patients with AD or iLBD. Studies examining in vivo measures of synuclein in the gut (ie, with colonic biopsies) have detected Lewy pathology in 21/29 (72%) and 9/10 (90%) of PD patients in two separate studies, but none in the control subjects.44,45 Interestingly, Lebouvier et al44 also noted a higher frequency of constipation reported in the Lewy neurite-positive patients, suggesting a potential link between the GI synuclein pathology and constipation, a potential premotor symptom in PD patients. Shannon et al46 have investigated a predictive role of gut synuclein pathology in PD and found synuclein-staining deposits in colonic tissue biopsies in three patients that were taken 2–5 years before their clinical diagnosis of PD. Other studies, however, have detected α-synuclein staining in the colonic mucosa of healthy controls as well as in PD patients.47 Differences in study results may be due to differing methodologies, such as depth of biopsy (full versus partial thickness), tissue source (use of autopsied versus biopsied samples), frequency of sampling, and longitudinal follow-up of healthy controls.
Regarding the olfactory system, α-synuclein pathology has been identified in mitral and tufted cells in the anterior olfactory nucleus of the olfactory bulb, which receive projections from primary olfactory epithelial neurons.40,48,49 Of note, the anterior olfactory nucleus has numerous connections with tertiary olfactory regions as well as nonolfactory structures, including the amygdala, substantia nigra, and other brainstem nuclei.50 In autopsy studies, α-synuclein staining in the olfactory bulb was found to effectively distinguish Lewy body disorders from healthy controls. Sensitivities and specificities, respectively, were 91% and 95% for PD versus controls, 97% and 91% for DLB versus controls, and 67% and 91% for iLBD versus controls.51 Based on these results, Beach et al51 propose olfactory bulb biopsy be considered to confirm PD diagnosis, though further research regarding this is needed.
Synuclein pathology also has been detected in the peripheral nerves in the skin, heart, and oropharynx. α-Synuclein pathology was identified in unmyelinated dermis fibers from skin biopsies of the abdominal wall and upper arm flexors in 20/85 (24%) autopsied subjects, with concurrent central nervous system Lewy body pathology, whereas no staining was found in 194 control subjects.52 Donadio et al53 distinguished idiopathic PD from other causes of parkinsonism (including vascular parkinsonism, tauopathy, and parkin mutation) by examining peripheral nerves obtained from cervical skin biopsies, finding phosphorylated α-synuclein deposits in 21/21 (100%) idiopathic PD subjects, but none in the 20 subjects with parkinsonism or 30 controls. In cardiac tissue studies, α-synuclein aggregates have been found in the distal axons of cardiac sympathetic nerves in 18/20 (90%) of subjects with idiopathic Lewy body disease, 6/10 (60%) of PD subjects, 3/20 (15%) of MSA subjects, but none of the controls.54 Cardiac synuclein pathology may underlie the autonomic symptoms that many PD and MSA patients experience. In addition, Mu et al55 observed synuclein positivity in pharyngeal sensory nerves (including CN 9, pharyngeal sensory branch of CN 10, and internal superior laryngeal nerve innervating the laryngopharynx) in all 10 neuropathologically confirmed PD patients, but none of the control subjects. Del Tredici et al56 described Lewy-related pathology in postmortem samples of submandibular glands and cervical superior ganglia in all nine PD cases, compared to none in controls. Cervical sympathetic trunk and peripheral vagus nerve inspection similarly revealed synuclein pathology among the PD subjects.56 Adler et al57 assessed in vivo Lewy-related pathology in submandibular gland biopsies in living PD patients, finding that 9/12 (75%) were positive for α-synuclein staining as well as that submandibular needle core biopsies are feasible to perform in PD patients.57
Taken together, these pathological studies suggest that peripheral tissue may have potential as a diagnostic, and in vivo, biomarker for PD. In their meta-analysis, Malek et al58 reviewed the sensitivities and specificities associated with different tissues in regards to detecting α-synuclein pathology: skin (19%, 80%), colonic mucosa (42%–90%, 100%), and submandibular salivary glands (100%, 100%) for sensitivity and specificity, respectively. Of the tissues discussed, some are more easily accessible (eg, colonic mucosa, submandibular gland, skin) compared to others (eg, cardiac tissue). Longitudinal studies are also needed to determine if tissue biopsies could serve as preclinical biomarkers of PD. Additionally, more in vivo studies are needed to ascertain the feasibility of obtaining tissue biopsies in PD patients at differing disease stages and in potentially at-risk PD populations. Furthermore, studies will need to examine how these tissue markers correlate with other biofluid or imaging measures of synuclein pathology and with clinical symptomatology. Nonetheless, given the high specificity for identifying α-synuclein pathology in peripheral nerve fibers and other target tissues, biopsies may serve as useful premortem diagnostic tests for PD.
Future
Many interesting theories are forming about the etiology and spread of PD. The “dual hit hypothesis” developed from Braak’s pathological staging studies and the descriptions of Lewy-related pathology in the peripheral nervous system, including the olfactory bulb and GI tract. Thus, it has been posited that a pathogen could enter the brain via two routes: nasal and gastric. For example, a pathogen could penetrate the GI epithelial lining and travel through Meissner’s plexus of the gut, vagus nerve, and ultimately reach the midbrain substantia nigra, at which point, typical motor symptoms of PD are manifested.59
Recent theories propose that PD is a prion-like disorder, with α-synuclein, specifically, acting like a prion protein such that the misfolded protein forms pathologic aggregates, which act as a template to promote conformational change in wild-type protein in a chain-reaction manner. The abnormal protein is transmitted from “infected” to healthy, unaffected nerve cells, thereby leading to neurodegeneration.39,60 This theory is supported by postmortem studies of PD patients who received grafts of fetal mesencephalic brain tissue 10–22 years prior to autopsy. The grafted neurons stained positive for α-synuclein and had reduced staining for DAT and tyrosine hydroxylase. These studies suggest that the misfolded α-synuclein formed β-sheets, which migrated from affected cells to unaffected graft neurons.61–63 In a recent study by Recasens et al,64 purified human nigral Lewy body-enriched extracts were injected into the nigrostriatal system of mice and two macaque monkeys. They observed time-dependent degeneration of dopaminergic fibers and nigral neurons in both the mice and monkeys during the 4–17 months postinoculation period, thereby demonstrating that pathological transport of Lewy body extracts can induce nigrostriatal degeneration in nonhuman primates.64,65 Many in vivo and in vitro laboratory studies suggest cell-to-cell transfer of α-synuclein pathology. Though the exact mechanism is unknown, it is possible that α-synuclein travels along long unmyelinated axons and acts as a “seed” that induces endogenous α-synuclein production in neighboring neurons.60,66
Both the dual hit and prion hypotheses operate under the premise that α-synuclein aggregation initially occurs in the periphery, before reaching the brainstem and cortex. If this holds true, then detection of PD pathology in peripheral tissues could serve as an important preclinical biomarker or diagnostic confirmatory test. Furthermore, if the exact mechanism of α-synuclein spread is elucidated, then targeted therapies could be developed to inhibit its spread and subsequent neurodegenerative processes.
Biomarker-driven diagnosis of PD
Past
Historically, biomarker studies, such as biofluids or neuroimaging, had limited use as diagnostic tools for PD. For example, neuroimaging of PD patients was typically characterized by “negative” or “normal” results. That is, magnetic resonance imaging (MRI) and computed tomography (CT) scans of the brain generally revealed normal age-related changes, but no structural abnormalities that were distinctive to PD. Thus, neuroimaging was primarily utilized to exclude secondary causes of parkinsonian symptoms such as vascular, neoplastic, infectious, or toxic/metabolic etiologies and thus, had minimal diagnostic relevance for PD.67,68 The field of biomarker studies has grown tremendously in recent years, with markers of diagnosis, progression, and prognosis in AD, oncology, and other disciplines, with carry over to investigations in PD.
Present
Advances in neuroimaging modalities and biofluid assays have led to a growing interest in their potential for diagnosis, monitoring progression, and establishing prognosis in PD. Here, several examples of biomarker studies focused on PD diagnosis and early disease detection will be highlighted. Among various neuroimaging modalities and techniques are positron emission tomography (PET), single photon emission computed tomography (SPECT), TCS, diffusion tensor imaging (DTI), and MR spectroscopy (MRS).68 In addition, blood and cerebrospinal fluid (CSF) assays measure biochemical markers that reflect disrupted dopamine metabolism, misfolded protein aggregation, and oxidative stress.69
Radioligands used in PET and SPECT studies for PD aim to capture dopaminergic function, such as presynaptic dopamine function, by binding DAT or with VMAT, AAAD, postsynaptic dopamine function (eg, dopamine receptor), or nondopaminergic systems (eg, serotonergic, cholinergic, noradrenergic, and inflammation/glial activation markers).68,70 SPECT imaging has been proposed as a highly sensitive indicator of early PD.70 [123I]-β-CIT SPECT imaging had 92% sensitivity and 100% specificity in diagnosing parkinsonian syndromes when compared to the gold standard of clinical diagnosis by a movement disorder specialist.71 SPECT may have a role in ruling out PD in clinically uncertain cases.71,72 In several studies, 123I-ioflupane [123I]FP-CIT has differentiated PD from ET, with high sensitivity and specificity.73 In 2011, the United States Food and Drug Administration (FDA) approved DAT SPECT imaging (DaTSCAN) using the [123I]FP-CIT ligand for use in suspected parkinsonian syndromes, based on two multicenter, Phase III studies.72,74 In early parkinsonian patients with or without tremor (designated possible and probable PD), compared to patients with non-PD tremor and healthy controls, the DaTSCANs had 79% sensitivity and 97% specificity, while clinical diagnosis in early PD had 98% sensitivity but 67% specificity. Among patients with the clinical diagnosis of PD, DAT scan identified striatal neurodegeneration with sensitivity of 88% and specificity of 91%; additionally, a striatal anterior posterior ratio >2 differentiated between clinically idiopathic PD and atypical parkinsonism syndromes, but with 63% sensitivity and 84% specificity.75 Moreover, since the sensitivity, specificity, and PPV of DAT imaging are similar to that of clinical assessment, the utility of DAT imaging for diagnostic purposes, especially among parkinsonian syndromes, has been questioned.76
Another puzzling aspect of DAT imaging is the SWEDD phenomenon (scans without evidence of dopaminergic deficit), found in patients with clinically apparent symptoms of PD but normal DAT scans. A study of subjects with uncertain parkinsonism had a 14% rate of SWEDDs. Of the 12 SWEDDs, 7 had a revised diagnosis by 3–6 months follow-up and the remaining 5 were younger than the PD patients with positive imaging (average age: 53.4 years versus 62.1 years).77 A 22-month longitudinal follow-up study found that SWEDD subjects had minimal change in DAT imaging and minimal progression of motor symptoms.78 A 5-year follow-up of SWEDD patients with dystonic tremor showed similar results; of the 16 subjects, 12.5% (2/16) had reduced nigrostriatal uptake on DAT scan at follow-up, whereas the remaining (87.5%) continued to have normal dopaminergic imaging and no change in clinical progression.79 Further studies and longitudinal follow-up of the SWEDD patients are needed to understand this phenomenon.
Combining the presence of hyposmia and positive DAT imaging may be helpful in predicting PD development. Sommer et al80 found that of 30 patients with hyposmia, 11 had substantia nigral hyperechogenicity and 7 of these 11 also had decreased DAT binding or borderline uptake ratios on SPECT scans. In PARS, 11% of hyposmic patients had abnormal DAT scans.25 Longitudinal studies show that hyposmic patients with abnormal DAT imaging have increased risk of developing PD. In a cohort of first-degree relatives of PD patients, the combination of hyposmia and abnormal DAT SPECT imaging at baseline was associated with 100% risk of PD development in 5 years, whereas hyposmia alone conferred a risk of only 12.5%.81 Borghammer et al82 similarly showed that the combination of positive dopamine SPECT scans and hyposmia predicted PD development with 91%–97% certainty at 18–42-month follow-up.
Transcranial ultrasound (TCS) permits the visualization of the echogenecity of the substantia nigra at the level of the mesencephalic brainstem. In PD, TCS demonstrates characteristic substantia nigral hyperechogenicity (SN+). Advantages to TCS include short test duration, ability to use in patients with tremor or involuntary movement, lack of radiation, ability to perform at bedside, and low cost, though drawbacks include dependence on user experience, obtaining an adequate bone window, lack of well-established cutoff for hyper- or hypoechogenecity, and a fairly high rate of false positives.67 TCS differentiates PD patients from healthy controls, with sensitivity of 95% and PPV of 85.7%, though having high sensitivity but low specificity when distinguishing PD from other neurodegenerative conditions.83,84 Laučkaitė et al85 noted that TCS was better at distinguishing PD from ET, but poor at distinguishing PD from atypical parkinsonian syndromes. Kim et al86 found a higher degree of SN+ in ET cases with premotor symptoms and PD risk factors (eg, constipation, RBD-like symptoms, family history) compared to ET subjects without such features; longitudinal studies will be needed to determine whether this ET group has increased risk of developing PD. Gaenslen et al87 followed 60 patients with new parkinsonian symptoms for 12 months and determined that baseline SN+ on TCS was able to predict the PD diagnosis with 90.7% sensitivity, 82.4% specificity, and 92.9% PPV.
Other imaging modalities that show diagnostic potential for PD include MR techniques (eg, DTI, MRS) and myocardial scintigraphy (MIGB). MRS measures the concentration of brain tissue metabolites as markers of neuron integrity, while DTI measures diffusion of water molecules through different tissues based on directionality, or anisotropy. Using MRS, Emir et al88 reported greater γ aminobutyric acid (GABA) elevation in the pons than putamen as consistent with the involvement of the lower brainstem structures and basal ganglia and a rostrocaudal progression of the disease as suggested by Braak et al.40,41 Using high-resolution DTI in early, untreated PD patients, Vaillancourt et al89 reported 100% sensitivity and specificity in distinguishing PD patients from healthy controls, with decreased fractional anisotropy in the substantia nigra in all 14 newly diagnosed PD cases. Originally developed to assess cardiac postganglionic presynaptic sympathetic nerve endings in heart failure and cardiomyopathy patients, MIGB scintigraphy demonstrates peripheral noradrenergic depletion. Reduced cardiac MIGB uptake has been demonstrated in patients with Lewy body disease, perhaps also reflecting their dysautonomia. In a meta-analysis of 13 studies, pooled data revealed that MIGB scanning differentiated PD from atypical parkinsonian disorders (including MSA, PSP, CBD), with >80% sensitivity and specificity.90
There are numerous potential biomarkers that may reflect disrupted protein misfolding, mitochondrial dysfunction, oxidative stress, inflammation, and altered gene expression in PD. α-Synuclein stands out given the deposition of this misfolded protein is a pathological hallmark of PD and a core component of Lewy bodies.69 Total α-synuclein levels have been decreased in the CSF of PD patients. A meta-analysis of nine studies showed that CSF α-synuclein levels were lower in patients with synucleinopathies (PD and MSA) compared to healthy controls and PSP subjects,91 though further study is needed. While blood is more easily obtained than CSF, serum α-synuclein detection has been inconsistent across studies. Differences may be due to variation in study methodologies, such as differing assays or type of α-synuclein protein detection (eg, total versus oligomeric, phosphorylated versus nonphosphorylated).69 Despite difficulties with using serum α-synuclein as a diagnostic marker of PD, it may have a role in monitoring disease progression. In a longitudinal study of newly diagnosed PD patients followed for 3–4 years, total plasma α-synuclein was found to increase over time in PD patients during the study period.92
Another potential biomarker that has been studied is urate, an antioxidant which has been investigated regarding a protective role against oxidative stress in PD. Two recent studies have demonstrated that elevated serum urate levels correlate with decreased likelihood of abnormal DAT imaging in newly diagnosed, medication-naïve PD or parkinsonian patients,93,94 though in one study, this association was only significant in male subjects.94 Additionally, high serum and CSF urate were associated with slower rates of clinical decline in PD.95 Moreover, lower urate concentrations may reflect worse nonmotor function as they were related to higher severity scores in attention, memory, cardiovascular, and sleep domains.96 However, serum urate levels did not affect risk of dementia.97 Genetic variability may account for differing serum urate concentrations in PD patients. Of note, single nucleotide polymorphisms (SNPs) in urate transporter gene SLC2A9 (GLUT9) were found to predict differences in serum urate among patients with early PD. Subjects who had SLC2A9 SNPs associated with lower serum urate had faster progression to disability, requiring dopaminergic treatment at 13-month follow-up.98
Neuroinflammatory markers have been noted in biofluids of PD patients.99 Numerous cytokines and chemokines have been found to be elevated in serum or CSF of PD subjects compared to non-PD controls, including interleukin (IL)-1β, IL-6, IL-8, IL-10, monocyte chemoattractant protein-1, macrophage inflammatory protein-1α, tumor necrosis factor-α, transforming growth factor-α, and RANTES.99–102 Interestingly, the degree of inflammatory marker elevation in CSF correlated with severity of nonmotor symptoms in PD patients, including depression, anxiety, fatigue, and cognitive impairment.103 The presence of such common inflammatory molecules in CSF or blood, however, may not be a specific marker for PD, given that elevations can occur in a variety of disease states, including infection, acute injury/trauma, malignancy, autoimmune disease, and other non-PD inflammatory conditions. Certain markers may show greater promise in diagnosing PD, as a recent study by Shi et al104 found that elevated Flt3 ligand (an inflammatory marker) in CSF differentiated PD from MSA with a sensitivity of 99% and specificity of 95%.
PD-associated gene products may also serve as biomarkers. DJ-1 gene mutation (PARK 7 locus) is a rare autosomal recessive form of PD, and the resulting DJ-1 protein is detectable in blood and CSF.69 Hong et al105 found significantly decreased DJ-1 levels in CSF of PD patients compared to healthy controls, distinguishing the PD patients with 90% sensitivity and 70% specificity. Also with CSF DJ-1, Herbert et al106 distinguished PD from MSA with 78% sensitivity and 78% specificity. However, as with serum and CSF α-synuclein, DJ-1 protein detection in blood has proven somewhat complicated, and CSF measurements may be more compelling at this time, though additional study is needed.69
Future
Several areas pertaining to PD biomarkers require further inquiry. Additional longitudinal studies are needed to determine which biomarker techniques will be most useful in the earlier detection of PD and for identifying preclinical, at-risk populations. Potential diagnostic imaging or biofluid assays may be compared to the gold standard of neuropathology, rather than solely clinical assessment, since in some settings, this diagnostic accuracy may be low.8 Biomarker study methodologies must be fine-tuned and standardized, especially relevant for biofluid assays and neuroimaging protocols, as methodological differences may account for inconsistent results. Lastly, novel biomarkers and analytic methods must be identified. Imaging methods to detect α-synuclein in the brain are in development.107 Multimodal datasets, such as with different types of neuroimaging, in combination with biofluid measurements, among other data, may advance our understanding of PD and, through various models, our ability to detect PD early and distinguish PD patients (or at-risk persons) from healthy controls, atypical parkinsonian syndromes, or other neurodegenerative conditions. Multiple consortia have developed as resources for biomarker research in PD, including Parkinson’s Progression Markers Initiative (PPMI), Fox Investigation for New Discovery of Biomarkers in Parkinson’s disease (BioFIND), and Parkinson’s Disease Biomarker Program (PDBP) among others, which are multicenter, international, shared databases with clinical, imaging, and biologic information made available to the larger scientific community.108–110 Such collaborative efforts will aid in identifying and validating markers of PD not only for diagnosis, but also for progression and prognostication.
Conclusion
Our understanding of PD – its pathogenesis and clinical spectrum – has evolved in the almost 200 years since James Parkinson first described this disorder. PD is now recognized as a complex multisystem disorder with multiple potential etiologies, including environmental triggers, genetic predisposition, and other risk factors. Moreover, PD is now recognized pathologically to have a topographical spread and potentially cellular spread as postulated in the prion-like hypotheses. As such, the progress and future prospects now take these factors into account (Table 2). These prospects provide avenues to expand our thinking regarding the many facets of PD and potentially redefine the PD syndrome and its diagnostic criteria, not only utilizing clinical, pathological, and biomarker-driven elements, but also developing models or algorithms that combine these features in order to diagnose PD at its very earliest stage and calculate individualized risk and personalized profiles. One could envision a diagnostic schema for PD in some ways similar to the revised criteria for a diagnosis of dementia due to AD by McKhann et al111 on behalf of the National Institute on Aging and the Alzheimer’s Association workgroup, while also capturing elements of the MDS Task Force introductory statement on redefining PD. These revised criteria provide diagnostic categories with “probable, possible and unlikely due to AD” categories, each with required clinical criteria and with varying degrees of biomarker probability (eg, uninformative, low, intermediate, or high) or biomarker availability (eg, unavailable, indeterminate, negative, or positive) for evidence of β-amyloid deposition on PET or decrease in CSF and of neuronal injury such as with elevated CSF tau, hippocampal atrophy, or positive FDG-PET scans.111 For PD, one such hypothesized schema could include several diagnostic levels or categories such as a purely clinical diagnosis without regard to biomarkers or underlying pathology; a clinical diagnosis with support of biomarkers (positive genetics, blood, CSF, tissue biopsy, or neuroimaging findings) that could be ranked by probability, availability, and combined effect to a similar degree as in the AD criteria; and a pathologically confirmed diagnosis. Furthermore, this schema could be extended to a category of premotor PD symptoms and how they fit in preclinical or prodromal subtypes: a pure clinical symptomatology or as a clinical diagnosis of premotor PD with supportive biomarker evidence. If biomarkers are to be included, research will be needed to standardize assays and analyses; determine appropriate cutoff values for quantitative variables; interpret qualitative and quantitative biomarker data; elucidate how they relate to clinical symptoms, underlying pathology, and disease progression; and identify ways to practically incorporate biomarkers into clinical practice and research settings in convenient, accessible, and affordable ways. In addition, future studies will be necessary to determine the best ways to incorporate clinical features, pathological findings, and biomarkers, individually and collectively, into PD diagnostic criteria and to capture the clinical breadth of the PD spectrum with not only premotor and motor symptoms but also PD’s different motor and nonmotor phenotypes.
 | Table 2 Factors to be considered in PD diagnosis present and future |
Disclosure
The authors report no conflicts of interest in this work.
References
Wright-Willis A, Evanoff BA, Lian M, Criswell SR, Racette BA. Geographic and ethnic variation in Parkinson disease: a population-based study of US Medicare beneficiaries. Neuroepidemiology. 2010;34(3):143–151. | |
Huse DM, Schulman K, Orsini L, Castelli-haley J, Kennedy S, Lenhart G. Burden of illness in Parkinson’s disease. Mov Disord. 2005;20(11):1449–1454. | |
Parkinson J. An essay on the shaking palsy. London Med Phys J. 1817:38. | |
Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatr. 1988;51(6):745–752. | |
Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatr. 1992;55(3):181–184. | |
Rajput AH, Rozdilsky B, Rajput A. Accuracy of clinical diagnosis in parkinsonism – a prospective study. Can J Neurol Sci. 1991;18(3):275–278. | |
Hughes AJ, Daniel SE, Ben-shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125(Pt 4):861–870. | |
Adler CH, Beach TG, Hentz JG, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology. 2014;83(5):406–412. | |
Ali K, Morris HR. Parkinson’s disease: chameleons and mimics. Pract Neurol. 2015;15(1):14–25. | |
Langston JW. The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann Neurol. 2006;59(4):591–596. | |
Mckeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. | |
Mathew R, Bak TH, Hodges JR. Diagnostic criteria for corticobasal syndrome: a comparative study. J Neurol Neurosurg Psychiatr. 2012; 83(4):405–410. | |
Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670–676. | |
Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9. | |
Wenning GK, Colosimo C. Diagnostic criteria for multiple system atrophy and progressive supranuclear palsy. Rev Neurol (Paris). 2010; 166(10):839–833. | |
Williams DR, de Silva R, Pavior DC, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain. 2005;128(Pt 6):1247–1258. | |
Goldman JG, Postuma R. Premotor and nonmotor features of Parkinson’s disease. Curr Opin Neurol. 2014;27(4):434–441. | |
Pont-sunyer C, Hotter A, Gaig C, et al. The onset of nonmotor symptoms in Parkinson’s disease (the ONSET PD study). Mov Disord. 2015;30(2):229–237. | |
Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 2015;14(1):57–64. | |
Noyce AJ, Bestwick JP, Silveira-Moriyama L, et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol. 2012;72(6):893–901. | |
Sixel-döring F, Trautmann E, Mollenhauer B, Trenkwalder C. Rapid eye movement sleep behavioral events: a new marker for neurodegeneration in early Parkinson disease? Sleep. 2014;37(3):431–438. | |
Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med. 2013;14(8):744–748. | |
Iranzo A, Tolosa E, Gelpi E, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol. 2013;12(5):443–453. | |
Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. 2009;72(15):1296–1300. | |
Jennings D, Siderowf A, Stern M, et al. Imaging prodromal Parkinson disease: the Parkinson Associated Risk Syndrome Study. Neurology. 2014;83(19):1739–1746. | |
Busse K, Heilmann R, Kleinschmidt S, et al. Value of combined midbrain sonography, olfactory and motor function assessment in the differential diagnosis of early Parkinson’s disease. J Neurol Neurosurg Psychiatr. 2012;83(4):441–447. | |
Walter U, Heilmann R, Kaulitz L, et al. Prediction of Parkinson’s disease subsequent to severe depression: a ten-year follow-up study. J Neural Transm. Epub September 14, 2014. | |
Brás J, Guerreiro R, Hardy J. SnapShot: genetics of Parkinson’s disease. Cell. 2015;160(3):570–570. e1. | |
Thenganatt MA, Jankovic J. Parkinson disease subtypes. JAMA Neurol. 2014;71(4):499–504. | |
Spatola M, Wider C. Genetics of Parkinson’s disease: the yield. Parkinsonism Relat Disord. 2014;20(Suppl 1):S35–S38. | |
Gaig C, Vilas D, Infante J, et al. Nonmotor symptoms in LRRK2 G2019S associated Parkinson’s disease. PLoS One. 2014;9(10):e108982. | |
Bonifati V. Genetics of Parkinson’s disease – state of the art, 2013. Parkinsonism Relat Disord. 2014;20(Suppl 1):S23–S28. | |
Le W, Pan T, Huang M, et al. Decreased NURR1 gene expression in patients with Parkinson’s disease. J Neurol Sci. 2008;273(1–2):29–33. | |
Berg D, Postuma RB, Bloem B, et al. Time to redefine PD? Introductory statement of the MDS task force on the definition of Parkinson’s disease. Mov Disord. 2014;29(4):454–462. | |
Berg D, Postuma R. Task Force Paper: Redefinition of Parkinson’s Disease. Milwaukee, WI: MDS International Secretariat; 2014. Available from: http://www.movementdisorders.org/MDS/News/Online-Web-Edition/In-This-Edition/Definition-of-PD-Overview.htm. Accessed May 30, 2015. | |
Holdorff B, Rodrigues e silva AM, Dodel R. Centenary of Lewy bodies (1912–2012). J Neural Transm. 2013;120(4):509–516. | |
Lennox G, Lowe J, Landon M, Byrne EJ, Mayer RJ, Godwin-austen RB. Diffuse Lewy body disease: correlative neuropathology using anti-ubiquitin immunocytochemistry. J Neurol Neurosurg Psychiatr. 1989;52(11):1236–1247. | |
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. | |
Olanow CW, Brundin P. Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord. 2013;28(1):31–40. | |
Braak H, Del tredici K, Rüb U, De vos RA, Jansen steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. | |
Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318(1):121–134. | |
Braak H, De vos RA, Bohl J, Del tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396(1):67–72. | |
Beach TG, Adler CH, Sue LI, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119(6):689–702. | |
Lebouvier T, Neunlist M, Bruley des varannes S, et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS One. 2010;5(9):e12728. | |
Shannon KM, Keshavarzian A, Mutlu E, et al. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord. 2012;27(6):709–715. | |
Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord. 2012;27(6):716–719. | |
Visanji NP, Marras C, Kern DS, et al. Colonic mucosal a-synuclein lacks specificity as a biomarker for Parkinson disease. Neurology. 2015;84(6):609–616. | |
Sengoku R, Saito Y, Ikemura M, et al. Incidence and extent of Lewy body-related alpha-synucleinopathy in aging human olfactory bulb. J Neuropathol Exp Neurol. 2008;67(11):1072–1083. | |
Hubbard PS, Esiri MM, Reading M, Mcshane R, Nagy Z. Alpha-synuclein pathology in the olfactory pathways of dementia patients. J Anat. 2007;211(1):117–124. | |
Ubeda-bañon I, Saiz-sanchez D, De la rosa-prieto C, Martinez-marcos A. α-Synuclein in the olfactory system in Parkinson’s disease: role of neural connections on spreading pathology. Brain Struct Funct. 2014; 219(5):1513–1526. | |
Beach TG, White CL, Hladik CL, et al. Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2009;117(2):169–174. | |
Ikemura M, Saito Y, Sengoku R, et al. Lewy body pathology involves cutaneous nerves. J Neuropathol Exp Neurol. 2008;67(10):945–953. | |
Donadio V, Incensi A, Leta V, et al. Skin nerve α-synuclein deposits: a biomarker for idiopathic Parkinson disease. Neurology. 2014;82(15):1362–1369. | |
Orimo S, Uchihara T, Nakamura A, et al. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain. 2008;131(Pt 3):642–650. | |
Mu L, Sobotka S, Chen J, et al. Parkinson disease affects peripheral sensory nerves in the pharynx. J Neuropathol Exp Neurol. 2013;72(7):614–623. | |
Del Tredici K, Hawkes CH, Ghebremedhin E, Braak H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol. 2010;119(6):703–713. | |
Adler CH, Dugger BN, Hinni ML, et al. Submandibular gland needle biopsy for the diagnosis of Parkinson disease. Neurology. 2014;82(10):858–864. | |
Malek N, Swallow D, Grosset KA, Anichtchik O, Spillantini M, Grosset DG. Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease – a systematic review. Acta Neurol Scand. 2014;130(2):59–72. | |
Hawkes CH, Del tredici K, Braak H. Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33(6):599–614. | |
George S, Rey NL, Reichenbach N, Steiner JA, Brundin P. α-Synuclein: the long distance runner. Brain Pathol. 2013;23(3):350–357. | |
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14(5):504–506. | |
Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14(5):501–503. | |
Kurowska Z, Englund E, Widner H, Lindvall O, Li JY, Brundin P. Signs of degeneration in 12-22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson’s disease. J Parkinsons Dis. 2011;1(1):83–92. | |
Recasens A, Dehay B, Bové J, et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75(3):351–362. | |
Kordower JH. The prion hypothesis of Parkinson’s disease: this hot topic just got hotter. Mov Disord. 2014;29(8):988. | |
Lamberts JT, Hildebrandt EN, Brundin P. Spreading of α-synuclein in the face of axonal transport deficits in Parkinson’s disease: a speculative synthesis. Neurobiol Dis. Epub July 15, 2014. | |
Berg D, Steinberger JD, Warren olanow C, Naidich TP, Yousry TA. Milestones in magnetic resonance imaging and transcranial sonography of movement disorders. Mov Disord. 2011;26(6):979–992. | |
Brooks DJ, Pavese N. Imaging biomarkers in Parkinson’s disease. Prog Neurobiol. 2011;95(4):614–628. | |
Henchcliffe C. Blood and cerebrospinal fluid markers in Parkinson’s disease: current biomarker findings. Current Biomarker Findings. 2015;5:1–11. | |
Godau J, Hussl A, Lolekha P, Stoessl AJ, Seppi K. Neuroimaging: current role in detecting pre-motor Parkinson’s disease. Mov Disord. 2012;27:634–643. | |
Jennings DL, Seibyl JP, Oakes D, Eberly S, Murphy J, Marek K. (123I) beta-CIT and single-photon emission computed tomographic imaging vs clinical evaluation in Parkinsonian syndrome: unmasking an early diagnosis. Arch Neurol. 2004;61:1224–1229. | |
Marshall VL, Reininger CB, Marquardt M, et al. Parkinson’s disease is overdiagnosed clinically at baseline in diagnostically uncertain cases: a 3-year European multicenter study with repeat [123I]FP-CIT SPECT. Mov Disord. 2009;24:500–508. | |
Cummings JL, Henchcliffe C, Schaier S, Simuni T, Waxman A, Kemp P. The role of dopaminergic imaging in patients with symptoms of dopaminergic system neurodegeneration. Brain. 2011;134:3146–3166. | |
Benamer TS, Patterson J, Grosset DG, et al. Accurate differentiation of parkinsonism and essential tremor using visual assessment of [123I]-FP-CIT SPECT imaging: the [123I]-FP-CIT study group. Mov Disord. 2000;15:503–510. | |
Ziebell M, Andersen BB, Thomsen G, et al. Predictive value of dopamine transporter SPECT imaging with [123I]PE2I in patients with subtle parkinsonian symptoms. Eur J Nucl Med Mol Imaging. 2012;39(2):242–250. | |
De la fuente-fernández R. Role of DaTSCAN and clinical diagnosis in Parkinson disease. Neurology. 2012;78(10):696–701. | |
Sixel-döring F, Liepe K, Mollenhauer B, Trautmann E, Trenkwalder C. The role of 123I-FP-CIT-SPECT in the differential diagnosis of Parkinson and tremor syndromes: a critical assessment of 125 cases. J Neurol. 2011;258(12):2147–2154. | |
Marek K, Seibyl J, Eberly S, et al. Longitudinal follow-up of SWEDD subjects in the PRECEPT Study. Neurology. 2014;82(20):1791–1797. | |
Batla A, Erro R, Stamelou M, et al. Patients with scans without evidence of dopaminergic deficit: a long-term follow-up study. Mov Disord. 2014;29(14):1820–1825. | |
Sommer U, Hummel T, Cormann K, et al. Detection of presymptomatic Parkinson’s disease: combining smell tests, transcranial sonography, and SPECT. Mov Disord. 2004;19(10):1196–1202. | |
Berendse HW, Ponsen MM. Diagnosing premotor Parkinson’s disease using a two-step approach combining olfactory testing and DAT SPECT imaging. Parkinsonism Relat Disord. 2009;15(Suppl 3): S26–S30. | |
Borghammer P, Knudsen K, Østergaard K, et al. Combined DaT imaging and olfactory testing for differentiating parkinsonian disorders. Int J Clin Pract. 2014;68(11):1345–1351. | |
Mehnert S, Reuter I, Schepp K, Maaser P, Stolz E, Kaps M. Transcranial sonography for diagnosis of Parkinson’s disease. BMC Neurol. 2010;10:9. | |
Prestel J, Schweitzer KJ, Hofer A, Gasser T, Berg D. Predictive value of transcranial sonography in the diagnosis of Parkinson’s disease. Mov Disord. 2006;21(10):1763–1765. | |
Laučkaitė K, Rastenytė D, Šurkienė D, et al. Specificity of transcranial sonography in parkinson spectrum disorders in comparison to degenerative cognitive syndromes. BMC Neurol. 2012;12:12. | |
Kim JS, Oh YS, Kim YI, Koo JS, Yang DW, Lee KS. Transcranial sonography (TCS) in Parkinson’s disease (PD) and essential tremor (ET) in relation with putative premotor symptoms of PD. Arch Gerontol Geriatr. 2012;54(3):e436–e439. | |
Gaenslen A, Unmuth B, Godau J, et al. The specificity and sensitivity of transcranial ultrasound in the differential diagnosis of Parkinson’s disease: a prospective blinded study. Lancet Neurol. 2008;7(5):417–424. | |
Emir UE, Tuite PJ, Öz G. Elevated pontine and putamenal GABA levels in mild-moderate Parkinson disease detected by 7 tesla proton MRS. PLoS One. 2012;7(1):e30918. | |
Vaillancourt DE, Spraker MB, Prodoehl J, et al. High-resolution diffusion tensor imaging in the substantia nigra of de novo Parkinson disease. Neurology. 2009;72(16):1378–1384. | |
Orimo S, Suzuki M, Inaba A, Mizusawa H. 123I-MIBG myocardial scintigraphy for differentiating Parkinson’s disease from other neurodegenerative parkinsonism: a systematic review and meta-analysis. Parkinsonism Relat Disord. 2012;18(5):494–500. | |
Sako W, Murakami N, Izumi Y, Kaji R. Reduced alpha-synuclein in cerebrospinal fluid in synucleinopathies: evidence from a meta-analysis. Mov Disord. 2014;29(13):1599–1605. | |
Foulds PG, Diggle P, Mitchell JD, et al. A longitudinal study on α-synuclein in blood plasma as a biomarker for Parkinson’s disease. Sci Rep. 2013;3:2540. | |
Moccia M, Pappatà S, Erro R, et al. Uric acid relates to dopamine transporter availability in Parkinson’s disease. Acta Neurol Scand. 2015;131(2):127–131. | |
Schwarzschild MA, Marek K, Eberly S, et al. Serum urate and probability of dopaminergic deficit in early “Parkinson’s disease”. Mov Disord. 2011;26(10):1864–1868. | |
Ascherio A, Lewitt PA, Xu K, et al. Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch Neurol. 2009;66(12):1460–1468. | |
Moccia M, Picillo M, Erro R, et al. Is serum uric acid related to non-motor symptoms in de-novo Parkinson’s disease patients? Parkinsonism Relat Disord. 2014;20(7):772–775. | |
González-aramburu I, Sánchez-juan P, Sierra M, et al. Serum uric acid and risk of dementia in Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(6):637–639. | |
Simon KC, Eberly S, Gao X, et al. Mendelian randomization of serum urate and parkinson disease progression. Ann Neurol. 2014;76(6):862–868. | |
Chao Y, Wong SC, Tan EK. Evidence of inflammatory system involvement in Parkinson’s disease. Biomed Res Int. 2014;2014:308654. | |
Rentzos M, Nikolaou C, Andreadou E, et al. Circulating interleukin-15 and RANTES chemokine in Parkinson’s disease. Acta Neurol Scand. 2007;116(6):374–379. | |
Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett. 1996;211(1):13–16. | |
Reale M, Iarlori C, Thomas A, et al. Peripheral cytokines profile in Parkinson’s disease. Brain Behav Immun. 2009;23(1):55–63. | |
Lindqvist D, Hall S, Surova Y, et al. Cerebrospinal fluid inflammatory markers in Parkinson’s disease – associations with depression, fatigue, and cognitive impairment. Brain Behav Immun. 2013;33:183–189. | |
Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol. 2011;69(3):570–580. | |
Hong Z, Shi M, Chung KA, et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain. 2010;133(Pt 3):713–726. | |
Herbert MK, Eeftens JM, Aerts MB, et al. CSF levels of DJ-1 and tau distinguish MSA patients from PD patients and controls. Parkinsonism Relat Disord. 2014;20(1):112–115. | |
Eberling JL, Dave KD, Frasier MA. Alpha-synuclein imaging: a critical need for Pakrinson’s disease research. J Parkinsons Dis. 2013;3(4):565–567. | |
Parkinson Progression Marker Initiative. The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol. 2011;95(4):629–635. | |
Kang U, Alcalay R, Goldman J, et al. The BioFIND study (Fox investigation for new discovery of biomarkers in Parkinson’s disease): design and methodology (P4.043). Neurology. 2014;82(10):P4-043. | |
National Institute of Neurological Disorders and Stroke. PDBP: Parkinson’s Disease Biomarkers Program. Bethesda, MD: National Institute of Neurological Disorders and Stroke; 2014. Available from: https://pdbp.ninds.nih.gov/index.jsp. Accessed March 30, 2015. | |
Mckhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.