Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 10
Diabetes and dyslipidemia: characterizing lipoprotein metabolism
Received 18 April 2017
Accepted for publication 7 June 2017
Published 28 July 2017 Volume 2017:10 Pages 333—343
DOI https://doi.org/10.2147/DMSO.S115855
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
GH Tomkin,1,2 D Owens1,2
1Diabetes Institute of Ireland, Beacon Hospital, 2Trinity College, University of Dublin, Dublin, Ireland
Abstract: Premature atherosclerosis in diabetes accounts for much of the decreased life span. New treatments have reduced this risk considerably. This review explores the relationship among the disturbances in glucose, lipid, and bile salt metabolic pathways that occur in diabetes. In particular, excess nutrient intake and starvation have major metabolic effects, which have allowed us new insights into the disturbance that occurs in diabetes. Metabolic regulators such as the forkhead transcription factors, the farnesyl X transcription factors, and the fibroblast growth factors have become important players in our understanding of the dysregulation of metabolism in diabetes and overnutrition. The disturbed regulation of lipoprotein metabolism in both the intestine and the liver has been more clearly defined over the past few years, and the atherogenicity of the triglyceride-rich lipoproteins, and – in tandem – low levels of high-density lipoproteins, is seen now as very important. New information on the apolipoproteins that control lipoprotein lipase activity has been obtained. This is an exciting time in the battle to defeat diabetic atherosclerosis.
Keywords: obesity, type 2 diabetes, dyslipidemia, low-density lipoprotein, fibroblast growth factor, forkhead transcription factor O1, farnesyl X transcription factors
Introduction
Diabetes is still often considered a sugar-related disease, but the disease might well have been named diabetes lipidus if only lipids instead of sugar could have been tasted in the urine, as suggested by Shafrir and Raz.1 Only in recent years has the devastating complication of the lipid-related disease atherosclerosis become more feared than the glucose-centric small vessel disease.2–5 Whereas small vessel disease is very much related to hyperglycemia, large vessel disease has been difficult to attribute to dysglycemia. Many studies have failed to reduce cardiovascular disease (CVD) events by improvement in blood sugar control.6–9 On the other hand, cholesterol-lowering treatment, in particular, statins, have been shown to have a major impact on cardiovascular events from the first statin trials in diabetic patients.10–12 The pathways by which insulin regulates fuel usage are still being discovered. It is clear that there is a switch from glucose to fat metabolism overnight when, in the fasting state, insulin deficiency results in not only high serum glucose but also high serum triglyceride levels. The triglycerides are packaged in lipoprotein particles driving the cascade through abnormal chylomicrons, very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL), and finally high-density lipoprotein (HDL) (Figure 1).
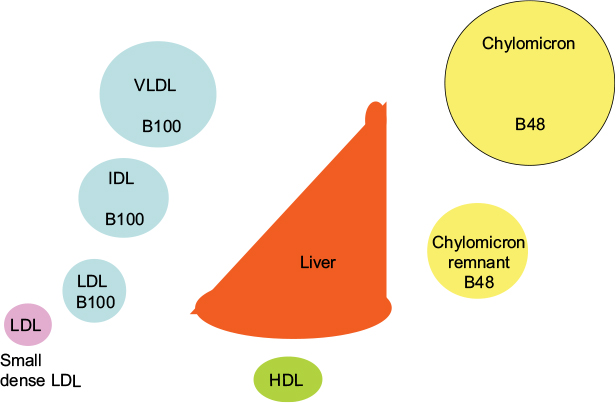 | Figure 1 Lipoprotein cascade. Notes: In the circulation, VLDL is gradually delipidated, resulting in increasingly smaller lipoprotein particles, ie, IDL, LDL, and small dense LDL. The intestinally derived chylomicron, characterized by presence of apoB48, is delipidated to form the chylomicron remnant, which is taken up by the liver. Abbreviations: HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low-density lipoprotein. |
Fasting hypertriglyceridaemis is significantly associated with cardiovascular events and death.13 A similar picture emerges when postprandial triglycerides are examined.14–16 Starvation and bariatric surgery both have a profound effect on serum lipids.17–19 Cholesterol is both absorbed and synthesized. Insulin regulates both these pathways, and since cholesterol synthesis is regulated through the bile acid cholesterol pathway, bile acids play a major part in cholesterol homeostasis.20
The purpose of this review is to explore the relationship among insulin resistance, diabetes, and dyslipidemia. We highlight areas of research that may lead to the discovery of possible new treatments to prevent premature heart disease in diabetes.
Insulin action
The secretion of insulin is glucose dependent. This is relevant in the fed state to prevent postprandial hyperglycemia. In fasting conditions, when the blood sugar is low, insulin is still needed; otherwise, free fatty acids will rise and hepatic glucose suppression will not occur, leading to hyperglycemia. In the fasting state, when blood sugars are low, fatty acids, not glucose, stimulate insulin secretion from the β cells.21
It has been shown that fatty acids acutely enhance insulin secretion, oxygen consumption rate, and extracellular acidification rate in human islets at fasting glucose concentrations, with monounsaturated fatty acids (MUFAs) being more potent than saturated fatty acids (SFAs).22 Cen et al22 suggest that the high fatty acids in their study may account for the hyperinsulinemia in patients who have raised fatty acids but normal blood sugars. In overnutrition, insulin initially manages to store the excess calories in the adipose tissue. This process breaks down at some stage and fatty acids collect in the liver and the muscle, leading to insulin resistance, and a vicious cycle arises in which the pancreas fails to deliver sufficient insulin to cope with the increased demands. This leads to even more difficulty in disposal of the fatty acids, and then the lack of inhibition of glucose release in the liver leads to hyperglycemia against a background of raised fatty acids. The high glucose level inhibits β-oxidation via a product of the glycolytic pathway, malonyl coenzyme A (Co-A), and fatty acids are directed toward formation of triglycerides.23 In the long term, the rise in free fatty acids has a detrimental effect on the β cells, leading to apoptosis.24
Diacylglycerol and insulin resistance
Diacylglycerol (DAG) is the precursor for triglyceride biosynthesis. The DAG kinases (DAGKs) are a group of kinases that regulate signal transduction via protein kinase C (PKC), Ras and Rho family proteins, and phosphatidylinositol 5-kinases.25 Elevated DAG content is linked with the development of insulin resistance in type 2 diabetes.26,27 DAGK delta activity and total DAG level are reduced in skeletal muscle from type 2 diabetic patients.28,29 Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a central regulator of energy metabolism. Metformin, the most commonly used drug to treat type 2 diabetes, activates AMPK to suppress gluconeogenesis.30 AMPK also suppresses gluconeogenesis by the downregulation of FoxO1 target genes.31 Transforming growth factor beta (TGF-b)/daf-16 (FoxO1) interact with AMPK to regulate metabolic and nutrient sensory pathways and glucose metabolism.32,33 Yadav et al33 have shown that that TGF-b1 signaling suppressed the liver kinase B1 (LKB1)–AMPK axis, thereby facilitating the nuclear translocation of FoxO1 and activation of key glucogenic genes regulating glucose-6 phosphatase and phosphoenolpyruvate carboxykinase both in the fasting state and in type 2 diabetes. PKC blocks AMPK activation.34 Nutrient excess in type 2 diabetes or obesity elevates DAG levels and PKC activity, in addition to impairing insulin sensitivity.35 AMPK activity is reduced in insulin-resistant and obese animal models.36 AMPK is involved in lipid metabolism through acetyl-CoA carboxylase and malonyl-CoA decarboxylase.37,38 Jiang et al39 have shown that DAGK delta deficiency impairs AMPK and lipid metabolism, as well as influencing skeletal muscle energetics. It seems that DAGK delta is a major player in the reduction in lipid oxidation and the insulin resistance found in type 2 diabetes (Figure 2).
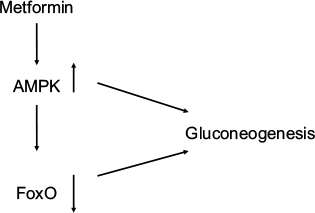 | Figure 2 Metformin stimulates AMPK, which downregulates gluconeogenesis both directly and through downregulation of FoxO. Abbreviation: AMPK, adenosine monophosphate-activated protein kinase. |
Bile acids
There is a third player in this process, namely, the bile acids. The two primary bile acids are chenodeoxycholic and cholic acids. They are synthesized in the liver, conjugated with taurine or glycine, and excreted in the bile.20 They aid fat absorption through their ability to form micelles, thus solubilizing fat and cholesterol.40 An increase in dietary cholesterol suppresses cholesterol synthesis and a decrease in dietary intake increases de novo synthesis in the liver. The bile acid-activated receptors play an important regulatory part in not only maintaining lipid, but also glucose, homeostasis.41–43 Chenodeoxycholic acid, which is an important farnesoid X receptor (FXR) agonist, lowers the biliary secretion of cholesterol, and reduces the cholesterol saturation of LDL through reduced clearance of plasma apolipoprotein B (apoB).44 Hepatic microsomal cholesterol 7 alpha hydroxylase (CYP7A1) and 3-hydroxy-3-methylglutaryl CoA (HMG CoA) reductase activities were reduced and specific LDL receptor binding was also reduced.45–47 Ghosh et al48 have shown that chenodeoxycholic acid reduces plasma clearance of LDL, somewhat mitigated by a decrease in LDL production. Proprotein convertase subtilisin/kexin type (PCSK9), apoA1, apoC111, lipoprotein (a), triglycerides, and insulin levels were reduced. This is of interest because FXR agonists have been shown to prevent the development of insulin resistance in animals.49
Glucose-dependent insulinotropic polypeptide (GIP) stimulates insulin secretion. The action of GIP is impaired in type 2 diabetes. GIP has been shown to lower nonesterified fatty acid (NEFA) concentration in obese type 2 diabetic patients despite diminished insulinotropic activity. GIP has also been shown to increase subcutaneous adipose tissue triglycerides. Reduction in NEFA concentration with GIP correlated with a reduction in adipose tissue insulin resistance.50
Fibroblast growth factors (FGFs)
FGF 15/19 and FGF 21 play an important role in metabolic regulation.51–53 Both molecules have demonstrated ability to lower serum glucose, triglyceride, and cholesterol levels; improve insulin sensitivity; and reduce body weight.54,55 FGF 19 activates FGF receptor 4 (FGFR4), the predominant receptor expressed in the liver, and regulates bile acid homeostasis.53,56–57 FGF 21 has recently been shown in mice to antagonize the action of FGF 15/19.53 Zhang et al53 have found, as expected, that overexpression of either FGF15 or FGF 21 reduced body weight, fasting glucose level, and insulin level, as well as decreasing plasma triglyceride and cholesterol levels. FGF 15 lowered the bile acid pool, but unexpectedly, the authors report that they found that FGF 21 increased the bile acid pool size through the beta-Klotho/FGFR4 complex. CYP7A1 catalyzes the first and rate-limiting step in the classic bile acid pathway.58 Cyp7A1 is tightly regulated by a negative feedback loop mediated by FGF 15/19.59,60 Overexpression of FGF15 significantly reduces Cyp7A1 mRNA.53 In contrast, FGF 21 overexpression results in CYP7A1 upregulation, suggesting that bile acid synthesis was the reason for the increased bile acid pool size in these animals. Serum FGF 21 has been shown to be increased in obesity.61 The authors have shown that there was a positive correlation between adiposity, fasting insulin, and triglycerides and a negative correlation with HDL cholesterol. Logistic regression analysis demonstrates an independent association between serum FGF 21 and the metabolic syndrome.61 FGF 21 has been shown to be raised in type 2 diabetic patients with nonalcoholic fatty liver disease.62 More recently, Alonge et al63 have shown that glucagon and insulin cooperatively stimulate FGF 21 gene transcription by increasing the expression of activating transcription factor 4. It has also been shown that FGF 21 is a superior biomarker to other adipokines.64 The authors suggested that serum FGF 21 might be considered an alternative to the oral glucose tolerance test.64 An FGF 21 analog has been shown to be superior to glargine insulin and a glucagon-like peptide-1 (GLP1) agonist liraglutide in reducing hemoglobin A1c (HbA1c) and improving glycemic control, insulin resistance, serum lipids, and liver function states in type 2 diabetic db/db mice (Figure 3).65
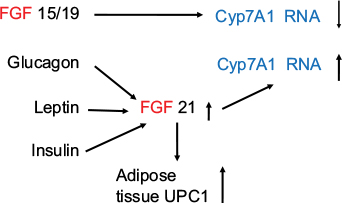 | Figure 3 FGF 15/19 and FGF 21 have opposing effects on bile acid synthesis through their effect on Cyp7A1. Note: Glucagon, leptin, and insulin increase FGF 21, which increases adipose tissue UCP1. Cyp7A1 is also termed cholesterol 7a-hydroxylase. Abbreviation: FGF, fibroblast growth factor. |
The FGF 21 analog LY2405319 has shown, in a 28-day proof-of-concept study66 in type 2 obese diabetic patients, significant improvement in lipids, with favorable effects on body weight, fasting insulin, and adiponectin. There was a trend toward glucose lowering.66 Another analog, PF-05231023, has been shown – in type 2 diabetes – to decrease body weight, improve lipoprotein profile, and increase adiponectin levels.The drug had no effect on glycemic control. The drug had effects on multiple markers of bone formation and resorption, and it increased insulin-like growth factor-1 (IGF-1). In adults, FGF 21 has been shown to be raised.64 In Chinese children aged between 6 and 18 years, the opposite has been described, with deficiency – rather than resistance – being found.67 The authors suggest that in children, FGF 21 deficiency – rather than resistance – contributes to insulin resistance and hypoadiponectinemia. Interestingly, leptin has recently been shown to increase FGF 21 levels in Wistar rats and in human-derived hepatoma HepG2 cells.68 Thus, the pathways between bile, cholesterol, glucose, and fat metabolic processes are linked, but there are still many discoveries yet to be made. Looking at the problem the other way, a deficiency of insulin leads to hyperglycemia, hypertriglyceridemia, and hypercholesterolemia, apart from abnormal bile acid metabolism, which affects the apoB-containing lipoproteins, and an interconnected decrease in HDL.
Serine/threonine protein kinase (STK25)
The networks controlling fat deposition and insulin responsiveness are very complex and attract much attention. The enzyme STK25 has been shown to influence intramyocellular lipid accumulation, impair skeletal muscle mitochondrial function and sarcomeric ultrastructure, and induce perimysial and endomysial fibrosis, thereby reducing endurance exercise capacity and muscle insulin sensitivity.69 The same group had previously shown that STK25 regulates lipid partitioning in human liver cells by controlling triglyceride synthesis as well as lipolytic activity and, thereby, NEFA release from lipid droplets for β-oxidation and triglyceride secretion.70
Forkhead transcription factors
FoxO1 plays an important role in orchestrating fuel metabolism and influences glucose, fat, and bile metabolic pathways through its effect on mitochondrial function and adipocyte differentiation.71–75 FoxO1 alters mitochondrial biogenesis, morphology, and function in the liver of insulin-resistant mice, while genetic ablation of FoxO1 significantly normalizes mitochondria and metabolism.73,76 In the adipocyte, silencing of FoxO1 inhibits cell differentiation and lipid accumulation, with changes in expression of mitochondrial respiration chain proteins.71,73,74 FoxO1 has been shown to control lipid droplet growth and adipose autophagy.77–81 Inhibition of autophagy leads to browning of white adipose tissue, which is characteristic of increased expression of uncoupling protein 1 (UCP1).78–81 UCP1 uncouples mitochondrial respiration from adenosine triphosphate (ATP) production/oxidative phosphorylation, dissipating energy as heat.82,83 Liu et al84 have recently shown that FoxO1 interacts with transcription factor EB (Tfeb), a key regulator of autophagosomes and lysosomes, and mediates the expression of UCP1, UCP2, and UCP3. However, the study84 showed that inhibition of FoxO1 suppressed Tfeb and autophagy, attenuated UCP2 and UCP3, but increased UCP1 expression (Figure 4). The enzyme protein deglycase (DJ-1) is involved in multiple physiological processes. Wu et al85 have recently shown that this protein is involved in maintaining energy balance and glucose homeostasis, regulating brown adipose tissue (BAT) activity. They showed that DJ-1-deficient mice had reduced body mass, increased energy expenditure, and improved insulin sensitivity. DJ-1 has been shown to inhibit FoxO1-dependent UCP1 expression in BAT. FoxO1 has also been shown to downregulate apoA1 gene activity in HepG2 cells under oxidative stress induced by hydrogen peroxide.86 ApoA1 forms HDL particles and has an antioxidant function.
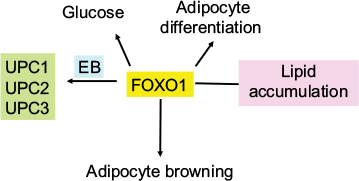 | Figure 4 Effect of FoxO1 on adipocyte differentiation and mitochondrial function. Notes: FoxO is a regulator of glucose metabolism, lipid accumulation, and adipocyte differentiation. It also increases adipocyte browning and interacts with Tfeb to regulate UCPs 1, 2, and 3. Abbreviation: Tfeb, transcription factor EB. |
Leptin is an important metabolic regulator. Leptin injections have been shown to increase plasma FGF21 in vivo in Wistar rats and in vitro using human-derived hepatocarcinoma HepG2 cells, mediated by STAT 3 activation.68 FoxO1, FoxO3, and FoxO4 have been shown to be involved in muscle proteasomal and autophagy–lysosomal degradation. Diabetes strongly affects protein metabolism, muscle wasting being a very significant finding in uncontrolled diabetes. Insulin and IGF-1 enhance muscle protein synthesis through their receptors.87 O’Neill et al88 have shown that both IGF-1 and the insulin receptor are involved in muscle proteostasis, the insulin receptor being more important than IGF-1. They found that muscle-specific deletion of FoxO1, FoxO3, and FoxO4 in double knockout of both insulin receptor and IGF-1 in mice completely rescued the muscle mass without changing the proteasomal activity.
FoxO1, rapamycin, and perilipin (PLIN)
Muscle is an important tissue for whole-body glucose homeostasis.89,90 Target of rapamycin (TOR) C2 is found in the insulin signaling pathway and is responsible for regulating muscle glucose metabolism.91–93 Acute inhibition of mTOR complexes increases lipid utilization, probably due to the effect of mTOR C2.91 PLIN 3 is a regulator of lipid storage.94–96 Knockdown of PLIN 3 in the liver of high-fat-diet-fed mice improves hepatic steatosis along with glucose homeostasis.97 PLIN 3 overexpression has been shown to increase muscle triglyceride.98 FoxO1 is a regulator of PLIN 1. AMPK modulates FoxO1 transcriptional activity.99 A FoxO1 antagonist has been shown to suppress autophagy and lipid droplet growth in adipocytes.77
Fibroblast activation protein (FAP)
FAP is a serine protease, and it has been shown to regulate the degradation of FGF 21100 Sánchez-Garrido et al101 have shown that inhibition of FAP using a known FAP inhibitor, talabostat, enhances levels of FGF21 in obese mice, reducing body weight, food consumption, and adiposity while increasing energy expenditure, glucose tolerance, and insulin sensitivity, as well as lowering cholesterol levels. The metabolic effect of FAP inhibition was markedly reduced in lean animals.101
Peroxisome proliferator-activated receptor (PPAR)
Insulin resistance in skeletal muscle plays a major role in obesity and type 2 diabetes.27 The PPAR superfamily of transcription factors includes the isoforms PPAR-alpha, which modifies insulin resistance in the liver; PPAR-γ, which regulates genes involved in fatty acid metabolism, inflammation, and macrophage homeostasis;102 and PPAR delta, which has been implicated in obesity-associated insulin resistance.103 It is highly expressed in muscle compared to PPAR alpha and gamma. A high-fructose diet-induced obesity results in insulin resistance in mice with hyperinsulinemia, hyperleptinemia, hyperlipidemia, and hypoadiponectinemia. The diet has been shown to impair insulin and AMPK signaling pathways and reduce glucose transporter type 4(GLUT-4) and GLUT-5 expressions. The study showed that a PPAR delta agonist GW0742 had no effect on control mice, but in the high-fructose-diet animals, it increased the expression of PPAR delta and significantly attenuated all the effects of the diet on the phosphorylation of insulin receptor substrate-1 (IRS-1), protein kinase B (PKB) or AKT, and glycogen synthase kinase 3 beta (GSK-3B). The agonist reduced skeletal muscle triglyceride and increased muscle glucose uptake. The drug increased phosphorylation of both AMPK and acetyl Co-A carboxylase (ACC) and increased protein expression of carnitine palmitoyl transferase-1 (CPT-1), all suggesting an increase in fatty acid oxidation. There was a dramatic increase of FGF-21 production in the muscle.104
DAG transferase
Hypertriglyceridemia is a major finding in uncontrolled diabetes. Indeed, many years ago,105 Shafrir and Gutman105 showed that as glucose intolerance increased from normal to diabetes through prediabetes, free fatty acids became much more markedly abnormal and preceded the glucose shift from normal to diabetes. Free fatty acids are converted to DAG through diglyceride acyltransferase (DGAT)-1 and then to triacylglycerol through DGAT2. The other major pathway of triglyceride synthesis is the glycerol phosphate pathway. In both pathways, fatty acyl-CoA and DAG are converted jointly to form triglyceride, catalyzed by DGAT. A novel DGAT1 inhibitor has been shown in mice to improve insulin resistance in adipose tissue, as well as systemic glucose metabolism, through a reduction in body weight.106
Triglyceride and cholesterol absorption in diabetes
Excess calories are first stored as triglyceride in adipose tissue to be released as fuel through the fatty acid cycle when carbohydrate is in short supply. Lipoprotein lipase (LpL) is suppressed by insulin, and therefore in insulin deficiency states, lipolysis increases even in a high-glucose environment. FoxA2 has been shown in the liver to regulate the LpL gene; thus, FoxA2 may be another important regulator of lipid and glucose metabolic pathways.107 Dietary fat is solubilized by bile acids in the intestine and, apart from the short-chain fatty acids, is absorbed by the lymphatic system passing to the liver. Triglyceride absorption is unregulated, so that fecal fat remains in very small quantities even in very-high-fat diets. Fatty acids stimulate synthesis of apoB100, which is edited to apoB48 in the intestine.108 ApoB48 is the solubilizing protein by which triglycerides and cholesterol are carried to the liver and then around in the circulation in the postprandial state. Although triglyceride absorption is unregulated, cholesterol absorption is tightly regulated. NPC1L1 is the regulating transporter protein in the first step in cholesterol absorption in the intestine. NPC1L1 mRNA is upregulated in diabetes.109 It has been shown that in a high- glucose environment, cholesterol absorption is increased.110
The dimer proteins ABCG 5/8 act together in the intestine to excrete excess cholesterol back into the lumen. These genes are downregulated in diabetes.109 Genetic variants in ABCs G5/8 have been shown to protect against myocardial infarction (MI) but also to increase the risk of symptomatic gallstone disease, demonstrating the interdependence between bile acid and cholesterol metabolic pathways.111 The final step in the absorption process is the attachment of the triglyceride and cholesterol onto apoB48 through MTP. MTP is upregulated in diabetes, and this is reflected in higher levels of apoB48 in serum (Figure 5).112 These particles are thought to be particularly atherogenic because of their large size and rapid turnover, so even though their cholesterol quantity per particle is low, the total carrying power of these particles is large; therefore, they are inherently atherogenic since the particles lodge in atheromatous plaques.113
 | Figure 5 Cholesterol absorption and chylomicron assembly and breakdown. Notes: Diacylglycerol is formed from free fatty acids under the influence of DGAT-1. Dietary cholesterol uptake from the intestine into the lymph is regulated by NPC1L1. ApoB48 is synthesized in the intestine. Triglyceride, cholesterol, and apoB48 are combined under the influence of MTP to form the chylomicron. In the circulation, the chylomicron is delipidated by LPL and cleared by the liver. Abbreviations: DGAT, diglyceride acyltransferase; LPL, lipoprotein lipase. |
The postprandial apoB48-containing particles and the VLDL apoB100 triglyceride-rich particles gather various apoproteins in the circulation. For example, apoC1 inhibits clearance of triglyceride by LpL. High levels of white adipose tissue apoC1 secretion has been shown to delay clearance of postprandial chylomicrons in overweight and obese subjects.114 ApoC11 is an obligatory cofactor for LpL. Recently, deficient cholesterol esterification has been found to occur in an apoC11-deficient zebrafish, which mimics the familial chylomicronemia syndrome in human patients, with a defect in apoC2 or LpL genes.115 ApoC111 inhibits the delipidation of triglyceride from the particle by inhibiting the action of LpL, thus delaying the clearance of the particle from the circulation.116 The Bruneck Study117 was designed to examine the importance of various apolipoproteins in the genesis of cardiovascular events over a 10-year period. The study found that apoC11, apoC111, and apoE were the apolipoproteins most significantly associated with incident CVD. These associations were independent of HDL and non-HDL cholesterol and extended to stroke and MI. Interestingly, these three apolipoproteins, apoC1, apoC11, and apoE, were implicated in de novo lipogenesis, glucose metabolism, complement activation, blood coagulation, and inflammation, through the lipidomic and proteomic profiles determined in the study.118 In the liver, NPC1L1 plays a part in the transport of cholesterol to the canaliculi, wherein the VLDL particle is assembled. The ABCs G5/8 play an important part in regulating the amount of cholesterol diverted to the bile for excretion. AUP1 is an endoplasmic reticulum-associated protein. Very recently, it has been shown to be involved in the regulation of apoB100, hepatic lipid droplet metabolism in the liver, and intracellular lipidation of VLDL particles.119 Its role in the intestine is so far unknown.
Diabetes disturbs the synthesis and metabolism of triglyceride-rich lipoprotein particles, increasing their atherogenicity. The specific role of triglycerides in atherogenesis has been difficult to tease out as the lipoprotein cascade is so interdependent and changes in the chylomicron influence VLDL assembly in the liver through the increase in delivery of both triglyceride and cholesterol to the liver.120 The increase in triglyceride content of the VLDL particle translates to an LDL particle with an increase in fatty acids. LDL atherogenicity is dependent at least in part on its oxidizability. The more the number of fatty acids with more-than-one double bond, the easier it is to oxidize, and it is the oxidized LDL that is taken up in an unregulated way by the macrophage, the hallmark of the atheromatous plaque.120 Small dense LDL particles are particularly associated with atheromatous risk and these particles arise from triglyceride-rich VLDL particles. An analysis of lipoprotein subfractions in 920 patients with and without type 2 diabetes confirmed the increase in concentration and size of smaller LDL particles.121
Free radical production is increased in the hyperglycemic state, so the diabetes environment increases the oxidation of LDL. In this context, delays in treatment intensification with oral antidiabetic drugs have been shown to increase the risk of major cardiovascular events.122
Diabetes dyslipidemia, atherosclerosis, and HDL
The hallmark of diabetes dyslipidemia is high triglycerides with low HDL.123,124 The interdependence of triglycerides and HDL has made it very difficult to separate the risk of atherosclerosis from one or the other. Until recently, HDL has come out on top and the triglyceride-rich lipoproteins have been undervalued as risk factors for accelerated atherosclerosis. Epidemiological studies in the 1970s established the strong inverse relationship between low HDL levels and coronary heart disease.125,126 More recently, the focus has been on the quality of HDL since functionality has been shown to be of major importance in predicting atherogenic risk.127,128 Hermans et al128 have suggested that the ratio of HDL-C/apoA1 might be a better way to predict angiopathic risk. Sun et al129 have shown that HDL from people with type 2 diabetes had the ability to stimulate secretion of tumor necrosis factor (TNF)-a, an inflammatory cytokine, in incubated human peripheral blood mononuclear cells to a greater extent as compared to HDL from control subjects. They showed that HDL from the patients with coronary artery disease (CAD) had a greater capacity to stimulate TNF-a as compared to HDL from the type 2 diabetic subjects who did not have coronary heart disease. The proinflammatory ability of HDL was a significant predictor for the presence of CAD in patients with diabetes. HDL particle number, rather than cholesterol content, may be a better predictor of atherogenicity. A multiethnic study130 of atherosclerosis has examined this in patients with the metabolic syndrome and diabetes. Tehrani et al130 found that HDL particle number in diabetes predicted coronary heart disease (CHD) and CVD. In those with metabolic syndrome, only LDL particle number was positively associated with CVD.
A retrospective study131 among >47,000 patients attending Italian diabetic centers investigated >15,000 patients with no evidence of renal disease. A 4-year follow up demonstrated that low HDL and high triglyceride levels were independent risk factors for the development of diabetic kidney disease over 4 years.131 Poor glycemic control in type 2 diabetes enhances functional and compositional alterations of small dense HDL3.132 Gomez Rosso et al132 showed that defective functionality of small dense HDL particles was present in patients with type 2 diabetes mellitus with poor glycemic control. The HDL had also diminished its antioxidant ability. One of the benefits of lifestyle intervention is the increase in HDL and, in particular, large HDL. It has recently been shown that lifestyle intervention can offset unfavorable genetic loading for most lipid traits, including the size of HDL.133 The understanding of functionality of HDL may become clearer following the description of the use of atomic force microscopy to examine the organization of apoA1.134
Conclusion
The dysregulation of metabolism when relative or absolute insulin deficiency appears has been more clearly defined in the past few years. The interplay between the bile, cholesterol, and carbohydrate metabolic pathways and the genes involved have opened up new possibilities of treatments to ameliorate the atherogenic potential of diabetic dyslipidemia. Overfeeding leads to obesity and insulin resistance. Hyperinsulinemia progresses to a relative, and then absolute, deficiency of insulin. It is difficult to dissect the metabolic disturbances that occur at each stage of the disease process. Dyslipidemia potentiates the disease process through oxidation of LDL, which further damages the β-cell. The abnormal HDL and the deficiency of its antioxidant functions in the defense of the β-cell have made for exciting speculations on treatments that might slow or stop β-cell destruction. Calorie excess, together with inadequate exercise, remains central to type 2 diabetes and diabetic dyslipidemia. Bariatric surgery and starvation both have shown how calorie restriction can ameliorate the metabolic dysfunction of type 2 diabetes, which includes dyslipidemia.
The most obvious lipid defect in uncontrolled diabetes is the elevated level of triglycerides. A consequence is the lowering of HDL. The triglyceride-rich lipoproteins have again come into fashion as important atherogenic particles. Although these particles carry much less cholesterol than LDL per particle, their actual load is similar to LDL if one takes into account their rapid half-life. LDL has a half-life of days rather than minutes in the case of chylomicrons. The influence of insulin on regulation of the apoB48-containing chylomicron in the intestine through a complex series of steps has helped to understand how dysregulation occurs in diabetes.
Disclosure
The authors report no conflicts of interest in this work.
References
Shafrir E, Raz I. Diabetes: mellitus or lipidus? Diabetologia. 2003;46:433–440. | ||
Despres JP, Lamarche B, Mauriège P, et al. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med. 1996;334(15):952–957. | ||
Zavaroni I, Bonini L, Gasparini P, et al. Hyperinsulinemia in a normal population as a predictor of non-insulin-dependent diabetes mellitus, hypertension, and coronary heart disease: the Barilla factory revisited. Metabolism. 1999;48(8):989–994. | ||
Gami AS, Witt BJ, Howard DE, et al. Metabolic syndrome and risk of incident cardiovascular events and death: a systematic review and meta-analysis of longitudinal studies. J Am Coll Cardiol. 2007;49(4):403–414. | ||
Reaven G. Insulin resistance and coronary heart disease in nondiabetic individuals. Arterioscler Thromb Vasc Biol. 2012;32(8):1754–1759. | ||
The Diabetes Control and Complications Trial Research Group, Nathan DM, Genuth S, et al. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329(14):977–986. | ||
Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ. 1998;316(7134):823–828. | ||
United Kingdom Prospective Diabetes Study Relative efficacy of randomly allocated diet, sulphonylurea, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ. 1995;310(6972):83–88. | ||
Gerstein HC, Miller ME, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes Study Group Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358(24):254525–254559. | ||
Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344(8934):1383–1389. | ||
Iso H, Naito Y, Sato S, et al. West of Scotland Coronary Prevention Study (WSCOPS): identification of high-risk groups and comparison with other cardiovascular intervention trials. Lancet. 1996;348(9038):1339–1342. | ||
Colhoun HM, Thomason MJ, Mackness MI, et al. The Collaborative Atorvastatin Diabetes Study (CARDS): design of the Collaborative Atorvastatin Diabetes Study (CARDS) in patients with type 2 diabetes. Diabet Med. 2002;19:201–211. | ||
Murad MH, Hazem A, Coto-Yglesias F, et al. The association of hypertriglyceridemia with cardiovascular events and pancreatitis: a systematic review and meta-analysis. BMC Endocr Disord. 2012;31(12):2. | ||
Iso H, Naito Y, Sato S, et al. Serum triglycerides and risk of coronary disease among Japanese men and women. Am J Epidemiol. 2001;153:490–499. | ||
Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Non-fasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308. | ||
Freiberg JJ, Tybjaerg-Hansen A, Jensen JS, Nordestgaard BG. Non-fasting triglycerides and risk of ischemic stroke in the general population. JAMA. 2008;300:2142–2152. | ||
Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011;54(10):2506–2514. | ||
Bays H, Kothari SN, Azagury DE, et al. Lipids and bariatric procedures Part 2 of 2: scientific statement from the American Society for Metabolic and Bariatric Surgery (ASMBS), the National Lipid Association (NLA), and Obesity Medicine Association (OMA). Surg Obes Relat Dis. 2016;12(3):468–495. | ||
Steven S, Carey PE, Small PK, Taylor R. Reversal of Type 2 diabetes after bariatric surgery is determined by the degree of achieved weight loss in both short- and long-duration diabetes. Diabet Med. 2015;32(1):47–53. | ||
Tomkin GH, Owens D. Obesity diabetes and the role of bile acids in metabolism. J Transl Int Med. 2016;4(2):73–80. | ||
Berne C. The metabolism of lipids in mouse pancreatic islets. The biosynthesis of triacylglycerols and phospholipids. Biochem J. 1975;152(3):667–673. | ||
Cen J, Sargsyan E, Bergsten P. Fatty acids stimulate insulin secretion from human pancreatic islets at fasting glucose concentrations via mitochondria-dependent and-independent mechanisms. Nutr Metab (Lond). 2016;13(1):59. | ||
Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J Biol Chem. 1992;267(9):5802–5810. | ||
Zhou YP, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest. 1994;93(2):870–876. | ||
Topham MK, Prescott SM. Mammalian diacylglycerol kinases, a family of lipid kinases with signaling functions. J Biol Chem. 1999;274(17):11447–11450. | ||
Kraegen EW, Cooney GJ. Free fatty acids and skeletal muscle insulin resistance. Curr Opin Lipidol. 2008;19(3):235–241. | ||
Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. | ||
Szendroedi J, Yoshimura T, Phielix E, et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A. 2014;111(26):9597–9602. | ||
Chibalin AV, Leng Y, Vieira E, et al. Downregulation of diacylglycerol kinase delta contributes to hyperglycemia-induced insulin resistance. Cell. 2008;132(3):375–386. | ||
Baur JA, Birnbaum MJ. Control of gluconeogenesis by metformin: does redox trump energy charge? Cell Metab. 2014;20(2):197–199. | ||
Sparks JD, Dong HH. FoxO1 and hepatic lipid metabolism. Curr Opin Lipidol. 2009;20(3):217–226. | ||
Li X, Wang L, Zhou XE, et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015;25(1):50–66. | ||
Yadav H, Devalaraja S, Chung ST, Rane SG. TGF-β1/Smad3 pathway targets PP2A-AMPK-FoxO1 signaling to regulate hepatic gluconeogenesis. J Biol Chem. 2017;292(8):3420–3432. | ||
Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. Nutrient excess in AMPK downregulation and insulin resistance. J Endocrinol Diabetes Obes. 2013;1(1):1008. | ||
Tomkin GH. Diabetic vascular disease and the rising star of protein kinase C. Diabetologia. 2001;44(6):657–658. | ||
Park SH, Gammon SR, Knippers JD, Paulsen SR, Rubink DS, Winder WW. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J Appl Physiol. 1985;92(6):2475–2482. | ||
Thomson DM, Winder WW. AMP-activated protein kinase control of fat metabolism in skeletal muscle. Acta Physiol (Oxf). 2009;196(1):147–154. | ||
Ruderman NB, Xu XJ, Nelson L, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298(4): E751–E760. | ||
Jiang LQ, de Castro Barbosa T, Massart J, et al. Diacylglycerol kinase-δ regulates AMPK signaling, lipid metabolism, and skeletal muscle energetics. Am J Physiol Endocrinol Metab. 2016;310(1):E51–E60. | ||
Albaugh VL, Banan B, Ajouz H, Abumrad NN, Flynn CR. Bile acids and bariatric surgery. Mol Aspects Med. Epub 2017 Apr 17. | ||
Evers SS, Sandoval DA, Seeley RJ. The physiology and molecular underpinnings of the effects of bariatric surgery on obesity and diabetes. Annu Rev Physiol. 2017;79:313–334. | ||
Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116(4):1102–1109. | ||
Fang S, Suh JM, Reilly SM, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med. 2015;21(2):159–165. | ||
Adler RD, Bennion LJ, Duane WC, Grundy SM. Effects of low dose chenodeoxycholic acid feeding on biliary lipid metabolism. Gastroenterology. 1975;68(2):326–334. | ||
LaRusso NF, Hoffman NE, Hofmann AF, Northfield TC, Thistle JL. Effect of primary bile acid ingestion on bile acid metabolism and biliary lipid secretion in gallstone patients. Gastroenterology. 1975;69(6):1301–1314. | ||
Haeusler RA, Astiarraga B, Camastra S, Accili D, Ferrannini E. Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes. 2013;62(12):4184–4191. | ||
Mazuy C, Helleboid A, Staels B, Lefebvre P. Nuclear bile acid signaling through the farnesoid X receptor. Cell Mol Life Sci. 2015;72(9):1631–1650. | ||
Ghosh LM, Eriksson M, Rudling M, Angelin B. Treatment with the natural FXR agonist chenodeoxycholic acid reduces clearance of plasma LDL whilst decreasing circulating PCSK9, lipoprotein(a) and apolipoprotein C-III. J Intern Med. 2017;281(6):575–585. | ||
Ma Y, Huang Y, Yan L, Gao M, Liu D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm Res. 2013;30(5):1447–1457. | ||
Thondam SK, Daousi C, Wilding JP, et al. Glucose-dependent insulinotropic polypeptide promotes lipid deposition in subcutaneous adipocytes in obese, type 2 diabetes patients: a maladaptive response. Am J Physiol Endocrinol Metab. 2017;312(3):E224–E233. | ||
Coskun T, Bina HA, Schneider MA, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149(12):6018–6027. | ||
Kharitonenkov A, Dunbar JD, Bina HA, et al. FGF-21/FGF-21 receptor interaction and activation is determined by beta Klotho. J Cell Physiol. 2008;2015(1):1–7. | ||
Zhang J, Gupte J, Gong Y, et al. Chronic over-expression of fibroblast growth factor 21 increases bile acid biosynthesis by opposing FGF15/19 action. EBioMedicine. 2017;15:173–183. | ||
Fu L, John LM, Adams SH, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145(6):2594–2603. | ||
Xu J, Stanislaus S, Chinookoswong N, et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models – association with liver and adipose tissue effects. Am J Physiol Endocrinol Metab. 2009;297(5):E1105–E1114. | ||
Wu X, Ge H, Gupte J, et al. Co-receptor requirements for fibroblast growth factor-19 signalling. J Biol Chem. 2007;282(40):29069–29072. | ||
Yang C, Jin C, Li X, Wang F, McKeehan WL, Luo Y. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PLoS One. 2012;7(3):e33870. | ||
Myant NB, Mitropoulos KA. Cholesterol 7 alpha-hydroxylase. J Lipid Res. 1977;18(2):135–153. | ||
Holt JA, Luo G, Billin AN, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17(13):1581–1591. | ||
Wu X, Ge H, Lemon B, et al. Selective activation of FGFR4 by an FGF19 variant does not improve glucose metabolism in Ob/Ob mice. Proc Natl Acad Sci U S A. 2009;106(34):14379–14384. | ||
Zhang X, Yeung DC, Karpisek M, et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes. 2008;57(5):1246–1253. | ||
Li X, Fan X, Ren F, et al. Serum FGF21 levels are increased in newly diagnosed type 2 diabetes with non-alcoholic fatty liver disease and associated with hsCRP levels independently. Diabetes Res Clin Pract. 2011;93(1):10–16. | ||
Alonge KM, Meares GP, Hillgartner FB. Glucagon and insulin cooperatively stimulate fibroblast growth factor 21 gene transcription by increasing the expression of activating transcription factor 4. J Biol Chem. 2017;292(13):5239–5252. | ||
Woo YC, Lee CH, Fong CH, et al. Serum fibroblast growth factor 21 is a superior biomarker to other adipokines in predicting incident diabetes. Clin Endocrinol (Oxf). 2017;86(1):37–43. | ||
Ye X, Qi J, Yu D, et al. Pharmacological efficacy of FGF21 analogue, liraglutide and insulin glargine in treatment of type 2 diabetes. J Diabetes Complications. 2017;31(4):726–734. | ||
Gaich G, Chien JY, Fu H, et al. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013;18(3):333–340. | ||
Li G, Yin J, Fu J, et al. FGF21 deficiency is associated with childhood obesity, insulin resistance and hypoadiponectinaemia: the BCAMS Study. Diabetes Metab. 2017;3636(16):30569–30569. | ||
Asrih M, Veyrat-Durebex C, Poher AL, Lyautey J, Rohner-Jeanrenaud F, Jornayvaz FR. Leptin as a potential regulator of FGF21. Cell Physiol Biochem. 2016;38(3):1218–1225. | ||
Chursa U, Nuñez-Durán E, Cansby E, et al. Overexpression of protein kinase STK25 in mice exacerbates ectopic lipid accumulation, mitochondrial dysfunction and insulin resistance in skeletal muscle. Diabetologia. 2017;60(3):553–567. | ||
Amrutkar M, Kern M, Nuñez-Durán E, et al. Protein kinase STK25 controls lipid partitioning in hepatocytes and correlates with liver fat content in humans. Diabetologia. 2016;59(2):341–353. | ||
Zou P, Liu L, Zheng L, et al. Targeting FoxO1 with AS1842856 suppresses adipogenesis. Cell Cycle. 2014;13:3759–3767. | ||
Cheng Z. FoxO1: mute for a tuned metabolism? Trends Endocrinol Metab. 2015;26(8):402–440. | ||
Cheng Z, Guo S, Copps K, et al. FoxO1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009;15(11):1307–1311. | ||
Munekata K, Sakamoto K. Forkhead transcription factor Foxo1 is essential for adipocyte differentiation. In Vitro Cell Dev Biol Anim. 2009;45(10):642–651. | ||
Higuchi M, Dusting GJ, Peshavariya H, et al. Differentiation of human adipose-derived stem cells into fat involves reactive oxygen species and Forkhead box O1 mediated upregulation of antioxidant enzymes. Stem Cells Dev. 2013;22(6):878–888. | ||
O-Sullivan I, Zhang W, Wasserman DH, et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun. 2015;6:7079. | ||
Liu L, Zheng LD, Zou P, et al. FoxO1 antagonist suppresses autophagy and lipid droplet growth in adipocytes. Cell Cycle. 2016;15(15):2033–2041. | ||
Singh R, Xiang Y, Wang Y, et al. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009;119(11):3329–3339. | ||
Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A. 2009;106(47):19860–19865. | ||
Baerga R, Zhang Y, Chen PH, Goldman S, Jin S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy. 2009;5(8):1118–1130. | ||
Armani A, Cinti F, Marzolla V, et al. Mineralocorticoid receptor antagonism induces browning of white adipose tissue through impairment of autophagy and prevents adipocyte dysfunction in high-fat-diet-fed mice. FASEB J. 2014;28(8):3745–3757. | ||
Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2(2):85–93. | ||
Rousset S, Alves-Guerra MC, Mozo J, et al. The biology of mitochondrial uncoupling proteins. Diabetes. 2004;53:S130–S135. | ||
Liu L, Tao Z, Zheng LD, et al. FoxO1 interacts with transcription factor EB and differentially regulates mitochondrial uncoupling proteins via autophagy in adipocytes. Cell Death Discov. 2016;2:16066. | ||
Wu R, Lui XM, Sun JG, et al. DJ-1 maintains energy and glucose homeostasis by regulating the function of brown adipose tissue. Cell Discov. 2017;3:16054. | ||
Shavva VS, Bogomolova AM, Nikitin AA, et al. FOXO1 and LXRα downregulate the apolipoprotein A-I gene expression during hydrogen peroxide-induced oxidative stress in HepG2 cells. Cell Stress Chaperones. 2017;22(1):123–134. | ||
Schiaffino S, Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle. 2011;1(1):1–4. | ||
O’Neill BT, Lee KY, Klaus K, et al. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J Clin Invest. 2016;126(9):3433–3446. | ||
Ferrannini E, Simonson DC, Katz LD, et al. The disposal of an oral glucose load in patients with non-insulin-dependent diabetes. Metabolism. 1988;37(1):79–85. | ||
Kelley D, Mitrakou A, Marsh H, et al. Skeletal muscle glycolysis, oxidation, and storage of an oral glucose load. J Clin Invest. 1988;81(5):1563–1571. | ||
Kleinert M, Sylow L, Fazakerley DJ, Krycer JR, Thomas KC, Oxboll AJ. Acute mTOR inhibition induces insulin resistance and alters substrate utilization in vivo. Mol Metab. 2014;3(6):630–641. | ||
Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC. Muscle-specific deletion of Rictor impairs insulin-stimulated glucose transport and enhances basal glycogen synthase activity. Mol Cell Biol. 2008;28(1):61–70. | ||
Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–768. | ||
Buers I, Robenek H, Lorkowski S, Nitschke Y, Severs NJ, Hofnagel O. TIP47, a lipid cargo protein involved in macrophage triglyceride metabolism. Arterioscler Thromb Vasc Biol. 2009;29(5):767–773. | ||
Bulankina AV, Deggerich A, Wenzel D, et al. TIP47 functions in the biogenesis of lipid droplets. J Cell Biol. 2009;185(4):641–655. | ||
Sztalryd C, Bell M, Lu X, et al. Functional compensation for adipose differentiation-related protein (ADFP) by Tip47 in an ADFP null embryonic cell line. J Biol Chem. 2006;281(45):34341–34348. | ||
Carr RM, Patel RT, Rao V, et al. Reduction of TIP47 improves hepatic steatosis and glucose homeostasis in mice. Am J Physiol Regul Integr Comp Physiol. 2012;302(8):R996–R1003. | ||
Kleinert M, Parker BL, Chaudhuri R, et al. mTORC2 and AMPK differentially regulate muscle triglyceride content via Perilipin 3. Mol Metab. 2016;5(8):646–655. | ||
Yun H, Park S, Kim MJ, et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J. 2014;281(19):4421–4438. | ||
Dunshee DR, Bainbridge TW, Kljavin NM, et al. Fibroblast activation protein cleaves and inactivates fibroblast growth factor 21. J Biol Chem. 2016;291(11):5986–5996. | ||
Sánchez-Garrido MA, Habegger KM, Clemmensen C. Fibroblast activation protein (FAP) as a novel metabolic target. Mol Metab. 2016;5(10):1015–1024. | ||
Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 2005;54(8):2460–2470. | ||
Benetti E, Patel NS, Collino M. The role of PPARβ/δ in the management of metabolic syndrome and its associated cardiovascular complications. Endocr Metab Immune Disord Drug Targets. 2011;11(4):273–284. Review. | ||
Benetti E, Mastrocola R, Rogazzo M, et al. High sugar intake and development of skeletal muscle insulin resistance and inflammation in mice: a protective role for PPAR- δ agonism. Mediators Inflamm. 2013;2013:509502. | ||
Shafrir E, Gutman A. Patterns of decrease in Free fatty acids in glucose tolerance test. Diabetes. 1969;14:77–83. | ||
Tomimoto D, Okuma C, Ishii Y, et al. JTT-553, a novel Acyl CoA:diacylglycerol acyltransferase (DGAT) 1 inhibitor, improves glucose metabolism in diet-induced obesity and genetic T2DM mice. J Pharmacol Sci. 2015;129(1):51–58. | ||
Kanaki M, Kardassis D. Regulation of the human lipoprotein lipase gene by the forkhead box transcription factor FOXA2/HNF-3β in hepatic cells. Biochim Biophys Acta. 2017;1860(3):327–336. | ||
Xiao C, Dash S, Morgantini C, Lewis GF. New and emerging regulators of intestinal lipoprotein secretion. Atherosclerosis. 2014;233(2):608–615. | ||
Lally S, Tan CY, Owens D, Tomkin GH. Messenger RNA levels of genes involved in dysregulation of postprandial lipoproteins in type 2 diabetes: the role of Niemann-Pick C1-like 1, ATP-binding cassette, transporters G5 and G8, and of microsomal triglyceride transfer protein. Diabetologia. 2006;49(5):1008–1016. | ||
Grenier E, Mailhot G, Dion D, et al. Role of the apical and basolateral domains of the enterocyte in the regulation of cholesterol transport by a high glucose concentration. Biochem Cell Biol. 2013;91(6):476–486. | ||
Stender S, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. The ABCG5/8 cholesterol transporter and myocardial infarction versus gallstone disease. J Am Coll Cardiol. 2014;63(20):2121–2128. | ||
Phillips C, Mullan K, Owens D, Tomkin GH. Intestinal microsomal triglyceride transfer protein in type 2 diabetic and non-diabetic subjects: the relationship to triglyceride-rich postprandial lipoprotein composition. Atherosclerosis. 2006;187(1):57–64. | ||
Proctor SD, Mamo JC. Intimal retention of cholesterol derived from apolipoprotein B100- and apolipoprotein B48-containing lipoproteins in carotid arteries of Watanabe heritable hyperlipidemic rabbits. Arterioscler Thromb Vasc Biol. 2003;23(9):1595–1600. | ||
Cyr Y, Wassef H, Bissonnette S, Lamantia V, Davignon J, Faraj M. WAT apoC-I secretion: role in delayed chylomicron clearance in vivo and ex vivo in WAT in obese subjects. J Lipid Res. 2016;57(6):1074–1085. | ||
Liu C, Gaudet D, Miller YI. Deficient cholesterol esterification in plasma of apoc2 knockout zebrafish and familial chylomicronemia patients. PLoS One. 2017;12(1):e0169939. | ||
Jensen MK, Rimm EB, Furtado JD, Sacks FM. Apolipoprotein C-III as a potential modulator of the association between HDL-cholesterol and incident coronary heart disease. J Am Heart Assoc. 2012;1(2):jah3–jah000232. | ||
Paige E, Masconi KL, Tsimikas S, et al. Lipoprotein(a) and incident type-2 diabetes: results from the prospective Bruneck study and a meta-analysis of published literature. Cardiovasc Diabetol. 2017;16(1):38. | ||
Pechlaner R, Tsimikas S, Yin X, et al. Very-low-density lipoprotein-associated apolipoproteins predict cardiovascular events and are lowered by inhibition of APOC-III. J Am Coll Cardiol. 2017;69(7):789–800. | ||
Zhang J, Zamani M, Thiele C, et al. AUP1 (ancient ubiquitous protein 1) is a key determinant of hepatic very-low-density lipoprotein assembly and secretion. Arterioscler Thromb Vasc Biol. 2017;37(4):633–642. | ||
Tomkin GH, Owens D. Dyslipidaemia of diabetes and the intestine. World J Diabetes. 2015;6(7):970–977. | ||
Zhao X, Zhang HW, Zhang Y, et al. Analysis of lipoprotein subfractions in 920 patients with and without type 2 diabetes. Heart Lung Circ. 2017;26(3):211–218. | ||
Folse HJ, Mukherjee J, Sheehan JJ, et al. Delays in treatment intensification with oral antidiabetic drugs and risk of microvascular and macrovascular events in patients with poor glycaemic control: an individual patient simulation study. Diabetes Obes Metab. Epub 2017 Feb 17:doi: 10.1111/dom.12913. | ||
Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia. 2003;46(6):733–749. | ||
Mooradian AD. Dyslipidemia in type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009;5(3):150–159. | ||
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62(5):707–714. | ||
Castelli WP, Doyle JT, Gordon T, et al. HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation. 1977;55(5):767–772. | ||
Simha V, Kudva YC. HDL cholesterol story is dead: long live HDL! Diabetes. 2016;65(10):2826–2828. | ||
Hermans MP, Amoussou-Guenou KD, Bouenizabila E, Sadikot SS, Ahn SA, Rousseau MF. Size density and cholesterol load of HDL predict microangiology, coronary artery disease and bcell function in men with T2 DM. Diabetes Metab Syndr. 2016;11(2):125–131. | ||
Sun JT, Liu Y, Lu L, et al. Diabetes-invoked high-density lipoprotein and its association with coronary artery disease in patients with type 2 diabetes mellitus. Am J Cardiol. 2016;118(11):1674–1679. | ||
Tehrani DM, Zhao Y, Blaha MJ, et al. Discordance of low-density lipoprotein and high-density lipoprotein cholesterol particle versus cholesterol concentration for the prediction of cardiovascular disease in patients with metabolic syndrome and diabetes mellitus (from the Multi-Ethnic Study of Atherosclerosis [MESA]). Am J Cardiol. 2016;117(12):1921–1927. | ||
Russo GT, De Cosmo S, Viazzi F, et al; AMD-Annals Study Group. Plasma triglycerides and HDL-C levels predict the development of diabetic kidney disease in subjects with type 2 diabetes: the AMD annals initiative. Diabetes Care. 2016;39(12):2278–2287. | ||
Gomez Rosso L, Lhomme M, Meroño T, et al. Poor glycemic control in type 2 diabetes enhances functional and compositional alterations of small, dense HDL3c. Biochim Biophys Acta. 2017;1862(2):188–195. | ||
Varga TV, Winters AH, Jablonski KA, et al; Diabetes Prevention Program Research Group. Comprehensive analysis of established dyslipidemia-associated loci in the diabetes prevention program. Circ Cardiovasc Genet. 2016;9(6):495–503. | ||
Gan C, Wang Z, Chen Y. In situ AFM imaging of apolipoprotein A-I directly derived from plasma HDL. Atherosclerosis. 2017;24(259):5–11. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.