Back to Journals » International Journal of Nanomedicine » Volume 13
Dexibuprofen nanocrystals with improved therapeutic performance: fabrication, characterization, in silico modeling, and in vivo evaluation
Authors Ullah N ![]() , Khan S
, Khan S ![]() , Ahmed S, Govender T, Faidah HS, de Matas M, Shahid M
, Ahmed S, Govender T, Faidah HS, de Matas M, Shahid M ![]() , Minhas MU
, Minhas MU ![]() , Sohail M
, Sohail M ![]() , Khurram M
, Khurram M
Received 13 September 2017
Accepted for publication 25 January 2018
Published 20 March 2018 Volume 2018:13 Pages 1677—1692
DOI https://doi.org/10.2147/IJN.S151597
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Naseem Ullah,1 Shahzeb Khan,1 Shaimaa Ahmed,2 Thirumala Govender,2 Hani S Faidah,3 Marcel de Matas,4 Muhammad Shahid,5 Muhammad Usman Minhas,6 Muhammad Sohail,7 Muhammad Khurram8
1Department of Pharmacy, University of Malakand, Chakdara, Pakistan; 2Discipline of Pharmaceutical Sciences, School of Health Sciences, University of KwaZulu-Natal, Durban, South Africa; 3Department of Microbiology, Faculty of Medicine, Umm Al Qura University, Makkah, Kingdom of Saudi Arabia; 4SEDA Pharmaceutical Development Services, The BioHub at Alderley Park, Cheshire, UK; 5Department of Pharmacy, Sarhad University of Science and Information Technology, Peshawar, Pakistan; 6Faculty of Pharmacy and Alternative Medicine, The Islamia University of Bahawalpur, Punjab, Pakistan; 7Department of Pharmacy, COMSATS, Abbottabad, Pakistan; 8Department of Pharmacy, Shaheed Benazir Bhutto University, Sheringal, Pakistan
Background: The aim of this study was to prepare and evaluate the impact of polymers on fabricating stable dexibuprofen (Dexi) nanocrystals with enhanced therapeutic potential, using a low energy, anti-solvent precipitation method coupled with molecular modelling approach.
Methods: Dexi nanocrystals were prepared using antisolvent precipitation with syringe pump. Crystallinity of the processed Dexi particles was confirmed using differential scanning calorimetry and powdered X-ray diffraction and transmission electron microscopy. Dissolution of Dexi nanocrystals was compared with raw Dexi and marketed tablets. Molecular modelling study was coupled with experimental studies to rationalise the appropriate polymers for stable Dexi nanocrystals. Antinociceptive study was carried out using balb mice.
Results: Combinations of hydroxypropyl methylcellulose (HPMC)–polyvinyl pyrrolidone (PVP) and HPMC–Eudragit (EUD) were shown to be very effective in producing stable Dexi nanocrystals with particle sizes of 85.0±2.5 nm and 90±3.0 nm, and polydispersity of 0.179±0.01, 0.182±0.02, respectively. The stability studies conducted for 90 days demonstrated that nanocrystals stored at 2°C–8°C and 25°C were more stable than those at 40°C. The maximum recovery of Dexi nanocrystals was observed from the formulations using the combination of HPMC–PVP and HPMC–EUD, which equated to 98% and 94% of the nominal active drug content respectively. The saturation solubility of the Dexi nanocrystals was substantially increased to 270.0±3.5 µg/mL compared to the raw Dexi in water (51.0±2.0 µg/mL) and stabilizer solution (92.0±3.0 µg/mL). Enhanced dissolution rate (P<0.05) was observed for the Dexi nanocrystals compared to the unprocessed drug substance and marketed tablets. Dexi nanocrystals produced the analgesic effect at much lower doses (5 mg/kg) than that of control standard, diclofenac sodium (20 mg/kg) and Dexi counterparts (40 mg/kg).
Conclusion: HPMC-PVP and HPMC-EUD were found the best polymer combination to stabilise Dexi nanocrystals. The Dexi nanocrystals exhibited significant dissolution, solubility and analgesic effect compared to the raw Dexi and the control standard diclofenac sodium.
Keywords: dexibuprofen, nanocrystals, dissolution, antinociceptive activity, molecular modeling, stability
Introduction
Poor aqueous solubility is known to limit the developability of a number of drug candidates, in spite of these molecules demonstrating suitable biologic activity and appropriate pharmacokinetic characteristics. The poor water solubility of a drug results in low dissolution rate, with subsequent partial and inconsistent absorption, which limits the drug exposure at its active site and constrains its clinical effectiveness.1
Additionally, poor patient compliance has been observed for drugs that are poorly water soluble because of the need to deliver higher doses, with consequent large unit dose sizes making them difficult to swallow. This also increases the cost of therapies and reduces their commercial attractiveness, while the variability in drug exposure typically associated with products of this nature might have negative consequences and benefit/risk profiles.2,3
Administering pharmaceutical dosage forms through the oral route is considered the most attractive and safest route of drug delivery. Solid dosage forms of active pharmaceutical ingredients, when administered orally, first undergo dissolution in the gastrointestinal fluids before absorption. Where permeability across the intestinal wall is good, dissolution is considered to be the rate-limiting step for poorly water soluble drug compounds, with a number of challenges being faced in developing suitable dosage forms for BCS Class II drugs.4 Additionally, BCS Class IV compounds, which also demonstrate limitations in intestinal permeability, provide significant challenges for drug development.5
Generally available techniques for addressing issues of low aqueous solubility in pharmaceutical development include particle size reduction, micronization,6 hot melt extrusion technology,7 solid dispersions,6 nanoemulsions,7 microencapsulation,8 micelles,9 salt formation, and complexation.10 More recently, the use of nanocrystals for enhancing oral bioavailability has received notable attention from the pharmaceutical industry, owing to reports of extensive increases in the dissolution rate and bioavailability.11–15 Surface area-to-volume ratio increases with a decrease in the size of particles, which in turn facilitates greater interaction between solute and solvent, resulting in improved solubility and dissolution.12 Two major methods are employed to reduce the particle size, which includes top–down and bottom–up methods.16–18
Dexibuprofen (Dexi) is an S (+)-isomer of ibuprofen, which is a chiral derivative of 2-arylpropionic acid, and demonstrates similar therapeutic behavior to other nonsteroidal anti-inflammatory drugs (NSAIDs). It has been established that administration of ~200 mg of Dexi produces equivalent analgesic effects to ibuprofen, but with a lower probability of producing adverse gastric effects.19
Dexi (Figure 1) belongs to BCS Class II of drug compounds, because of its poor water solubility, which leads to variability in drug absorption and erratic bioavailability. It is mainly used to manage mild-to-moderate pain and inflammatory conditions, such as headache, postoperative pain, dysmenorrhea, dental pain, and soft tissue rheumatism.19,20 A dose of 400 mg is effective to treat inflammatory conditions, with the biologic half-life of the Dexi being 1.8–3.5 hours.21–23 When considering a target product profile for a dosage form with enhanced bioavailability, a system giving adequate clinical performance at markedly reduced dose would be attractive. Until now, however, there has been no report of Dexi being developed in nanocrystals form. The aim of this study was, therefore, to produce stable Dexi nanocrystals using a low-energy method, with a view to enhance in vitro dissolution rate and deliver enhanced efficacy at lower doses. Moreover, this study aimed to comparatively evaluate the in vivo performance of the produced nanocrystals with the available marketed counterparts. The primary intent of this study was to couple experimental evaluation of formulations with molecular modeling to rationalize polymer selection to design optimal nanocrystal preparations. From previous experience, molecular modeling studies (Figure 2) are helpful in providing mechanistic insight at the molecular level, which could not be ascertained from traditional experiments.24,25
 | Figure 1 Chemical structure of dexibuprofen. |
 | Figure 2 Graphical abstract showing nanocrystals formation and their interaction with polymers. |
Materials and methods
Materials
Dexi (Batch No: C 102-1509014M) was purchased from Hubei Biocause Pharmaceutical Co. Ltd, China. Polyvinyl pyrrolidone (PVP) K-30 (Batch No: 08297052G0), hydroxypropyl methyl cellulose (HPMC) 6cps (Batch No: 8028213), polyvinyl alcohol (PVA) (Batch No: MKBR5960V), and Eudragit (EUD) RS100 (Batch No: 150313) were gifted by Navigal Pharmaceuticals, Pakistan, and analytical grade purified water was produced in laboratory using a purifier systems (Millipore, USA). Molecular Modeling Software, AutoDockVina, Marven Sketch, and Maestro were used for the extensive simulation studies. Mice (BALB/c) of both sexes weighing 22–28 g were purchased from the National Institute of Health Islamabad.
Methods
Preparation of Dexi nanosuspension
Antisolvent precipitation with syringe pump (APSP) method
The APSP method was employed with slight modification to produce nanocrystals of Dexi.11 The solution of the drug (30 mg/mL) was prepared in ethanol (EtOH) and filled into the syringe, and with the help of a syringe pump was quickly injected at a constant flow rate of 2–8 mL/minute into the polymer solutions that served as the antisolvent phase, with continuous stirring rate at 400–1,000 rpm. Different combinations and concentrations of the polymers were used to investigate their impact on the particle size of the produced Dexi nanocrystals. The polymer solutions were composed of 1% (w/v) of each of the polymers, which included PVP, EUD, HPMC, and PVA. EUD RS100 was dissolved in the drug solution in ethanol and was then injected into the polymeric solutions containing HPMC, PVP, and PVA. The nanocrystals were obtained by repaid evaporation of the solvent and antisolvent, using a rotary evaporator R201BL (SENCO, Shanghai, China). To achieve rapid drying of nanocrystals at low temperatures, in a way that does not damage the produced nanocrystals, specialized drying technology, vacuum drying, was employed. The vacuum drying of Dexi nanocrystals was carried out at room temperature for 24 hours with other conditions, such as a chamber capacity; 64 L with a pressure of 75 cm Hg. Moreover, for complete dryness of the produced Dexi nanocrystals, the drying process was carried out for 24 hours in a convection oven at three different temperatures, which include 40°C, 60°C, and 80°C.14
Characterization
Particle size measurements
Particle size measurements of the produced Dexi nanosuspensions were carried out using dynamic light scattering (DLS) (Zetasizer Nano-ZS, Malvern Instruments, UK). All the samples were analyzed without further dilution, the temperature of the system was kept at 25°C, and water was chosen as a dispersant, with the samples being analyzed with mean ± SD.
Assay of Dexi in nanosuspension
The produced nanosuspensions of Dexi were evaluated for active agent content using the high-performance liquid chromatography (HPLC) system (Waters 2695) connected to the ultraviolet detector. The produced Dexi nanocrystals were dissolved in the appropriate solvent, diluted with the mobile phase, and then injected into the HPLC system with the Ultra™ II column (C18 5 μm, 250×4.6 mm column). The mobile phase consisted of potassium dihydrogen orthophosphate buffer and acetonitrile, with its flow rate being set at 1 mL/minute with analysis at a wavelength of 222 nm. Moreover, after a series of dilutions, the final sample consisted of 60 μg of Dexi nanocrystals injected into the HPLC system and analyzed in triplicate.
Morphologic studies
Scanning electron microscopy (SEM)
Dexi images were taken at a range of magnifications, with the particles being sputter coated with gold prior to the morphologic studies using Joel JSM5910 scanning electron microscope at an operation voltage of 30 mA for a duration of 2 minutes and accelerating voltage of 20 kV.
Differential scanning calorimetry (DSC)
The heat of fusion and melting points of the samples of Dexi (nanocrystals and unprocessed drug substance), HPMC, PVP K-30, and EUD RS100 were carried out using the Mettler Tolado Differential Scanning Calorimeter (MettlerTolado®, USA). The samples of each of the raw Dexi nanocrystals and the optimized polymers (2–3 mg) were separately placed in an empty aluminum pan and sealed, and were then heated at the rate of 10°C min−1 from 25 to 120°C, with all samples being analyzed in triplicate.
Powder X-ray diffraction (PXRD) studies
The crystallinity of the unprocessed drug substance and prepared nanocrystals were determined by PXRD powder diffractometer (D8 ADVANCE, Bruker, Germany). In addition, for a control, the XRD analysis of the optimized polymers including HPMC, PVP K-30, and EUD RS100 was also carried out. Silicon-well sample holder was employed for the nanocrystals, while plastic sample holder was used for the unprocessed drug and polymers. The samples were scanned in triplicate in the range of 0°≤2θ≤50° by using copper Kα as a radiation source with 1 mm slit at 1.542 Å wavelength, while the step size was 0.05° and the time lapse between the steps was 2 seconds.
Fourier transform infrared (FTIR) spectroscopy analysis
Small quantities (2–3 mg) of both unprocessed Dexi and Dexi nanocrystals were mixed with 200–300 mg potassium bromide (KBr) separately, and then compressed to produce transparent pellets using a hydraulic press. The resulting pellets were scanned at a scanning range of 4,000–400 cm−1 using IR Prestige-21, Shimadzu Japan. Moreover, FTIR analysis of the optimized polymers including HPMC, PVP K-30, and EUD RS100 was also carried out as control samples.
Transmission electron microscopy (TEM) studies
TEM (TEM-1200Ex; Japan Electron Optics Laboratory Corporation, Tokyo, Japan) was used to evaluate Dexi nanocrystals at 120 kV. Nanosuspensions of Dexi were deposited on 200-mesh copper grid, which was coated with formar/carbon (code no: S162), followed by drying the samples at room temperature. The samples were stained negatively with the magnesium uranyl acetate (2%) solution because of the low conductivity of the API.
Solubility studies
The solubility studies of Dexi nanocrystals, unprocessed Dexi in optimized polymer solutions, and water were carried out. The Dexi nanocrystal samples were isolated from nanosuspensions by centrifugation using the method previously reported by Shah et al.12 Approximately 1.5 mL of the Dexi nanosuspension was filled into a centrifugation tube (1.5 mL) and stored for 24 hours. The sample was then centrifuged using a Sigma centrifuge (Scientific Lab supplier, Model: Sigma 0II5982IIII) at 14,800 rpm for 1 hour. The supernatant layer was taken and filtered through a 0.02-μm filter (Syringe Filter: 20 nm, Whatmananotop, Germany) to ensure that any undissolved API larger than 0.02 μm (eg, the nanoparticles) was separated from dissolved drug. The supernatant was then analyzed for the Dexi content using the HPLC method as previously described. The solubility study of unprocessed Dexi in pure water and optimized stabilizer solutions including HPMC–PVP and EUD–PVP was also carried out to assess the nanocrystals effect on saturation solubility of Dexi. Sufficient quantity of Dexi in pure water and stabilizer solutions was placed in vials and sonicated for 2 hours, followed by the use of the same procedure as mentioned for nanoparticles.
Statistical analysis
All the tests were run in triplicate and the results were given as mean ± standard error of the mean. Mean values were compared using analysis of variance (ANOVA) test, and the difference was considered statistically significant at level P<0.05 using Statistics 8.1 software.
Molecular modeling studies
The structures of Dexi and polymers were built up and energy minimized using Schrodinger’s Maestro Molecular Modeling suite.26 First, pKa calculations were performed at the experimental pH to assign the correct protonation states for the studied molecules. Two different tools were used to compute the pKa at the experimental pH (neutral); Marven Sketch27 and Maestro,28 to ensure the validity and reliability of the results, with both tools predicting the same results. A short MD run was performed to obtain relaxed energy conformers before docking, which was performed using OPLS 2005 force field without changing the ionization state, with AutoDock Vina being used for the docking calculation.29 Geister partial chargers were allocated during docking. AutoDock graphical user interface provided by MGL tools was used to outline the AutoDock atom types.30 The grid box was set to cover the entire polymer to ensure that all possible interactions with the drug were being searched. The Lamarckian Genetic Algorithm was used for docking calculations,35 with Auto Dock calculating the binding free energy based on the following energy terms: van der Waal (WvdW), hydrogen bonding (WH-bond), electrostatic (Welec), desolvation (Wdesolv), and torsional free energy change due to binding (Wtor).31
Stability studies
Physical stability of the Dexi nanocrystals was monitored for 10, 15, 30, 45, 60, 75, and 90 days by measuring the particle size and polydispersity index (PDI) using DLS. The chemical stability of the produced Dexi nanocrystals was monitored by quantifying the active agent content of the stored suspensions using HPLC. The samples manufactured at optimized process parameters were stored at 2°C–8°C, 25°C, and 40°C for the duration of the study.
Dissolution study
Dissolution studies on unprocessed Dexi, Dexi microsuspension, marketed tablets (200 mg), and Dexi nanocrystals were carried out to investigate the drug release behavior. The microsuspension was prepared by crushing the tablet using pestle and mortar, followed by sonication using the same dispersion medium that was used to fabricate the Dexi nanocrystals. The USP apparatus II was used to dissolve the selected formulations, with a phosphate buffer (pH: 7.2) being used as the dissolution medium (900 mL). The rotation speed of the paddles was set at 100 rpm, while keeping the temperature of the dissolution medium at 37°C±0.5°C. At predetermined intervals, 5 mL of sample was drawn from each vessel using a syringe filter (0.02 μm) and the same amount of fresh buffer added to maintain sink conditions. The syringe filter with the very small pore size (0.02 μm) has previously been used and recommended for dissolution studies of nanocrystals.38 The collected samples were properly diluted and analyzed by HPLC to determine the drug concentration.
Antinociceptive activity
Animals
Mice (BALB/c) of both sexes weighing 22–28 g were purchased from the National Institute of Health Islamabad. The maintenance temperature for the animals was 20°C–24°C, 12/12 hours light/dark cycle, and 40%–70% humidity. To maintain the environment from allergens, metabolic gases, debris, and odor buildup, adequate ventilation was provided to the experimental animals.
To avoid frown, variation in the light setting, and the formation of heat clusters, the cages were provided with uniformly dispersed light. The noise was minimized by keeping the animals in the place where the level of noise was not more than the human range, with the environmental factors being frequently observed for the animals on a cage level.
These experimental animals were kept in the environment for a period of 7 days prior to the experiments commencing, with free access to water and food for the full duration of the experiment, with each animals being given specific markings to enable identification. The protocol for this study was carried out according to standard requirements that have been approved by the Ethical Committee of the University of Malakand and relevant bye-laws (2008) (Scientific Procedure Issue-1). To perform experiments on animals, all the procedures were adapted according to the guidelines provided by National Institute of Health with respect to use and care of the animals. Furthermore, for all the experiments and testing on animals, the recommendation and policies of the international association for the study of pain were adhered to.
Acetic acid-induced writhing test
The antinociceptive activity of the nanocrystals was determined by the acetic acid-induced abdominal constriction assay.22 The animals were withdrawn from food 2 hours before the start of the experiment, with all the drugs being administered orally through an oral gavage tube. Diclofenac sodium was used as the positive control, and after 30 minutes of drug/standard treatment, individual animals were given an intraperitoneal injection of 1% acetic acid. The abdominal constrictions (writhes) were counted after 5 minutes of the acetic acid injection, with the observation continuing for the next 20 minutes. The animals were randomly assigned to the following groups (n=6). Group 1: saline control, group 2: diclofenac sodium (20 mg/kg), group 3: Dexi (40 mg/kg), group 4: Dexi-nano (5 mg/kg), group 5: Dexi-nano (10 mg/kg), group 6: Dexi-nano (15 mg/kg). The mean incidence of constrictions expressed as percentage protection across all experiments was normalized relative to the untreated controls. The percent protection from the tonic visceral chemical-induced nociception was calculated using the following formula:
|

Statistical analysis
Data were expressed as the mean ± standard error, with the total number of writhes being compared among treatment groups for the antinociceptive effect. Statistical comparisons were carried out by one-way ANOVA, followed by Dunnett’s post hoc test using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA), with statistical significance being deduced at P≤0.05.
Results and discussion
Preparation and characterization of Dexi nanosuspensions
The stable Dexi nanosuspension was fabricated using the APSP method. A range of polymer solutions were used as the antisolvent phase to elucidate the impact on particle size and polydispersity. The combination of HPMC–PVP and HPMC–EUD was shown to be the combination, as it produced Dexi nanocrystals with the smallest particle size of 85.0±2.5 and 90.0±3.0 nm, respectively (Table 1; Figure 3). This shows that these two polymers were sufficiently adsorbed onto the surfaces of the Dexi nanocrystals, which resulted in adequate particle–particle repulsion to prevent the growth of the particles during the nucleation process.32 In addition, the PDI of the abovementioned combinations was 0.17±0.01 and 0.18±0.02, respectively, both results being in good agreement with the PDI values of <0.5 reported for nanosuspensions with narrow size distributions.33 The nanocrystals produced using HPMC–PVP were subjected to further characterization, including the quantification of the active agent content using HPLC. Assay results were shown to be 98%, which indicates that the controlled ASPS method for producing nanocrystals is effective in delivering the target concentrations of suspensions with minimal losses.
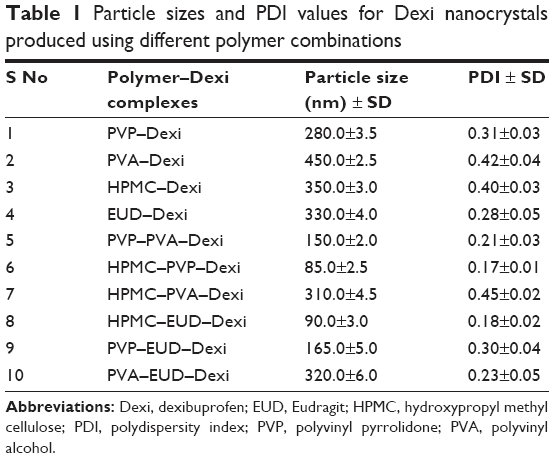 | Table 1 Particle sizes and PDI values for Dexi nanocrystals produced using different polymer combinations |
 | Figure 3 Particle size measurements of Dexi–HPMC–polyvinyl pyrrolidone (A) and Dexi–HPMC–Eudragit (B) nanocrystals. |
Morphology studies
The morphology studies of the unprocessed and processed Dexi were carried out using SEM and TEM. SEM micrographs of the unprocessed Dexi showed that the particles were of proper shapes having smooth surfaces, the average particle size being ~10–20 μm. Most of the particles were identified as rectangular, cuboidal, and prism shape crystals. Moreover, the particles were found to be homogeneously distributed with limited agglomeration (Figure 4A). The Dexi nanocrystals were analyzed using TEM, all the particles being below 100 nm, which is in good agreement with the Zetasizer data. In addition, the micrographs showed similar results to the SEM, indicating that all the particles were homogeneously distributed, with rectangular and cuboidal crystalline shapes (Figure 4B).
 | Figure 4 Scanning electron microscopy micrographs for unprocessed Dexi (A) and transmission electron microscopy images of Dexi nanocrystals (B). |
The comparative DSC and XRD studies demonstrated that the produced Dexi nanoparticles were crystalline in nature and maintained the physical form associated with the unprocessed drug substance. However, any prominent impact of the optimized polymers (HPMC, PVP, and EUD) was not observed on structure of the Dexi nanocrystals (Figure 5E, A and D). A sharp melting endotherm was observed for both the unprocessed and processed Dexi (Figure 5B and C). The melting temperature of the unprocessed Dexi was, however, observed to show a slightly higher melting temperature (52.5°C) compared to the Dexi nanocrystals (50°C). In addition, the endothermic peak of the Dexi nanocrystals appeared with a slightly broadened neck, which is caused by the small particle size and packing density of the processed samples compared to the raw Dexi.34,35 Moreover, broadening of the Dexi nanocrystals can potentially be caused by traces of the adsorbed polymers onto the surfaces of Dexi nanocrystals.35
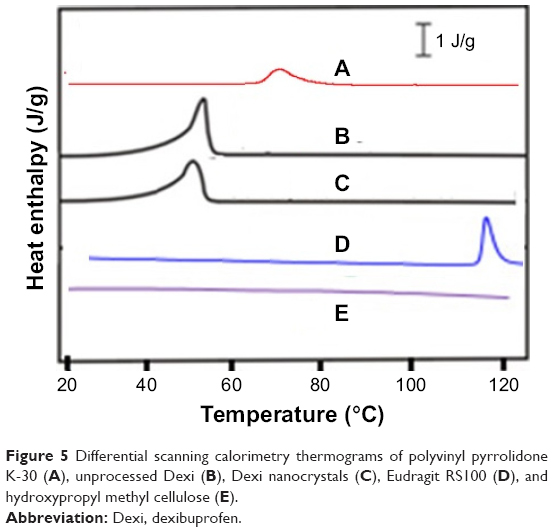 | Figure 5 Differential scanning calorimetry thermograms of polyvinyl pyrrolidone K-30 (A), unprocessed Dexi (B), Dexi nanocrystals (C), Eudragit RS100 (D), and hydroxypropyl methyl cellulose (E). |
Figure 6 shows that XRD studies of both raw and Dexi nanocrystals resulted in sharp X-ray diffractograms, which confirmed that the crystalline nature of Dexi was maintained, with no transformation to alternative physical forms after processing. A difference in the intensities and sharpness of the peaks was, however, observed between the unprocessed and processed Dexi nanocrystals (Figure 6). The PXRD studies of the polymers were also carried out as control to evaluate their impact on crystalline structure of the produced Dexi nanocrystals. However, any dominant effect of the polymers on diffractograms of the produced nanocrystals was not witnessed.
 | Figure 6 Powder X-ray diffraction diffractograms of Dexi nanocrystals, unprocessed Dexi, and chosen polymers. |
The appearance of the peaks for Dexi nanocrystals (Figure 6), with reduced intensity, might be attributed to the small particle size, which can impact on the reflection of the X-rays at small angles. Additionally, the presence of amorphous polymers adsorbed onto the surfaces of Dexi nanocrystals could produce a diffused scatter.36,37 The smaller the particle size, the lower the anticipated sharpness of the X-ray diffractogram, with subsequent disappearance of some peaks.34
FTIR spectroscopy
The comparative FTIR studies demonstrated that the process and optimized polymers (HPMC, PVP K-30, and EUD RS100) did not result in any structural changes of the produced Dexi nanocrystals. The FTIR spectra of both unprocessed and processed samples were compared and analyzed (Figure 7), and showed some specific structural characteristics. Peaks from 700 to 1,300 cm−1 represent skeletal C−C vibrations, while the peak at 935.48 cm−1 represents C−C bond, 1,008.27 cm−1 cyclohexane ring vibration, 1,379.10 cm−1 C−H methyl, 1,417.68 cm−1 O−H bond, 1,462.04 cm−1 C−H scissoring, 1,506.41 cm−1 C−C stretch in aromatic ring, 1,705.07 cm−1 C=O, 2,922.16 cm−1 C−H, and 2,954.95 cm−1 represents CH3 methyl. From the FTIR spectra of the samples, it is evident that the preparation process and polymers did not confer any changes to the structure of the produced Dexi nanocrystals (Figure 7C), their structure being almost similar to that of the FTIR spectrum of the unprocessed Dexi (Figure 7B).
 | Figure 7 Fourier transform infrared spectra of hydroxypropyl methyl cellulose (A), unprocessed Dexi (B), Dexi nanocrystals (C), polyvinyl pyrrolidone K-30 (D), and Eudragit RS100 (E). |
Solubility studies
The solubility profiles of Dexi nanocrystals and unprocessed Dexi in pure water and stabilizer solutions have been shown in Figure 8. In contrast to the water solubility of unprocessed Dexi (51±2.0 μg/mL), both Dexi nanocrystals, HPMC–EUD–Dexi and HPMC–PVP–Dexi, exhibited ~5-fold increase in their solubility profiles, which include 270.0±3.5 and 251.0±4.0 μg/mL, respectively. In addition, the solubility of unprocessed Dexi in the optimized polymeric solutions of 1% HPMC–PVP (92.0±3.0 μg/mL) and 1% HPMC–EUD (104±4.0 μg/mL) was found higher compared to the solubility in pure water (51.0±2.0 μg/mL). The nanocrystals have, however, substantially increased saturation solubility of Dexi and indicted that solubility of Dexi has been purely increased because of small particle size of the particles not due to the polymers.
 | Figure 8 Solubility studies of Dexi nanocrystals, unprocessed Dexi in pure water and stabilizer solutions. |
Owing to increased surface area of the produced nanocrystals, a significant difference (P<0.05, paired t-test, one-way ANOVA) was observed in solubility of the processed Dexi compared to the solubility in water and stabilizer solution. It has been previously reported that water-soluble polymers and surfactants (HPMC, PVP, and SLS) can increase solubility of drug compounds.12,38–40 We also observed an increase in solubility of Dexi in stabilizer solution (HPMC, PVP, and EUD). The nanocrystals have, however, substantially increased saturation solubility of Dexi and indicted that solubility of Dexi has been purely increased, because of small particle size of the particles not due to the polymers and surfactants. Generally, enhancement of drug saturation solubility could be attained by reducing drug particle size or by altering drug crystalline state, such as forming amorphous particles.
Molecular modeling studies
The binding energies for the Dexi–polymer complexes are shown in Table 2, which indicates that the docking results, and copolymeric structures HPMC–PVP and HPMC–EUD exhibited the highest binding with the drug (−4.7 and −4.6 kcal/mol, respectively), whereas PVA showed the least binding (−2.3. kcal/mol). The three-dimensional molecular representations of Dexi–polymeric structures are depicted in Figures 9 and 10. It can be concluded that the inclusion of PVP in the copolymeric forms tends to improve the binding of the drug, compared to the corresponding monopolymeric forms (eg, −4.7 kcal/mol versus −3.1 kcal/mol for HPMC–PVP and HPMC, respectively), which might be due to the contribution of the hydrophobic interactions with the PVP side chains rather than hydrogen bonding interactions (Figure 9).
 | Table 2 The binding energies for Dexi–polymer complexes obtained from docking calculations |
 | Figure 9 Interactions of HPMC–Dexi (top) and HPMC–PVP–Dexi (bottom) complexes showing hydrogen bonding as black lines. |
 | Figure 10 Interactions of EUD–Dexi (top) and HPMC–EUD–Dexi (bottom) complexes showing hydrogen bonding as black lines. |
It is also observed that the HPMC–EUD copolymer has enhanced drug binding compared to the respective individual polymers (−4.6, −3.1, and −3.2 kcal/mol, for HPMC–EUD, HPMC, and EUD, respectively) (Figure 10). As shown in Figure 10, the Dexi forms two hydrogen bonds with both the polymeric units, HPMC and EUD, which may have improved the binding (synergistic effect).
Dissolution studies of Dexi
Figure 11 shows the comparative dissolution studies of the Dexi nanocrystals with marketed tablets, unprocessed Dexi, and microsuspension. A substantial increase in dissolution rate was observed of the dexi nanocrystals compared to the raw Dexi, marketed tablets (200 mg), and Dexi microsuspension (10.0±2.5 μm; Figure 11).
 | Figure 11 Comparative dissolution studies of Dexi nanocrystals and its counterparts including tablets, microsuspension, and raw Dexi. |
Additionally, it was evident from the dissolution studies that ~70% and 65% of the Dexi nanocrystals HPMC–EUD–Dexi and HPMC–PVP–Dexi were dissolved, respectively, in the first 2 minutes. The dissolution rate of the HPMC–EUD–Dexi nanocrystals was slightly higher than the HPMC–PVP–Dexi nanocrystals, which is attributed to a slightly higher binding free energy of HPMC–PVP–Dexi (−4.7 kcal/mol) than the HPMC–EUD–Dexi (−4.6 kcal/mol; Table 2). The high binding energy can cause a slow release of the encapsulated drugs.25 In contrast, the dissolution rates of the unprocessed Dexi and marketed tablets were observed to be very low at 0.6% and 2.5%, respectively. A 117- and 108-fold increase in dissolution rates of the Dexi nanocrystals HPMC–EUD–Dexi and HPMC–PVP–Dexi, respectively, compared to the unprocessed Dexi, was observed. Additionally, the HPMC–EUD–Dexi and HPMC–PVP–Dexi nanocrystals exhibited 28- and 26-fold increases, respectively, in the dissolution rates compared to the marketed tablets (200 mg).
Stability studies
Nanocrystals, owing to their large surface area, have a high interfacial tension and increased free energies, making it challenging to control the particle growth in nanosuspension.17,41 The nanocrystals produced in this study possess a high tendency for agglomeration to reduce the overall system energy. Ostwald ripening is a well-known phenomenon that occurs in suspensions, where the small particles grow onto the surface of large particles.42 In this regard, uncontrolled particle growth has been reported as a major issue with nanocrystals produced by typical bottom–up methods.
Nucleation is an important step in antisolvent crystallization methods and can be exploited to tailor the particle size of crystallized materials,39–43 in addition to manipulating the polymeric medium, to facilitate surface stabilization and consequently control the particle size during the nucleation process.44 Table 1 shows the impact of different polymers on particle size and PDI values of the Dexi nanocrystals. The combination of HPMC–PVP was found to be the most suitable combination of polymers to control the particle growth of Dexi nanocrystals, which remained stable for 90 days when stored at room temperature (25°C) (Table 3). This study demonstrated that sufficient adsorption of the polymers occurred onto the surfaces of the produced nanocrystals, which resulted in strong repulsion of the particles, and subsequent colloidal stabilization. The molecular modeling studies also suggested that HPMC–PVP–Dexi gave higher binding free energy (−4.7 kcal/mol) than other complexes, which in turn provided higher levels of surface polymer adsorption and more effective stabilization. HPMC–EUD–Dexi nanocrystals were shown to be more stable for 90 days than other nanocrystals, but were less stable than their HPMC–PVP counterparts.
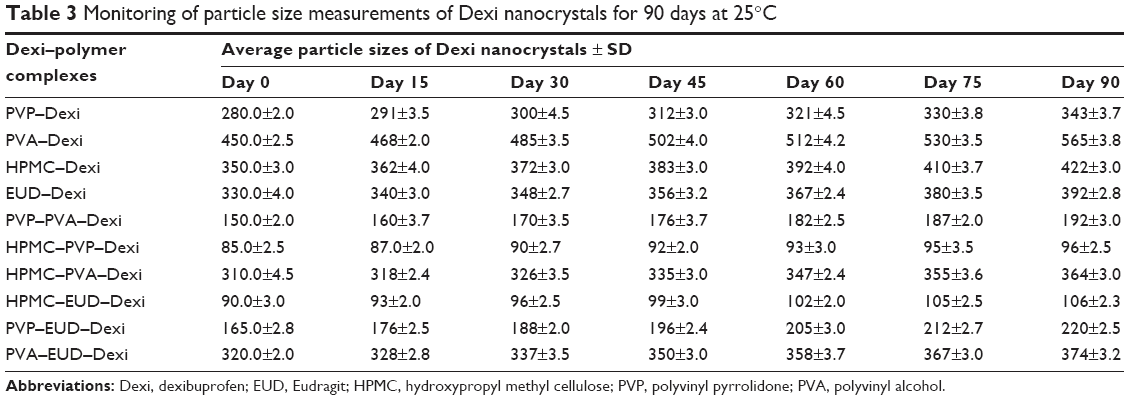 | Table 3 Monitoring of particle size measurements of Dexi nanocrystals for 90 days at 25°C |
The stability studies of Dexi nanocrystals at 2°C–8°C, 25°C, and 40°C for 90 days showed that when the samples were stored at 2°C–8°C and 25°C (Figure 12A and B), they demonstrated a higher stability than the samples stored at 40°C (Figure 12C). The Dexi nanocrystals stored at 2°C–8°C demonstrated improved stability (Figure 12A), with no substantial changes in the key attributes of the produced nanosuspensions. The statistical analysis (one-way ANOVA and paired t-test) showed insignificant difference (P>0.05) in particle sizes and PDI values after 90 days, which is indicative of a homogeneous particle size distribution of the Dexi nanocrystals, with the potential for avoiding particle growth by Ostwald ripening.45
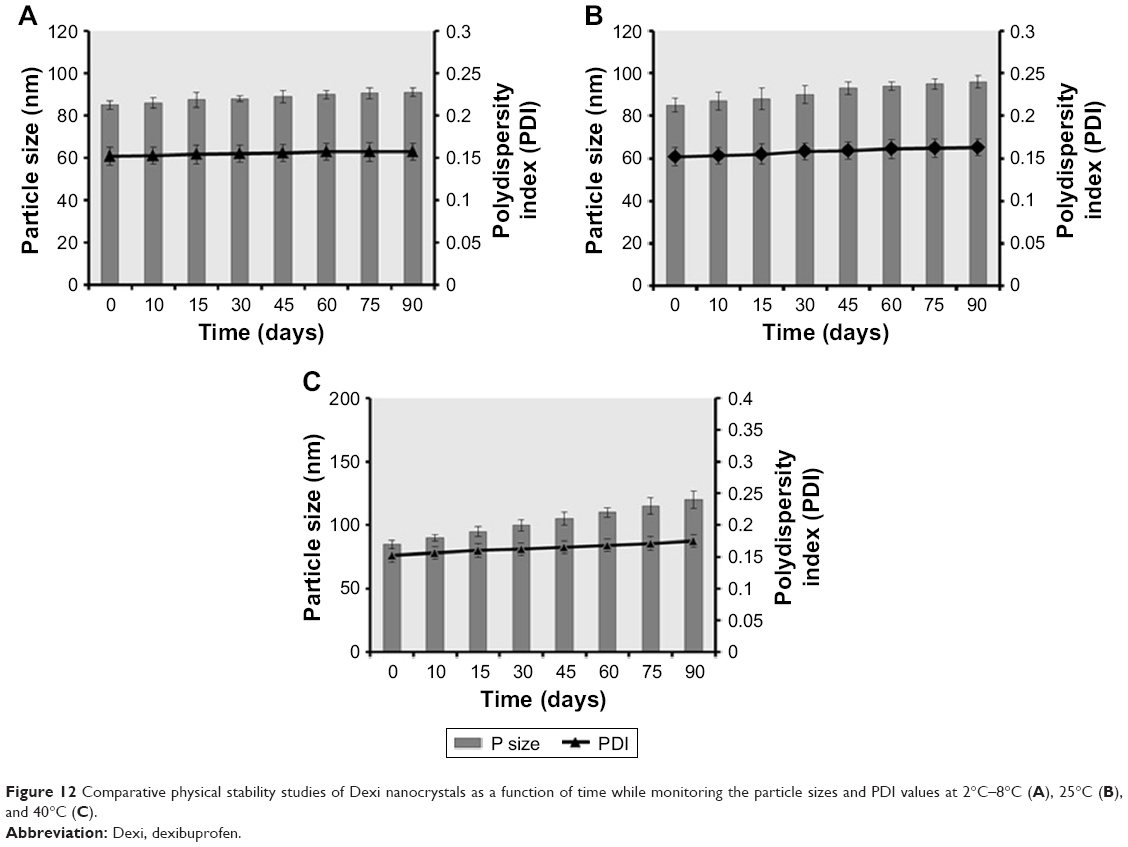 | Figure 12 Comparative physical stability studies of Dexi nanocrystals as a function of time while monitoring the particle sizes and PDI values at 2°C–8°C (A), 25°C (B), and 40°C (C). |
Previous reports have concluded that higher temperatures can cause instability of the nanosuspensions, because of the elevated Brownian motion and kinetic energy of the suspended particles, which results in increased particle collisions and the consequent aggregation and agglomeration of the particles.32 In addition, at a higher temperature, viscosity is decreased and solubility of the suspended particles is increased, which potentially leads to increased particle mobility, elevated interparticulate interactions, and consequent particle growth. For maximum stability of the nanosuspensions, Freitas and Müller recommended storing them at a temperature range of 2°C–8°C.46 In addition, the HPMC–PVP–Dexi nanocrystals were found to be chemically stable after 90 days while stored at room temperature, with active agent content being maintained at levels >90.0%.
In vivo study
Antinociceptive activity
Pain is associated with injury of the neuronal or non-neural tissue, being termed as neuropathic or nociceptive pain, respectively.44 Various mechanisms have been implicated in pain processing that includes mediators for inflammation; immunocompetent cells; adenosine triphosphate; cytokines, including protons; growth factors (neurotrophins); nitric oxide; activation of non-neuronal glial; and a variety of “exotic” species in reaction to tissue damage and inflammation.45 Glutamate is released in response to activation of the N-methyl-D-aspartate receptors, which are released from the nocisponsive afferent fibers, and finally decrease the inhibitory interneuronal tone in the dorsal horn.47 Despite the availability of a range of drugs, managing pain with safe therapeutic agents remains a challenge.48
In medicine, the application of nanotechnology has the potential to deliver several advantages, including improved safety profile and accumulation of drugs at their target site, enhanced biocompatibility and reduced toxicity.49
For the purposes of this study, the sensitivity of the acetic acid-induced writhing method to analgesic and sensory afferents in the peritoneum bear α1/2-adrenoceptors, β-adrenoceptors, and opioid receptors on their terminals has been recorded. The pain impulse production is deactivated when these receptors are activated by suitable agonists.
In our study, the standard diclofenac sodium (20 mg/kg) produced a robust antinociceptive response by producing a significant inhibition (68.08%) of acetic acid-induced writhes. Diclofenac sodium, which is a phenylacetic acid derivative and an important NSAID, is used in a number of inflammatory conditions and to treat pain.50
It was observed in our study that the oral ingestion of Dexi (40 mg/kg) provided efficient protection (58.87%) from nociception, as it significantly alleviated (P<0.001) the acetic acid-induced tonic visceral nociception during an observation time of 20 minutes (Figure 13). Dexi is considered to be a more effective and safer NSAID than other anti-inflammatory and analgesic drugs, as it causes markedly reduced gastric damage.
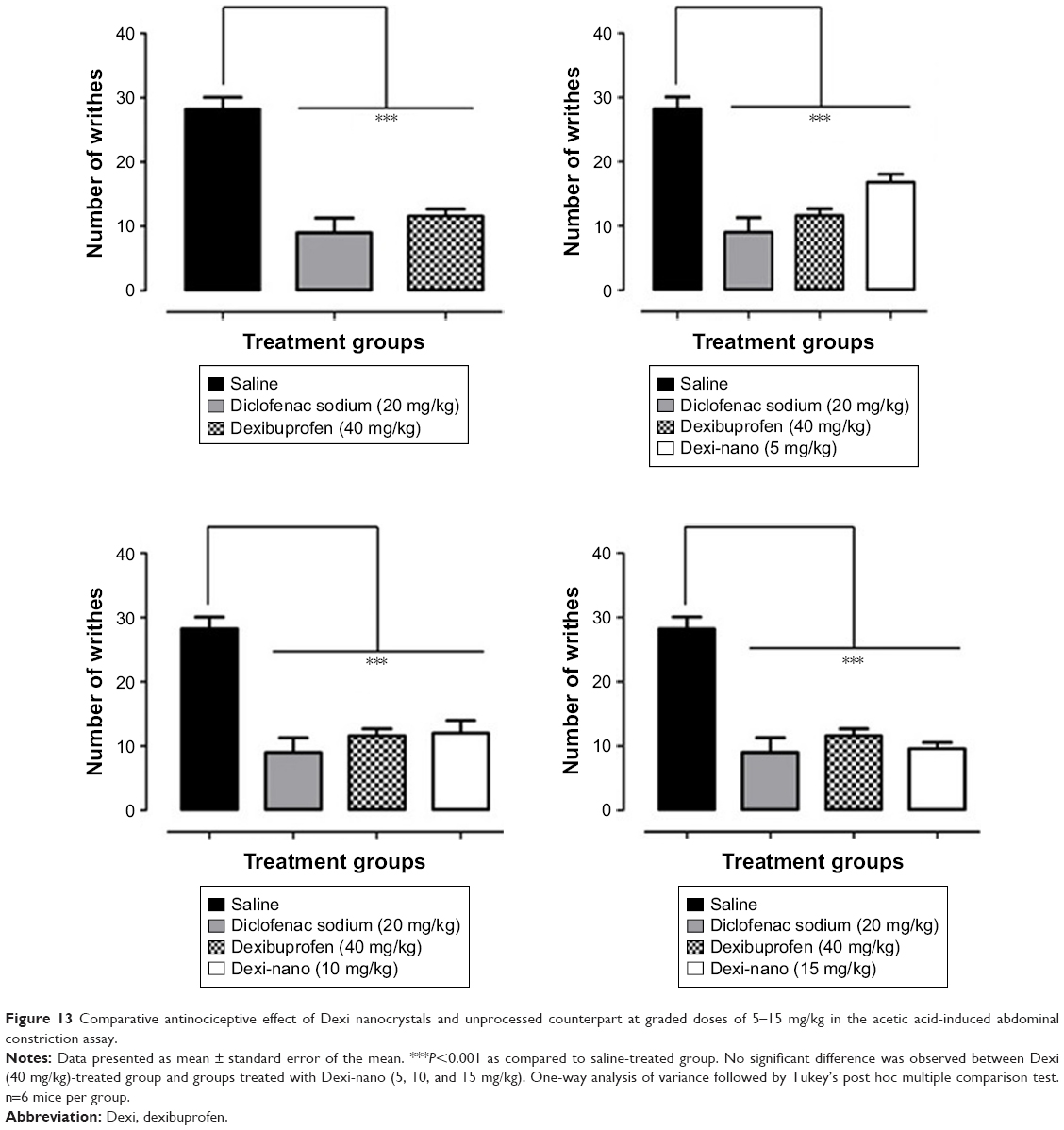 | Figure 13 Comparative antinociceptive effect of Dexi nanocrystals and unprocessed counterpart at graded doses of 5–15 mg/kg in the acetic acid-induced abdominal constriction assay. |
In this study, the Dexi nanocrystals produced a high-grade antinociceptive effect at much lower doses than the unprocessed Dexi formulation. The Dexi-nano drug, at a dose of 5 mg/kg, afforded significant analgesia (40.43%) with the same magnitude as that of the standard. A significant reduction in the writhes was observed at the lowest dose (5 mg/kg; P<0.001), as shown in Figure 13. Similarly, the 10 mg/kg dose also produced equipotent analgesia (57.44%), as the reduction in the abdominal constriction was significant (P<0.001) compared to the saline-treated animals (Figure 13). Additionally, the higher dose of Dexi-nano (15 mg/kg) also ameliorated the nociceptive response (65.95%) by producing an efficient suppression (P<0.001) of acetic acid-induced tonic visceral nociception, as shown in Figure 13.
The findings in the nociceptive assay showed that the nanoformulation provided significant analgesia comparable to that of Dexi and the standard diclofenac sodium. However, the analgesic effect was produced at much lower doses than that of the standard Dexi. These data suggest that the nano-drug-delivery systems could offer a clinically useful formulation for delivering Dexi.
Conclusion
A low-energy bottom–up method (ASPS) was found to be very effective for producing stable Dexi nanocrystals (85.0±2.5 nm), which had a high dissolution rate and enhanced therapeutic potential. This study supported the rational selection of polymer combinations to facilitate optimal control of the particle size of Dexi nanocrystals formed through antisolvent precipitation. The combination at 1% (w/v) HPMC–PVP and HPMC–EUD was found to produce Dexi nanocrystals with the smallest particle sizes of 85.0±2.5 and 90±3.0 nm, respectively. Dexi nanocrystals, with particle size at these levels, gave 117- and 28-fold increases in the dissolution rate compared to the unprocessed drug substance and the marketed tablets of the Dexi. A substantial increase in therapeutic performance of the Dexi nanocrystals compared to the unprocessed drug substance was observed, with the 10 mg/kg dose of the Dexi nanocrystals demonstrating equivalent therapeutic response to that of the unprocessed drug substance at the 40 mg/kg dose. This is very beneficial and could provide an opportunity for developing a cost-effective approach to achieve optimal therapeutic performance at a much lower dose than is currently used. Additionally, this study provided molecular insight into the mechanisms of binding of the optimal polymers to the surface of the Dexi nanocrystals, which supports the apparent avoidance of Ostwald ripening, with consequent particle stabilization.
Acknowledgments
The authors acknowledge Abdullah Jan at Central Research Laboratory, University of Peshawar, for scanning electron microscope, DSC, and FTIR spectroscopy instrumentation time and assistance by Mr Saddiq Afridi at Pakistan Council of Scientific and Industrial Research laboratory Peshawar KP. The authors also extend thanks to Mr Iqbal of the Central Research Laboratory Peshawar University for his contribution in carrying out the X-ray diffraction analysis. We are very grateful to Shaimaa Ahmed and Prof Thirumala Govender for conducting the extensive MD simulation studies, a part of this project, while using the established computational lab based at UKZN.
Disclosure
The authors report no conflicts of interest in this work.
References
Lipinski C. Poor aqueous solubility–an industry wide problem in drug discovery. Am Pharm Rev. 2002;5(3):82–85. | ||
Bittner B, Mountfield RJ. Formulations and related activities for the oral administration of poorly water-soluble compounds in early discovery animal studies: an overview of frequently applied approaches. Part 1. Drugs Made Ger. 2002;45(1):18–24. | ||
Bittner B, Mountfield R. Intravenous administration of poorly soluble new drug entities in early drug discovery: the potential impact of formulation on pharmacokinetic parameters. Curr Opin Drug Discov Devel. 2002;5(1):59–71. | ||
Speiser P. Poorly soluble drugs, a challenge in drug delivery. In: Emulsions and Nanosuspensions for the Formulation of Poorly Soluble Drugs. 1st ed. Stuttgart: Medpharm. 1998:15–28. | ||
Kasim NA, Whitehouse M, Ramachandran C, et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharm. 2004;1(1):85–96. | ||
Modi A, Tayade P. Enhancement of dissolution profile by solid dispersion (kneading) technique. AAPS PharmSciTech. 2006;7(3):E87–E92. | ||
Sarker DK. Engineering of nanoemulsions for drug delivery. Curr Drug Deliv. 2005;2(4):297–310. | ||
Jyothi NVN, Prasanna PM, Sakarkar SN, Prabha KS, Ramaiah PS, Srawan G. Microencapsulation techniques, factors influencing encapsulation efficiency. J Microencapsul. 2010;27(3):187–197. | ||
Kedar U, Phutane P, Shidhaye S, Kadam V. Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine. 2010;6(6):714–729. | ||
Serajuddin AT. Salt formation to improve drug solubility. Adv Drug Deliv Rev. 2007;59(7):603–616. | ||
Gao L, Liu G, Ma J, Wang X, Zhou L, Li X. Drug nanocrystals: in vivo performances. J Control Release. 2012;160(3):418–430. | ||
Shah SMH, Ullah F, Khan S, et al. Smart nanocrystals of artemether: fabrication, characterization, and comparative in vitro and in vivo antimalarial evaluation. Drug Des Devel Ther. 2016;10:3837. | ||
Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm. 2011;420(1):1–10. | ||
Kumar S, Jog R, Shen J, Zolnik B, Sadrieh N, Burgess DJ. In vitro and in vivo performance of different sized spray-dried crystalline itraconazole. J Pharm Sci. 2015;104(9):3018–3028. | ||
Kumar S, Jog R, Shen J, Zolnik B, Sadrieh N, Burgess DJ. Formulation and performance of danazol nano-crystalline suspensions and spray dried powders. Pharm Res. 2015;32(5):1694–1703. | ||
Walt DR. Nanomaterials: top-to-bottom functional design. Nat Mater. 2002;1(1):17–18. | ||
Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3(9):785–796. | ||
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3):278–287. | ||
Kaehler S, Phleps W, Hesse E. Dexibuprofen: pharmacology, therapeutic uses and safety. Inflammopharmacology. 2003;11(4):371–383. | ||
Di Pierro F, Settembre R. Safety and efficacy of an add-on therapy with curcumin phytosome and piperine and/or lipoic acid in subjects with a diagnosis of peripheral neuropathy treated with dexibuprofen. J Pain Res. 2013;6:497–503. | ||
Eller N, Kollenz C, Schiel H, Kikuta C, Mascher H. Pharmacokinetics of dexibuprofen administered as 200 mg and 400 mg film-coated tablets in healthy volunteers. Int J Clin Pharmacol Ther. 1998;36(8):414–417. | ||
Zhang X, Liu X, Gong T, Sun X, Zhang Z-R. In vitro and in vivo investigation of dexibuprofen derivatives for CNS delivery. Acta Pharmacol Sin. 2012;33(2):279–288. | ||
Evans AM. Comparative pharmacology of S (+)-ibuprofen and (RS)-ibuprofen. Clin Rheumatol. 2001;20(1):9–14. | ||
Anwar J. From virtual molecule to formulate medicine; a review of the potential of molecular simulation in drug delivery and formulation. LPT. 2007;10(7):66–72. | ||
Seedat N, Kalhapure RS, Mocktar C, et al. Co-encapsulation of multi-lipids and polymers enhances the performance of vancomycin in lipid–polymer hybrid nanoparticles: in vitro and in silico studies. Mater Sci Eng C Mater Biol Appl. 2016;61:616–630. | ||
Schrödinger L. Schrödinger Release 2015-1: Maestro (version 10.1). New York, NY; 2015. | ||
Fahlman BD. Materials Chemistry. Berlin: Springer; 2007. | ||
Maestro – The completely reimagined all-purpose molecular modeling environment. Available from: https://www.schrodinger.com/maestro. Accessed April 13, 2016. | ||
Trott O, Olson AJ. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Morris GM, Huey R, Lindstrom W, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem. 2009;16:2785–2791. | ||
Fernández-Urrusuno R, Calvo P, Remuñán-López C, Vila-Jato JL, José Alonso M. Enhancement of nasal absorption of insulin using chitosan nanoparticles. Pharm Res. 1999;16(10):1576–1581. | ||
Wu L, Zhang J, Watanabe W. Physical and chemical stability of drug nanoparticles. Adv Drug Deliv Rev. 2011;63(6):456–469. | ||
Patravale V, Date AA, Kulkarni R. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56(7):827–840. | ||
Bunjes H, Koch MH, Westesen K. Effect of particle size on colloidal solid triglycerides. Langmuir. 2000;16(12):5234–5241. | ||
Valleri M, Mura P, Maestrelli F, Cirri M, Ballerini R. Development and evaluation of glyburide fast dissolving tablets using solid dispersion technique. Drug Dev Ind Pharm. 2004;30(5):525–534. | ||
O’Mahony M, Leung AK, Ferguson S, Trout BL, Myerson AS. A process for the formation of nanocrystals of active pharmaceutical ingredients with poor aqueous solubility in a nanoporous substrate. Org Process Res Dev. 2014;19(9):1109–1118. | ||
Khan S, Matas MD, Zhang J, Anwar J. Nanocrystal preparation: low-energy precipitation method revisited. Cryst Growth Des. 2013;13(7):2766–2777. | ||
Ali HS, York P, Ali AM, Blagden N. Hydrocortisone nanosuspensions for ophthalmic delivery: a comparative study between microfluidic nanoprecipitation and wet milling. J Control Release. 2011;149(2):175–181. | ||
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. | ||
Rahim H, Sadiq A, Khan S, et al. Aceclofenac nanocrystals with enhanced in vitro, in vivo performance: formulation optimization, characterization, analgesic and acute toxicity studies. Drug Des Dev Ther. 2017;11:2443. | ||
Verma S, Kumar S, Gokhale R, Burgess DJ. Physical stability of nanosuspensions: investigation of the role of stabilizers on Ostwald ripening. Int J Pharm. 2011;406(1–2):145–152. | ||
Voorhees PW. The theory of Ostwald ripening. J Stat Phys. 1985;38(1):231–252. | ||
Dalvi SV, Dave RN. Analysis of nucleation kinetics of poorly water-soluble drugs in presence of ultrasound and hydroxypropyl methyl cellulose during antisolvent precipitation. Int J Pharm. 2010;387(1):172–179. | ||
Anwar J, Khan S, Lindfors L. Secondary crystal nucleation: nuclei breeding factory uncovered. Angew Chem. 2015;54(49):14681–14684. | ||
Deng J, Huang L, Liu F. Understanding the structure and stability of paclitaxel nanocrystals. Int J Pharm. 2010;390(2):242–249. | ||
Freitas C, Müller RH. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™) dispersions. Int J Pharm. 1998;168(2):221–229. | ||
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133. | ||
Suri SS, Fenniri H, Singh B. Nanotechnology-based drug delivery systems. J Occup Med Toxicol. 2007;2(1):16. | ||
Mogil JS. Animal models of pain: progress and challenges. Nat Rev Neurosci. 2009;10(4):283–294. | ||
Dionne RA, McCullagh L. Enhanced analgesia and suppression of plasma β-endorphin by the S (+)-isomer of ibuprofen. Clin Pharmacol Ther. 1998;63(6):694–701. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.