Back to Journals » Research and Reports in Urology » Volume 10
Diabetic bladder dysfunction is associated with bladder inflammation triggered through hyperglycemia, not polyuria
Authors Inouye BM, Hughes FM Jr ![]() , Jin H
, Jin H ![]() , Lütolf R
, Lütolf R ![]() , Potnis KC, Routh JC
, Potnis KC, Routh JC ![]() , Rouse DC, Foo WC, Purves JT
, Rouse DC, Foo WC, Purves JT ![]()
Received 19 June 2018
Accepted for publication 21 August 2018
Published 16 November 2018 Volume 2018:10 Pages 219—225
DOI https://doi.org/10.2147/RRU.S177633
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jan Colli
Brian M Inouye,1 Francis M Hughes Jr,1,2 Huixia Jin,1 Robin Lütolf,3 Kunal C Potnis,1 Jonathan C Routh,1,4 Douglas C Rouse,5 Wen-Chi Foo,6 J Todd Purves1,2,4
1Department of Surgery, Division of Urology, Duke University Medical Center, Durham, NC, USA; 2Department of Bioengineering, Clemson University, Clemson, SC, USA; 3Department of Health Science and Technology, ETH Zurich, Zürich 8092, Switzerland; 4Department of Pediatrics, Duke University Medical Center, Durham, NC, USA; 5Division of Laboratory Animal Medicine, Duke University Medical Center, Durham, NC, USA; 6Department of Pathology, Duke University Medical Center, Durham, NC, USA
Purpose: Diabetes is a grave and progressive condition characterized by debilitating complications. Diabetic bladder dysfunction (DBD) is a very common complication with no specific treatments currently available. Unlike other tissues affected by this disease, the bladder is subjected to two independent insults; 1) polyuria, created by the osmotic effects of glucose in the urine, and 2) hyperglycemia itself. Based on our understanding of inflammation as a major contributor to the underlying organ damage in several other diabetic complications, its presence in the bladder during DBD and the contribution of polyuria and hyperglycemia to its development were assessed.
Methods: Awake, restrained cystometry was performed on wild type C57BL/6 mice and diabetic (Akita) mice on a C57BL/6 background at 15 weeks of age. A subgroup of the Akita mice were treated with phlorizin, an inhibitor of sodium-glucose linked transporter types 1 and 2 that prevents glucose reabsorption in the kidney. All groups were assessed for serum glucose, 4-hour voiding totals, and inflammation in the bladder (Evans blue assay).
Results: Akita mice develop cystometrically-defined DBD by 15 weeks of age, as evidenced by an increase in urinary frequency, a decrease in voiding volume, and an increase in post-voiding residual volume. Phlorizin effectively normalized serum glucose in these animals while increasing the urine output. Inflammation in the bladder was present in the diabetic animals at this time point, but not detectable in animals receiving phlorizin.
Conclusion: Inflammation in the bladder of diabetic mice correlates with the development of DBD and is triggered by hyperglycemia, not polyuria.
Keywords: diabetes, inflammation, urinary bladder, urodynamics, cystitis, immunity, innate
Introduction
The WHO has estimated that, in 2014, there were 422 million people worldwide living with diabetes.1 Among those afflicted, somewhere between 25% and 90% had diabetic uropathy, a complication more common than neuropathy or nephropathy,2,3 and one that has been recognized since 1935.4 The wide range of this estimate is most likely due to the absence of a validated and standardized method for diagnosis and the selected referral of patients.5,6 Diabetic uropathy consists of diabetic bladder dysfunction (DBD), a more inclusive term than the previously en vogue diabetic cystopathy,7 sexual or erectile dysfunction, and recurrent urinary tract infection. In recent years, DBD has come to be recognized as a progressive set of symptoms, characterized in the early stages by increased frequency, urgency, and even urge incontinence.8 These changes are also characteristic of the irritative symptoms associated with overactive bladder (OAB).6 As diabetes develops, chronic DBD becomes characterized by an insensate, decompensated bladder that results in overflow incontinence and high post-void residual volumes.6,8 However, it must be stated that, clinically, not all patients will progress smoothly through these stages or present at the beginning of their disease.
In the early stages of diabetes, hyperglycemia overwhelms the capacity for renal reabsorption of glucose, leading to glucosuria and osmotic polyuria. Initially the bladder compensates, leading to increased urinary frequency. Contemporaneously in the bladder, as in all insulin-independent tissues, hyperglycemia drives enhanced oxidative phosphorylation that causes significant oxidative stress. Oxidative stress is an imbalance between the production of reactive oxygen species (free radicals) and antioxidant defenses, resulting in excess radicals that react and damage all components of the cell including proteins, lipids, and DNA. Increased oxidative stress has been implicated in a vast number of diseases including heart disease,9 cancer,10 and, critically, diabetes,11 and DBD in particular.6,12 It is this oxidative stress that triggers an inflammatory response in many target tissues.13–15 While for many years there has been general evidence that inflammation plays an important role in the development of several diabetic complications,16,17 there has been little evidence that it is occurring in the bladder. Suggestive indications were provided when Wang and Kuo18 recently found that mast cells in the urothelium and suburothelial layer were increased in diabetic patients. In this study, we have examined the presence of inflammation in the bladder of the Akita diabetic mice at a time where they demonstrate DBD symptoms. To assess the contribution of polyuria and hyperglycemia to the activation of inflammation, we have treated a group of these diabetic mice with phlorizin. Phlorizin is a dihydrochalcone (a bicyclic flavonoid) that functions as a competitive inhibitor of the sodium-glucose linked transporter types 1 and 2 (SGLT1 and SGLT2) in the kidney and gut. Treatment with phlorizin prevents glucose reabsorption and results in increased glucose in the urine.19,20 Thus, phlorizin results in significantly lower blood glucose coupled with an enhanced polyuria (due to osmotic effects of glucosuria) allowing for discrimination between the effects of polyuria vs hyperglycemia on the formation of an inflammatory response. Similar SGLT1 and 2 inhibitors have been developed for human use and are marketed under the trade names Invokana® and Farxiga®,21 and, while phlorizin has been used in many studies for lowering blood glucose,19,22 it has not been used previously to distinguish the contribution of polyuria and hyperglycemia to DBD.19,22
Methods
Animals
The Akita mouse strain contains a spontaneous mutation in the insulin 2 (Ins2) gene, which causes it to fold incorrectly, leading to an inactive protein as well as producing toxicity in pancreatic β cells.23 Mice heterozygous for the Ins2 mutation are viable and fertile and begin developing diabetes around the time of weaning (4 weeks). Akita mice on a C57BL/6 background (C57BL/6-Ins2Akita/J, stock number: 003548), and the wild type C57BL/6J strain (stock number: 000664) were purchased from Jackson labs and bred in the mouse breeding core at Duke University Medical Center. Wild type and heterozygote (ie, diabetic) female mice were provided to the lab upon weening (≈ 4 weeks of age). Mice were housed socially, as required by the Guide for the Care and Use of Laboratory Animals published by the US Public Health Service. Animals were housed 2–5 per cage, depending on how they were transferred from the breeding core. Individual cages were not necessarily composed of one genotype or experimental group or targeted for one endpoint. The exception to social housing were with the animals implanted with a suprapubic tube for urodynamics. These animals were housed individually to prevent them from chewing one another’s tubes. Within 3 days of arrival one group of mice were anesthetized (80 mg/kg ketamine/5 mg/kg xylazine, ip), treated prophylactically with carprofen (5 mg/kg, sc) and enrofloxacin (0.5 mL/kg of a 2.27% solution), and implanted with osmotic mini-pumps (Alzet, Cupertino, CA; cat# 2004; release rate 0.25 µL/h for 4 weeks) containing phlorizin (40% w/v in propylene glycol).24 The effective dose was 120 mg/kg/day. Bupivacaine (0.25%) was applied topically to the sutures after the procedure. Pumps were replaced every 4 weeks. Prior to any surgery mice were given preemptive analgesic (carprofen, 5 mg/kg). All animals were maintained on a normal light:dark cycle (12 hours on, 12 hours off) until 15 weeks of age, when they were used for various studies. Animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at Duke University Medical Center and conformed to the Guide for the Care and Use of Laboratory Animals published by the US Public Health Service.
Cystometry
For cystometry, week 14 mice were anesthetized (80 mg/kg ketamine; 5 mg/kg xylazine), treated prophylactically with carprofen (5 mg/kg, sc) and enrofloxacin (0.5 mL/kg of a 2.27% solution), and had a suprapubic catheter (PE-10 tubing with a flared end) implanted in the dome of the bladder and secured with a purse string suture (6-0 silk). The tubing was tunneled to the back of the neck and secured to interscapular tissue (6-0 silk). The end was hermetically sealed and allowed to extend 2–3 cm out of the body. The abdominal wall and skin were closed separately (6-0 PGA) and bupivacaine (0.25%) was applied once topically to the incision. One week later, after appropriate training, the mouse was placed in a Ballman-type restrainer (Natsume Seisakusho Co., Tokyo, Japan), which was put inside of a Small Animal Cystometry Lab Station (Med Associates, St. Albans, VT) and situated above an analytical balance used to measure voided volume. A small piece of cardboard was bent to form a tent-like shape and affixed to the pan on the scale and covered with absorbent material. This had the effect of deflecting stool, while absorbing urine, although any stool not deflected was easily discernable from a void based on weight. Sterile saline was infused at 15 µL/min and intravesicular pressure was measured with an in-line transducer. Infusion continued for 60–120 minutes, and all data recorded with Med-CMG software (Med Associates, St. Albans, VT). At the end of the experiment, and immediately after a void, infusion was stopped and the post-void residual volume was measured by attaching the catheter to a 3 mL syringe and withdrawing the plunger for 10–15 seconds. The volume recovered was then measured gravimetrically.
Analysis of urodynamics data was performed using CMG Analysis software (version 1.06) provided by Med Associates (St. Albans, VT). One micturition cycle was defined as the time it took for intravesicular pressure to return to baseline after a previous void until it returned to baseline following the next void. Typically, three successive cycles were quantitated. During each cycle, voiding pressure was recorded as the peak intravesicular pressure, void volume as the amount of change on the scale, and frequency calculated as the number of voids per hour.
Blood glucose
Blood was obtained from the submandibular vein in the early afternoon and assessed immediately using the AimStrip Plus Blood Glucose Testing system (Germaine Laboratories, San Antonio, TX).
Four hour voiding assay
Fifteen week-old mice were placed into a Cystometry Animal Holder for Mouse (Med Associates, St. Albans, VT), which is essentially a small acrylic cage with a screen bottom. The screen was treated with NeverWet superhydrophobic water-resistant coatings (NeverWet LLC, Leola, PA) to minimize urine retention. The cage was fitted with a water bottle and situated inside of a Small Animal Cystometry Lab Station (Med Associates, St. Albans, VT) and above an analytical balance with the aforementioned tent-like stool-deflecting apparatus. Readings from the balance were recorded for 4 hours, and the volume of all voids added for a cumulative volume. Any stool that was not deflected was easily discernable from a void, based on weight. Because voiding can vary dramatically over the course of a 24-hour period,25 all experiments were initiated in the morning before 11:30.
Evans blue extravasation
Fifteen week-old mice were given Evans blue in saline (10 mg/Kg) intravenously. One hour later, animals were sacrificed and bladders weighed and placed in 1 mL formamide overnight at 56°C. Absorbance (620 nM) was measured and the amount of dye in solution calculated from a standard curve and normalized to bladder weight.
Statistics
All assays and parameters were assessed by either a two-tailed Students t-test (Figure 1) or a one-way ANOVA followed by a Tukey’s post-hoc analysis using GraphPad InStat software (La Jolla, CA). Statistical significance was defined as P<0.05. No correction for multiple comparisons was performed.
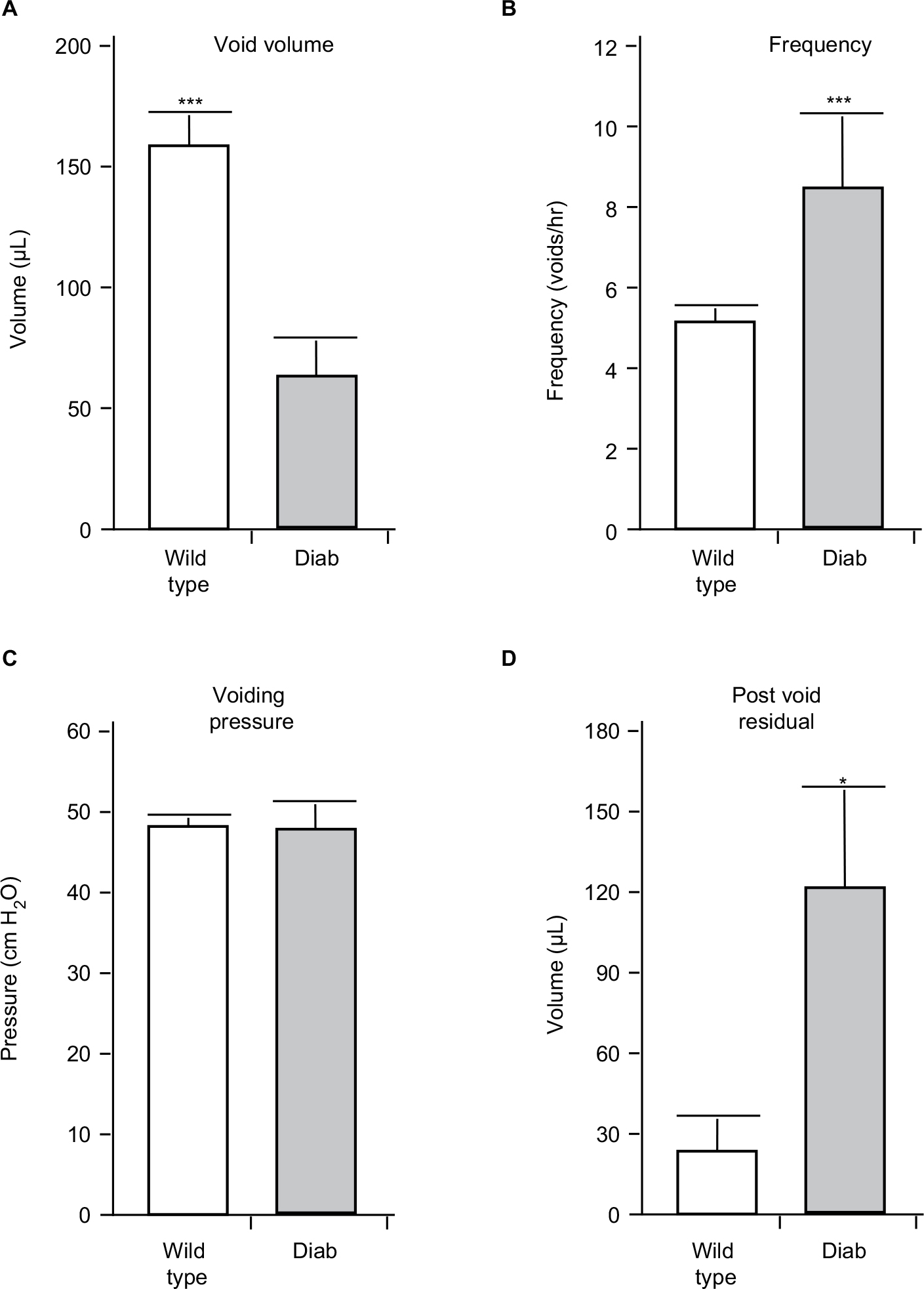 | Figure 1 The results of urodynamics at 15 weeks of age in wild type and Akita diabetic (Diab) mice. Animals were implanted with a suprapubic catheter 1 week prior to analysis. Results shown are the mean of the (A) voiding volume, (B) frequency of voiding, and (C) voiding pressure. (D) The post-void residual volume is the volume of urine remaining in the bladder immediately after the last void, as described in the methods section. Notes: Results are presented as the mean±SEM. N=8 and 4, respectively. * P<0.05, *** P<0.005 by a Student’s two-tailed t-test. Abbreviation: SEM, standard error of the mean. |
Results
Diabetic Akita mice develop DBD by week 15
As shown in Figure 1, Akita mice at 15 weeks of age show a pronounced increase in urinary frequency (Figure 1A) and a decrease in voided volume (Figure 1B). There was no change in voiding pressure between the groups (Figure 1C), but there was a significant increase in post-void residual volume in the diabetic mouse compared to wild type (Figure 1D).
Phlorizin lowers serum glucose and increases 4-hour voided volumes in Akita mice
In order to differentiate the effect of hyperglycemia and polyuria, animals were treated with phlorizin (via osmotic mini-pump) during the course of DBD development (week 4 to week 15 of life). As shown in Figure 2, serum glucose was increased in the diabetic mouse. Phlorizin, however, reduced blood glucose to levels not significantly different from wild type mice, clearly indicating the dosing regime was effective in reducing hyperglycemia. Conversely, as shown in Figure 3, phlorizin induced an increase in polyuria (total volume voided over 4 hours) compared to the diabetic mice, which themselves were elevated over wild type.
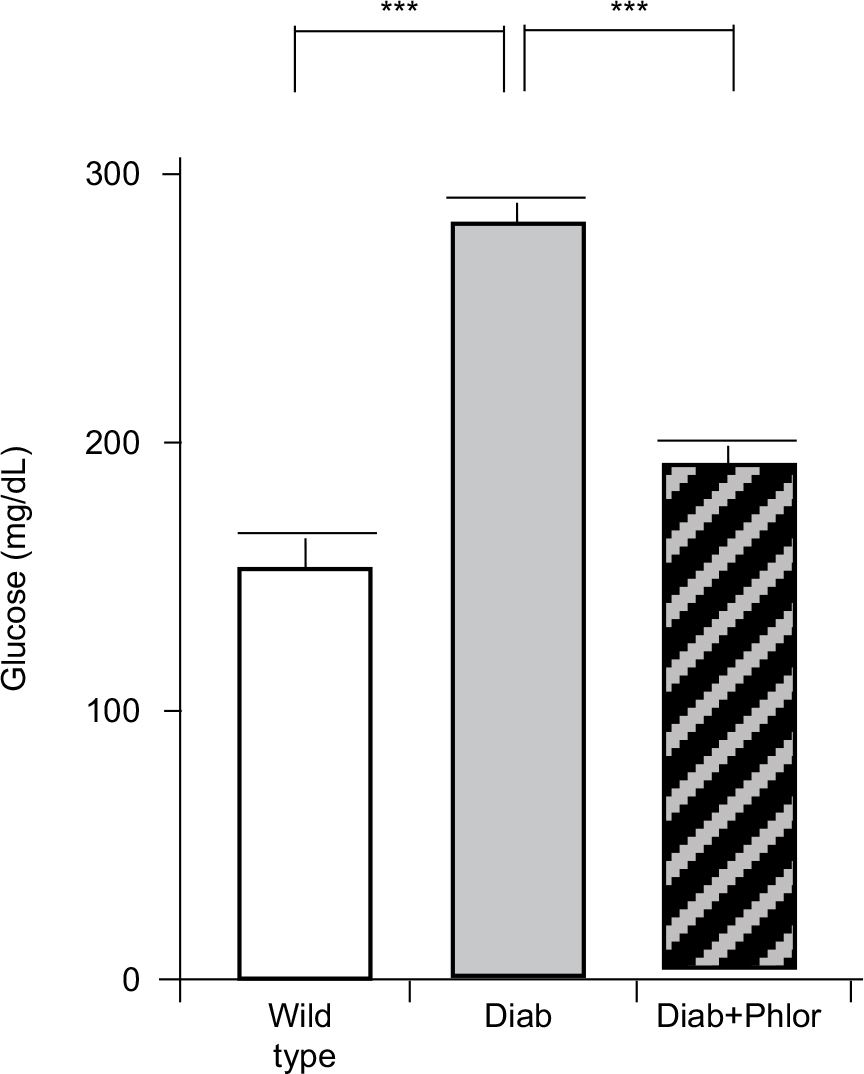 | Figure 2 Serum glucose levels at 15 weeks of age in wild type, Akita diabetic mice (Diab) and Akita diabetic mice treated with phlorizin (Diab+Phlor), as described in the Methods section. Notes: Results are presented as the mean±SEM. *** P<0.005 by ANOVA followed by Tukey’s post-hoc test. N=18, 20, and 10, respectively. Abbreviation: SEM, standard error of the mean. |
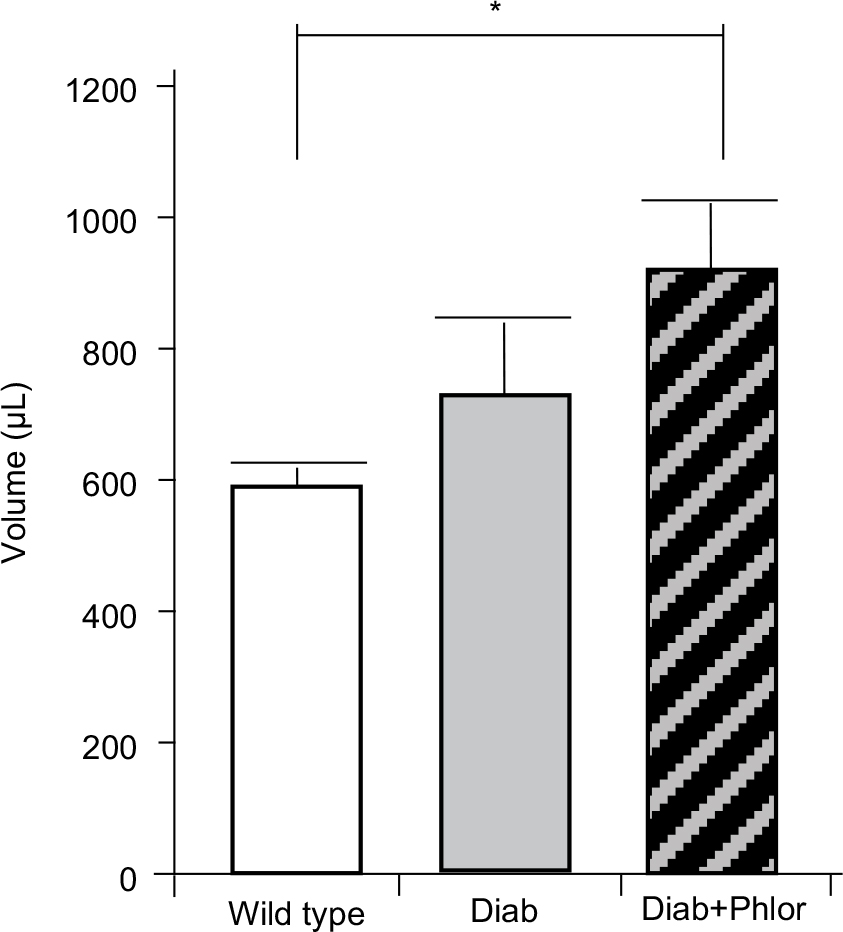 | Figure 3 Four-hour voiding assay. Total amount voided over 4 hours in wild type, Akita diabetic mice (Diab) and Akita diabetic mice treated with phlorizin (Diab+Phlor), as described in the Methods section. Notes: Results are presented as the mean±SEM. N=8, 5, and 5, respectively. *p<0.05 by ANOVA followed by Tukey’s post-hoc tes Abbreviation: SEM, standard error of the mean. |
Phlorizin limits Evans blue extravasation in diabetic mouse bladders
To determine if DBD may be associated with inflammation, we utilized the Evans blue dye extravasation assay, a well validated assay that we have employed in several studies of bladder inflammation.26,27 As shown in Figure 4, there was a highly significant increase in Evan’s blue dye in the bladder tissue of the diabetic mouse at 15 weeks of age. Importantly, extravasation of this dye was completely blocked with phlorizin treatment.
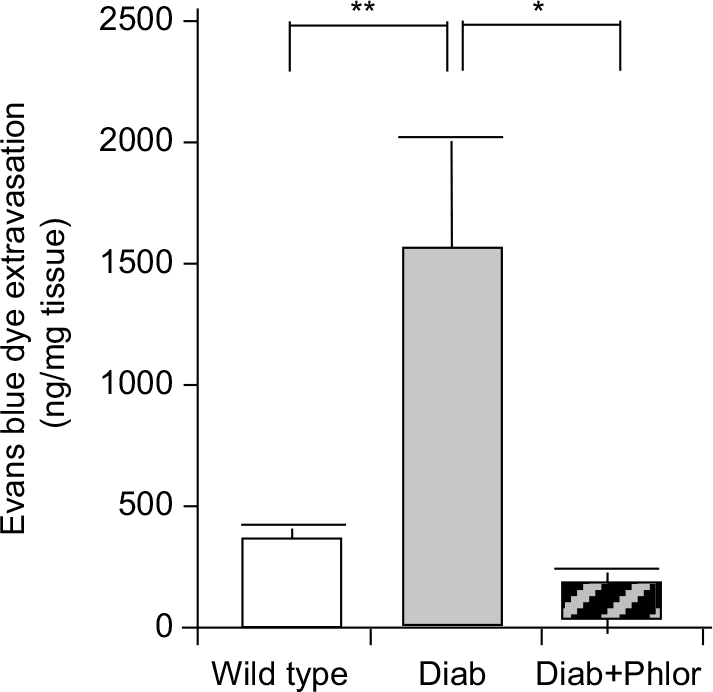 | Figure 4 Extravasation of Evans blue into the bladder of wild type, Akita diabetic mice (Diab), and Akita diabetic mice treated with phlorizin (Diab+Phlor), as described in the Methods section. Notes: Results are presented as the mean±SEM. N=13, 8, and 5, respectively. * P<0.05 and ** P<0.01 by ANOVA, followed by Tukey’s post-hoc test. Abbreviation: SE, standard error of the mean. |
Discussion
The early stages of DBD recapitulate the classic symptoms of OAB such as urgency, frequency, and urge incontinence,6,8,28 commonly documented in rodent models as an increase in urinary frequency and a decrease in voiding volume. Later stages of DBD demonstrate insensate bladders with increases in micturition volume, residual volume, and bladder capacity.8 Interestingly, some commonly used models, such as the streptozotocin-induced model, appear to bypass the early stages altogether.29–33 Since we sought to explore a possible role for diabetic inflammation in the bladder, and inflammation typically causes irritative volume symptoms identical to those that define OAB, we sought a model with a clearly-defined OAB-like early stage of DBD.
The Akita diabetic mouse model, which has a spontaneous mutation in the Ins2 gene, has been used previously in studies of DBD,34 although at the age those mice were used (20 weeks) the urodynamic changes appear to reflect the latter stages of this complication. In an attempt to identify an earlier stage we performed cystometry at several different time points (data not shown), and discovered that OAB-like changes are reliably produced at 15 weeks of age (Figure 1). Thus, this model appears to be quite suitable to study DBD, because it progresses through the temporal changes of early and late stages. It should be noted that there was a significant increase in post-void residual volume in the 15-week diabetic Akita mouse, which is typically a hallmark of later stages of DBD. Thus, mice at 15 weeks may already be progressing past the early stage. Since we did not identify a time point with clear and reproducible OAB-like symptoms, but no increase in post-void residual volumes (data not shown), completely separating early and late stages may not be feasible as the stages overlap and merge into one another. This may explain the failure of other studies to find clearly defined early DBD symptoms, and may, ultimately, be responsible for the mixed clinical picture of this complication.
As noted, inflammation during diabetes has been described in many different tissues that are associated with specific complications, including the retina, kidney, heart, blood vessels, and neurons, where it has been proposed to underlie the diabetic injury found in these tissues.16,17,35 However, little attention has been focused on the bladder, with only a single recent study drawing a correlation between inflammation, urothelial dysfunction, and diabetes in patients.18 To our knowledge, this is the first study to directly address bladder inflammation in an animal model of diabetes, and we found direct evidence that the bladder is indeed inflamed.
The bladder is unique among organs affected by diabetes, in that it is subjected to enhanced activity simply due to the osmotic effects of the sugar in urine preventing water reuptake, resulting in polyuria and an increase in urinary frequency. The result, of course, is an organ that is working harder than normal. In addition, the bladder urothelium is insulin-independent, so this tissue will experience hyperglycemic conditions, a second harmful stimulus that acts independently from polyuria. Numerous studies have sought to differentiate the effects of polyuria from hyperglycemia on various endpoints associated with DBD. In this study, we have addressed this topic by treating one group of diabetic mice with phlorizin, a competitive inhibitor of the SGLT1 and SGLT2 glucose transporters in the kidney and gut. Blocking glucose reabsorption in the kidney prevents reuptake of glucose, resulting in lowering blood sugar levels, but also increases osmotic urine output and enhances the overwork of the bladder. The effect of this inhibitor is strongly seen on blood glucose, which was significantly enhanced in the diabetic mouse but completely normal in the diabetic mouse treated with phlorizin. While a group of wild type mice with phlorizin would help identify the effects of the SGLT inhibitor, we believed that it would also cause lethal hypoglycemia in these mice and that we could already appreciate the effects in the diabetic mice with phlorizin. When examining total urine output, there was an expected increase in diabetes compared to wild type, although the variability of the assay prevented it from reaching significance. However, treatment with phlorizin did significantly increase total urine output over wild type, creating the desired phenotype of a mouse with polyuria but normal blood glucose levels. Excitingly, while inflammation was strongly present in the diabetic bladder, phlorizin completely blocked its appearance, clearly demonstrating that hyperglycemia is responsible for inducing diabetic bladder inflammation, with polyuria playing little or no role at the time point under investigation.
These results are consistent with previous studies that attempt to parcel out diuresis and hyperglycemia, while not excluding important roles for polyuria. Previously, Xiao et al29 concluded that polyuria controlled bladder hypertrophy, and that hyperglycemia was responsible for substantial oxidative stress in the bladder. Oxidative stress is an imbalance between the production of ROS (free radicals) and antioxidant defenses, resulting in excess radicals that react and damage all components of the cell including proteins, lipids, and DNA. Increased oxidative stress has been implicated in a vast number of diseases, including heart disease,9 cancer,10 and diabetes,11 and DBD in particular.6,12 Moreover, they hypothesized that oxidative stress may have pathological roles in late stage DBD. We would further hypothesize that such a pathological role is brought about by the induction of inflammation. Indeed, enhanced oxidative stress is known in other tissues to initiate an inflammatory response by engaging the NLRP3 inflammasome to trigger a proinflammatory cascade. Recent studies have shown the presence of NLRP3 in the urothelia36 and its importance in the pathology associated with diseases of sterile inflammation such as bladder outlet obstruction.26 Moreover, in those studies NLRP3 activation directly leads to denervation,37 an important change that may contribute to the late stage DBD where the bladder is insensate and decompensated, unable to adequately empty.38 Further studies will explore the role of NLRP3 and NLRP3-mediated denervation in the development of DBD.
Disclosure
The authors report no conflicts of interest in this work.
References
Roglic G, World Health Organization. Global report on diabetes. Geneva, Switzerland: World Health Organization; 2016. | ||
Panigrahy R, Singh B, das SK. Diabetic uropathy and bladder dysfunctions. Diabetes Metab Syndr. 2017;11(1):81–82. | ||
Daneshgari F, Moore C. Diabetic uropathy. Semin Nephrol. 2006;26(2):182–185. | ||
Jordan WR, Crabtree HH. Paralysis of the bladder in diabetic patients. Arch Intern Med. 1935;55(1):17–25. | ||
Brown JS. Diabetic cystopathy--what does it mean? J Urol. 2009;181(1):13–14. | ||
Yuan Z, Tang Z, He C, Tang W. Diabetic cystopathy: A review. J Diabetes. 2015;7(4):442–447. | ||
Abrams P, Cardozo L, Fall M, et al. The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Neurourol Urodyn. 2002;21:167–178. | ||
Daneshgari F, Liu G, Birder L, Hanna-Mitchell AT, Chacko S. Diabetic bladder dysfunction: current translational knowledge. J Urol. 2009;182(6 Suppl):S18–S26. | ||
Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. 2018:457–470. | ||
Calaf GM, Urzua U, Termini L, Aguayo F. Oxidative stress in female cancers. Oncotarget. 2018;9(34):23824–23842. | ||
Ma X, Chen Z, Wang L, et al. The pathogenesis of diabetes mellitus by oxidative stress and inflammation: its inhibition by berberine. Front Pharmacol. 2018;9:782. | ||
Liu G, Daneshgari F. Diabetic bladder dysfunction. Chin Med J. 2014;127(7):1357–1364. | ||
Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16(8):485–497. | ||
Naquet P, Giessner C, Galland F. Metabolic adaptation of tissues to stress releases metabolites influencing innate immunity. Curr Opin Immunol. 2016;38:30–38. | ||
Mcmaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. 2015;116(6):1022–1033. | ||
King GL. The role of inflammatory cytokines in diabetes and its complications. J Periodontol. 2008;79(8 Suppl):1527–1534. | ||
Navarro JF, Mora C. Role of inflammation in diabetic complications. Nephrol Dial Transplant. 2005;20(12):2601–2604. | ||
Wang CC, Kuo HC. Urothelial dysfunction and chronic inflammation in diabetic patients with overactive bladder. Low Urin Tract Symptoms. 2017;9(3):151–156. | ||
Kawanami D, Matoba K, Takeda Y, et al. SGLT2 inhibitors as a therapeutic option for diabetic nephropathy. Int J Mol Sci. 2017;18(5):1083. | ||
Vallon V, Thomson SC. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia. 2017;60(2):215–225. | ||
Filippas-Ntekouan S, Filippatos TD, Elisaf MS. SGLT2 inhibitors: are they safe? Postgrad Med. 2018;130(1):72–82. | ||
Pei F, Li BY, Zhang Z, et al. Beneficial effects of phlorizin on diabetic nephropathy in diabetic db/db mice. J Diabetes Complications. 2014;28(5):596–603. | ||
Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46(5):887–894. | ||
Lee DL, Sasser JM, Hobbs JL, et al. Posttranslational regulation of NO synthase activity in the renal medulla of diabetic rats. Am J Physiol Renal Physiol. 2005;288(1):F82–F90. | ||
Yu W, Ackert-Bicknell C, Larigakis JD, et al. Spontaneous voiding by mice reveals strain-specific lower urinary tract function to be a quantitative genetic trait. Am J Physiol Renal Physiol. 2014;306(11):F1296–F1307. | ||
Hughes FM, Hill HM, Wood CM, et al. The NLRP3 inflammasome mediates inflammation produced by bladder outlet obstruction. J Urol. 2016;195(5):1598–1605. | ||
Hughes FM, Vivar NP, Kennis JG, et al. Inflammasomes are important mediators of cyclophosphamide-induced bladder inflammation. Am J Physiol Renal Physiol. 2014;306(3):F299–F308. | ||
Daneshgari F, Liu G, Hanna-Mitchell AT. Path of translational discovery of urological complications of obesity and diabetes. Am J Physiol Renal Physiol. 2017;312(5):F887–F896. | ||
Xiao N, Wang Z, Huang Y, Daneshgari F, Liu G. Roles of polyuria and hyperglycemia in bladder dysfunction in diabetes. J Urol. 2013;189(3):1130–1136. | ||
Fathollahi A, Daneshgari F, Hanna-Mitchell AT. Effect of polyuria on bladder function in diabetics versus non-diabetics: an article review. Curr Urol. 2015;8(3):119–125. | ||
Daneshgari F, Huang X, Liu G, Bena J, Saffore L, Powell CT. Temporal differences in bladder dysfunction caused by diabetes, diuresis, and treated diabetes in mice. Am J Physiol Regul Integr Comp Physiol. 2006;290(6):R1728–R1735. | ||
Daneshgari F, Liu G, Imrey PB. Time dependent changes in diabetic cystopathy in rats include compensated and decompensated bladder function. J Urol. 2006;176(1):380–386. | ||
Moss HE, Lincoln J, Burnstock G. A study of bladder dysfunction during streptozotocin-induced diabetes in the rat using an in vitro whole bladder preparation. J Urol. 1987;138(5):1279–1284. | ||
Dolber PC, Jin H, Nassar R, Coffman TM, Gurley SB, Fraser MO. The effects of Ins2(Akita) diabetes and chronic angiotensin II infusion on cystometric properties in mice. Neurourol Urodyn. 2013. | ||
Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. Faseb J. 2004;18(12):1450–1452. | ||
Hughes FM, Turner DP, Todd Purves J, Purves JT. The potential repertoire of the innate immune system in the bladder: expression of pattern recognition receptors in the rat bladder and a rat urothelial cell line (MYP3 cells. Int Urol Nephrol. 2015;47(12):1953–1964. | ||
Lütolf R, Hughes FM, Inouye BM, et al. NLRP3/IL-1β mediates denervation during bladder outlet obstruction in rats. Neurourol Urodyn. 2018;37(3):952–959. | ||
Aizawa N, Igawa Y. Pathophysiology of the underactive bladder. Investig Clin Urol. 2017;58(Suppl 2):S82–S89. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.