Back to Journals » Cancer Management and Research » Volume 6
Developments in Burkitt's lymphoma: novel cooperations in oncogenic MYC signaling
Received 9 August 2013
Accepted for publication 23 September 2013
Published 9 January 2014 Volume 2014:6 Pages 27—38
DOI https://doi.org/10.2147/CMAR.S37745
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Lindsay C Spender, Gareth J Inman
Division of Cancer Research, Medical Research Institute, Jacqui Wood Cancer Centre, University of Dundee, Ninewells Hospital and Medical School, Dundee, UK
Abstract: Burkitt's lymphoma (BL) is an aggressive disorder associated with extremely high rates of cell proliferation tempered by high levels of apoptosis. Despite the high levels of cell death, the net effect is one of rapid tumor growth. The tumor arises within the germinal centers of secondary lymphoid tissues and is identifiable by translocation of the c-MYC gene into the immunoglobulin gene loci, resulting in deregulation of the proto-oncogene. Many of the major players involved in determining the development of BL have been characterized in human BL cell lines or in mouse models of MYC-driven lymphomagenesis. Both systems have been useful so far in characterizing the role of tumor suppressor genes (for example, p53), prosurvival signaling pathways, and members of the B-cell lymphoma-2 family of apoptosis regulators in determining the fate of c-MYC overexpressing B-cells, and ultimately in regulating lymphoma development. Signaling through phosphoinositide (PI)3-kinase stands out as being critical for BL cell survival. Recurrent mutations in ID3 or TCF3 (E2A) that promote signaling through PI3-kinase have recently been identified in human BL samples, and new therapeutic strategies based on coordinately targeting both the prosurvival factor, B-cell lymphoma-XL, and the PI3-kinase/AKT/mammalian target of rapamycin (mTOR) signaling pathway to synergistically induced BL apoptosis have been proposed. Now, engineering both constitutive c-MYC expression and PI3-kinase activity, specifically in murine B-cells undergoing the germinal center reaction, has revealed that there is synergistic cooperation between c-MYC and PI3-kinase during BL development. The resulting tumors phenocopy the human malignancy, and acquire tertiary mutations also present in human tumors. This model may, therefore, prove useful in further studies to identify functionally relevant mutational events necessary for BL pathogenesis. This review discusses these cooperating interactions, the possible influence of BL tumor-associated viruses, and highlights potential new opportunities for therapeutic intervention.
Keywords: Burkitt's lymphoma, c-MYC, PI3-kinase, AKT, mTOR, Epstein–Barr virus
Introduction
Molecular pathology of Burkitt’s lymphoma
Burkitt’s lymphoma (BL) was originally identified by Dr Denis Burkitt in 1958 as an aggressive tumor of the jaw found in a cohort of children living in malarial areas of Africa (endemic BL).1 Today, this lymphoma is subdivided into three different subclasses with some distinct pathological features evident in each subtype. The second subgroup, sporadic BL, accounts for a high proportion of western pediatric lymphoma cases. These commonly arise within the ileum, but progress to involve the bone marrow or blood where circulating lymphoma cells are evident. The third subgroup is associated with immunodeficiency, either as a consequence of human immunodeficiency virus (HIV) infection, or as a result of immunosuppressive therapy following organ transplantation. BL accounts for approximately 30% of those lymphomas related to HIV infection. Concurrent infection with the γ-herpesvirus, Epstein–Barr virus (EBV) is found in the majority of endemic BL cases, but EBV-positive lymphoma cells are also detected, to a variable degree, in sporadic (approximately 15%) and immunodeficiency-associated cases (between 40% and 50%) depending upon the demographic.2 The common features of all subtypes are that they arise from differentiating B-cells within germinal center (GC) reactions and carry somatic hyper-mutations of the B-cell receptor (BCR) immunoglobulin (Ig) heavy chain variable region, indicative of this site of origin. Histologically, the lymphomas have a characteristic “starry sky” appearance and are comprised of densely packed, CD10/CD20/IgM-positive cells with a highly proliferative Ki-67-positive fraction of greater than 95%. Unlike follicular lymphoma, BLs are typically negative for the antiapoptotic protein, B-cell lymphoma (BCL)-2, again indicative of the cell of origin. All three subtypes also have a common genetic abnormality that is critically required for tumorigenesis. Deregulation and overexpression of the proto-oncogene c-MYC in BL is a result of translocation of the c-MYC gene on chromosome 83,4 to either the heavy, light, or kappa chain Ig loci (t[8;14], t[2;8], or t[8;22] translocation) with t(8;14) being the most frequently observed. How malaria and EBV infections contribute to BL pathogenesis is not entirely understood, but they presumably help to establish a cellular- or micro-environment conducive for c-MYC translocation and survival of the c-MYC overexpressing B-cell, eventually leading to lymphoma development. Their involvement in BL pathogenesis may well involve establishing a state of chronic B-cell proliferation and activation (as reviewed in the case of malaria by Moormann et al).5 The extraordinary ability of EBV to drive B-cell proliferation (interestingly, also involving a similar increase in c-MYC expression) while simultaneously abrogating apoptosis has also been reviewed elsewhere.6 This review will discuss both the effects of oncogenic c-MYC deregulation in BL cells, and the subsequent cooperating events required for BL tumorigenesis.
c-MYC
The proto-oncogene c-MYC is commonly deregulated in human cancers. It is a member of the helix–loop–helix leucine zipper family of nuclear transcription factors (TCFs), regulating the expression of a whole host of genes (and micro ribonucleic acids [micro RNAs])7,8 involved in signal transduction, cell cycle regulation, metabolism, apoptosis, cell adhesion, and protein biosynthesis. The carboxyl-terminus of the 430aa protein contains a helix–loop–helix deoxyribonucleic acid (DNA)-binding domain. The dimerization domain mediates heterodimerization with MYC associated factor X (MAX), and this complex formation is required for the conversion of MYC into an active polypeptide capable of binding enhancer box (E-box) target DNA sequences (5’-CACGTG-3’).9,10 MYC/MAX activity may itself be inhibited by the competitive binding of MAX by MAD1.11
Regulation of c-MYC expression is complex, occurring both at the level of gene transcription from one of four potential transcriptional promoters, and also via posttranslational modifications affecting protein stability. Moreover, c-MYC promoters P1 and P2 lie in the noncoding exon 1 of the gene, upstream of the coding exons 2 and 3. The vast majority (80%–90%) of MYC in normal cells is transcribed from P2; however, a switch to P1 promoter usage is observed following gene translocation when transcription falls under the control of Ig enhancer elements.12 The normal c-MYC allele is silent in BL.13,14 The c-MYC protein itself is normally degraded very rapidly, having a half-life of only 20–30 minutes. MYC is usually difficult to detect by Western blot analysis in normal cells, however, high protein expression levels are observed (and tolerated) in BL cell lines which necessitates mutational events and crosstalk with other signaling pathways (which will be discussed).
Functionally, in normal cells, c-MYC has an important role in increasing the mass of a cell prior to its division – a process required to maintain the size of the two resulting daughter cells. Hypofunctioning MYC in drosophila, results in a small-body phenotype (minutes) caused by decreased cell size (as opposed to fewer cell numbers).15 This phenotype correlates with a reduction in the expression of ribosomal protein genes required for protein synthesis.16 MYC expression also subsequently promotes the cell division through the upregulation of cyclin D, E2F, and the cyclin-dependent kinase (CDK)4,17 while repressing genes involved in cell cycle arrest (p27, p15, p21, p57, growth arrest and DNA damage [GADD]45, GADD34, and GADD153).
Deregulation of MYC in BL is undoubtedly a potent promoter of lymphocyte proliferation; however, overexpression of this proto-oncogene also induces powerful antiproliferative, apoptotic stress responses (Figure 1), which ultimately must be overcome for lymphomagenesis.
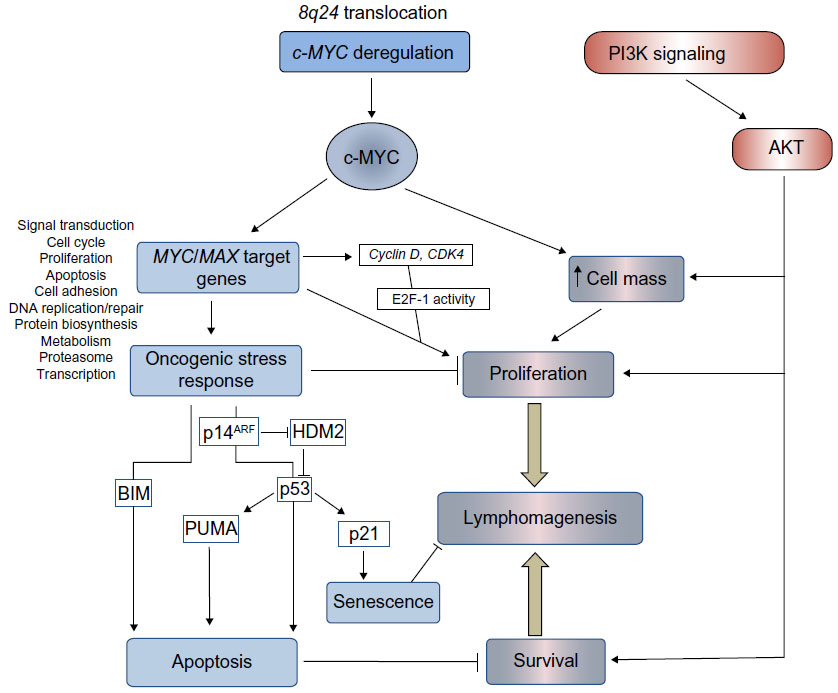 | Figure 1 Pathways regulating proliferation, survival, and cell death in BL. |
Functions of c-MYC in BL
The role of MYC in BL development, as well as the tumor suppressor pathways employed in an attempt to control lymphocyte proliferation, have been studied in depth using various experimental approaches. These include the analysis of both established BL cell lines and the engineered c-MYC-inducible cell line; P493-6, as well as the study of mouse models of MYC-driven lymphomas (Eμ-Myc and iMYCEμ).18
It is clear that continuous MYC activity is required for BL lymphomagenesis. Pharmacological inhibition of MYC/MAX heterodimer formation and MYC function in BL cell lines (using 10058-F4; Sigma-Aldrich, St Louis, MO, USA)19 has demonstrated conclusively that c-MYC is essential for BL cell proliferation20,21 and survival.22 Using alternative approaches, conditional MYC expression systems have also been analyzed both in vitro and in vivo. Conditional activation of MYC in the P493-6 cell line, which contains an estrogen-regulatable estrogen receptor–c-MYC fusion protein, demonstrated that in BL cells (as in normal cells), c-MYC regulates cell growth through an increase in metabolism and protein synthesis. Following serum starvation, cell growth and division become uncoupled since the starved cells increase in size, but are unable to progress through the cell cycle. Proliferation itself is, therefore, dependent on serum factors.23,24 Conditional Tet-off MYC expression in murine lymphoid cells has also demonstrated that loss of transgenic MYC expression following treatment with doxycycline results in a G1 cell cycle arrest and apoptosis of the MYC-driven lymphoma cells.25 MYC, however, does not appear to play a significant role in driving cells through stages of B-cell development since lymphomas in Eμ-MYC or MYC insertion iMYCEμ mice arise at the pre-B-cell or mature stages (a deviation from a faithful phenocopy of the GC-derived human disease). More mechanistic studies have examined the transcriptome associated with BL and MYC overexpression, either using the conditional systems mentioned above,26–28 or by genome-wide chromatin immunoprecipitation analysis of MYC DNA-binding sites in BL cell lines.29,30 What is evident from these studies is that MYC deregulation establishes a generalized increase in metabolic activity, gene transcription, and protein synthesis in the afflicted cell, rather than selective upregulation of certain cancer-causing genes (Figure 1). Since many of the MYC target genes are TCFs themselves,29 waves of time-dependent transcription are established through direct and indirect target gene regulation.31 The critical question to ask in order for us to understand how lymphomas develop following c-MYC translocation is what cooperating genetic alterations and mutations enable a cell to tolerate such extensive transcriptional deregulation and oncogene-induced stress?
Cooperating mutations
Determining which cooperating mutations can promote the survival of MYC-driven lymphoma may help in the identification of more selective targeted therapies. Genomic analysis has revealed that BLs are genomically heterogeneous, with 70 genes identified as being recurrently mutated (and as many as 17 mutated per lymphoma).32 Recent advances in our understanding of these mutations will be discussed in the following sections.
Mutations in MYC
Levels of c-MYC, even in proliferating normal cells, are usually relatively low. Since MYC has a short half-life,33 any change in the transcriptional/translational output of the gene would therefore be “sensed” rapidly by the cell – a sensible precaution for such a potent metabolic and transcriptional activator. BL cells, which are dependent on continuous MYC expression for survival, select for cooperating events, which have the effect of increasing the availability of the transforming protein. These may involve crosstalk with activated signaling pathways (such as phosphoinositide 3-kinase [PI3K], discussed later) or mutations within MYC itself. The amino-terminus transactivation domain of c-MYC contains two conserved, functionally critical MYC family regions called box 1 and box 2.34 Mutations within box 2 identified within the BL samples affect the transforming ability of MYC and its ability to promote apoptosis.35 Box 1 contains phosphorylation sites involved in the proteolysis of c-MYC by the ubiquitin–proteasome pathway.36 Residues within this region are mutated in approximately 20% of BLs, especially at the hotspot residues 39, 57, and 58. The threonine 58 (Thr58) to alanine mutation results in inefficient ubiquitination, a decrease in proteasomal degradation, and thus, an increase in protein stability (Figure 2).37–39 Thr58 is a target of glycogen synthase kinase (GSK)3β phosphorylation, an event that is dependent on phosphorylation of Serine (Ser)62 and the proline residue at position 57. Therefore, any mutation of Proline (Pro)57 would also inhibit Thr58 modification.40 The observation that the Thr58 mutation of c-MYC and the inactivating mutations of the p53 tumor suppressor pathway are mutually exclusive in BL, however, also implies that the mutation of MYC has a function in the avoidance of oncogene-induced apoptotic responses.
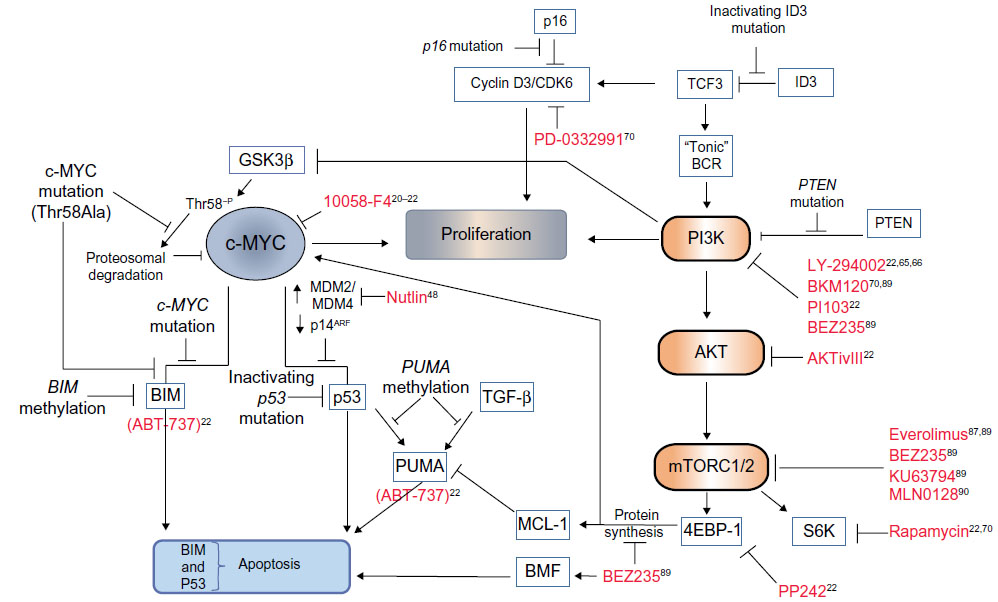 | Figure 2 Cooperating mutational events in MYC-driven lymphomagenesis and novel therapeutic opportunities. |
Avoiding oncogene-induced stress responses
Various mechanisms for avoiding such responses have been identified in BL and are represented schematically in Figure 2. Analysis of the murine lymphoma model indicates that acute MYC expression while inducing cell proliferation also induces apoptosis, provided that two requirements are met – the first being the activation of the p53 tumor suppressor pathway.
To activate the p53 pathway, p53 is stabilized through MYC/Forkhead box protein (FOXO) transcription of the CDKN2A locus encoding p14ARF (named p19ARF in mice),41 which inhibits degradation of p53 by mouse double minute (MDM)2 homolog. The resulting induction of apoptosis42 and senescence43 in premalignant Eμ-Myc B-cells is rate limiting for lymphoma development. As might be predicted, p53 is frequently a target for mutation in BL.44,45 Elevated levels of MDM4,46 MDM2 (called human double minute 2 homolog in humans) or loss of p14ARF expression have also been detected in BL cells carrying wild type p53,47 which would also have the effect of inactivating p53 tumor suppressor function. The activity of p53 may be reactivated in cases of elevated MDM2/4 expression using inhibitors of MDM2 activity to induce apoptosis.48
The second requirement for oncogene-induced apoptosis is the induction of BIM. BIM is a pro-apoptotic Bcl-2 homology 3 (BH3)-only member of the BCL-2 family, which acts via the mitochondrial membrane proteins Bcl-2 associated X protein (BAX) and Bcl-2 homologous antagonist/killer (BAK) to induce intrinsic mitochondrial apoptosis (Figure 2). The loss of a single allele of BIM in Eμ-MYC mice accelerates the development of lymphomas.49 This decision point50 where both p53 and the mitochondrial apoptotic machinery must be engaged for commitment to apoptosis is presumably a safeguard to ensure cell destruction only occurs when absolutely necessary. Lymphoma development applies a selective pressure such that at least one of these pathways must be inactivated. The evidence suggests that a loss of function of one of the arms of the stress response precludes any need for mutational inactivation of the other.51
If, for example, BAX is experimentally inactivated (BAX activity being required for the BIM-induced apoptosis), the selection of p53 mutations in MYC-driven lymphoma is circumvented since the intrinsic pathway is now nonfunctional.52 Similarly the MYC Thr58 mutation, as discussed above, is seen only in BL with an intact p53 pathway, and so it must provide a function that is not needed in the context of inactive p53. Analysis of lethally irradiated mice reconstituted with stem cells transfected with wild type or mutant MYC showed that this mutation, in fact, disables the ability of MYC to induce transcription of BIM, subsequently negating the intrinsic apoptosis arm of the oncogene-induced stress response. Consequently, only cells carrying wild type MYC developed inactivating p53 mutations. In biopsies of EBV-positive BLs, the BIM promoter may also be methylated at CpG dinucleotides, suggesting that epigenetic silencing of BIM could play an important role in tumorigenesis.53,54
Interestingly, p53-upregulated modulator of apoptosis (PUMA), a BH3 protein functionally akin to BIM, is also involved in the p53-induced apoptotic response.55 While this protein is not mutated in BL, it may be subject to decreased expression through promoter methylation – even in tumors carrying mutant p5356 – suggesting that there are p53-independent regulators of PUMA active in BL. We recently showed that PUMA is a downstream effector of a transforming growth factor (TGF)-β-induced apoptosis pathway active in both human BL cell lines and murine Eμ-MYC-derived lymphoma cells.57 TGF-β also negatively regulates the BL survival factor, BCL-XL.58 The selective pressure used to reduce PUMA expression in lymphomas, therefore, may arise from either the induction of p53, or through activation of the TGF-β tumor suppressor pathway (Figure 2).
So far, the studies mentioned describe the impact of genetic changes directly affecting two rate-limiting tumor suppressor pathways acting through p53 and intrinsic apoptosis. There is, however, evidence to implicate mutational changes in other signaling pathways that indirectly impact on these tumor suppressor pathways. For instance, further mutations have been identified in BL samples which upregulate signaling through PI3K. This signaling pathway has a critical role in cooperating with MYC during lymphomagenesis and promoting BL survival. The involvement of PI3K signaling in BL pathogenesis and opportunities for therapeutic intervention will be discussed.
The role of PI3K: MYC is not the only one “AKT”ing up to cause BL
BL development has only recently, for the first time, been modeled faithfully in mice by Sander et al,59 who investigated the effects of coordinately targeting both c-MYC and constitutively active PI3K. The idea that PI3K activity may be required for survival of BL cells harboring high levels of c-MYC expression was certainly not new, given that there have been numerous previous studies linking c-MYC-mediated transformations with the requirement for PI3K activity.60–64 The dependence of human BL cells on PI3K activity for proliferation and survival was also demonstrated by Curnock and Knox65 over a decade ago using PI3K inhibitors, and by others since.22,66 However, the new mouse model advances our understanding in that MYC and PI3K activity were targeted specifically to cells undergoing the GC reaction. Thus, the model more accurately recapitulates oncogenic processes occurring at the site of origin of BL, and these data unequivocally link PI3K signaling with MYC as cooperating factors in the development of BL. The lymphomas that formed in this model were a phenocopy of human tumors in terms of histology, cell surface marker expression, and ongoing somatic hypermutations of Ig-variable regions (indicative of GC B-cells). Importantly, the tumors exhibited gene and protein expression profiles similar to human BL, which distinguished them from those seen in diffuse large B-cell lymphoma (DLBCL). The tumors also developed tertiary stabilizing mutations in cyclin D3, an important regulator of GC B-cell proliferation,67,68 and frequently detected in BL samples.69 In addition to this advance, however, there have been exciting new discoveries as to how PI3K might be activated in BL, and what downstream effectors could be targeted effectively in new therapeutic strategies.
Significantly, Schmitz et al70 have identified recurrent mutations within approximately 70% of BL that increase signaling through PI3K. RNA sequencing on sporadic BL and BL cell lines revealed monoallelic mutations within TCF3 (also called E2A), biallelic mutations within its highly expressed negative regulator Inhibitor of DNA binding-3 (ID3), as well as infrequent (7%) mutations of the inhibitory phosphatase, PTEN (see Figure 2). In normal B-cells, E2A is involved in antigen-dependent B-cell responses and is expressed within the dark zone of GCs.71 It is itself a target for translocation events in B-cell acute lymphoblastic leukemia. The mutant transcription factor 3 (TCF3) proteins identified in BLs escape from the inhibitory effects of the ID proteins to upregulate expression of the heavy and light Ig chains of the BCR. The authors suggest a dependency of BL cells on “tonic” BCR signaling for survival of the tumor cells. Such so-called tonic BCR signaling is a state of low-level BCR activation required for the survival of resting mature B cells in vivo. It involves constitutive signaling via Syk, but does not require binding of the BCR to its cognate antigen (antigen-independent signaling). Like those seen in the Sander et al59 mouse model, mutations in BL samples were also identified within the TCF3 target gene cyclin D3. Furthermore, using the CDK4/6 inhibitor PD 0332991 the study demonstrated that the BL cells were dependent on cyclin D3/CDK6 activity (regardless of the mutational status of the cyclin D3 gene), and that the cyclin D3 mutations augmented cell cycle progression. Similar recurrent ID3 mutations were independently identified in BL (but notably not in diffuse large BCL also carrying MYC translocations);32,72 however, no global gene expression differences between cells carrying wild type or mutant ID3 were evident in this study.72
Interestingly, Love et al,32 in addition to identifying ID3 mutations in BL, also identified recurrent mutations in PI3KR1. The class 1 PI3Ks are comprised of a heterodimer made up of a catalytic subunit (p110α, p110β, or p110δ) and one of three regulatory subunits (p85α, p55α, and p50α, which are all derived from the same gene, PI3KR1). It is noteworthy that the p55α regulatory subunit is induced following infection of primary B-cells with the BL-associated virus EBV, and is required for the survival and proliferation of lymphoblastoid cell lines.73 However, the functional significance of the mutations in PI3KR1, or the differential induction of regulatory subunits by EBV, have not been investigated in the context of lymphoma and warrant further study.
As in other cancer types,74 signaling through PI3K may also be involved in stabilizing MYC through regulation of GSK3β activity.75 The MYC p-Thr58 modification, mediated by GSK3β and required for MYC degradation, can be blocked via a PI3K-dependent inhibitory phosphorylation of GSK3β on Ser9. Potentially, constitutive PI3K activation in BL carrying wild type MYC would help promote its stability and may contribute to its tumorigenic effects. In addition, PI3K/AKT activation in the context of MYC-induced transformation interferes with the oncogene-induced apoptotic response, again through an inhibitory modification, but this time, by phosphorylation of FOXO TCFs downstream of MYC. Although described in an artificial in vitro MycER system, inhibition of FOXO activity by AKT is required for MYC-induced transformation and proliferation,64 and it has been reported that PI3K/AKT and MYC together cooperatively repress PUMA and GADD45a expression by blocking their FOXO-dependent transcription (independently of PUMA induction by p53).76 If inhibition of FOXO-mediated transcription by PI3K/AKT signaling also occurs in MYC-driven lymphomas, it could potentially contribute to some of the cooperation seen between MYC and PI3K. Indeed, using a dominant negative FOXO protein to inhibit FOXO transcription accelerates lymphomagenesis in Eμ-MYC mice by attenuating MYC-induced apoptosis.41 In this case, the authors attribute the effect to a loss of transcription of the p53 regulator p19ARF, which correlated with reduced p53 and PUMA induction. In either event, posttranslational modifications of AKT substrates involved in oncogene-induced apoptosis are likely, at least in part, to account for the cooperative effects of MYC and AKT seen during lymphomagenesis. The alternative potential prosurvival mechanisms of AKT, which could be involved in subverting MYC-induced apoptosis include phosphorylation of HDM2, which increases its ligase activity and results in enhanced degradation of p53,77 blocking caspase activity,78 and increasing expression of prosurvival BCL-2 family members, such as induced myeloid leukemia cell differentiation protein MCL-1 (discussed in the following section).
The role of the AKT substrate mTOR and therapeutic opportunities
In a recent in vitro mechanistic study using selective pharmacological inhibitors to treat BL cell lines, we analyzed which effectors downstream of PI3K are critical for BL survival.22 In agreement with previous work,66 apoptosis of BL cell lines is induced by the PI3K inhibitor (LY-294002) and also by the dual PI3K/mTOR inhibitor PI103. The AKT inhibitor (AKTi-VIII) also induces BL apoptosis as a single agent, confirming that PI3K signaling via AKT is important for BL survival. Data on Eμ-MYC mice treated with the allosteric AKT inhibitor MK-2206 have now also demonstrated that AKT activity is required for lymphoma progression in vivo.79 However, further analysis using inhibitors of the downstream effectors of AKT activity have identified functions of mTOR in both BL survival and chemoresistance.
It should be noted that mTOR exists as two complexes: mTORC2 (acting upstream of AKT), and the AKT substrate mTORC1, which regulates the AKT effectors p70 S6-kinase/S6 ribosomal protein, and 4E-BP1/eIF4E. Using PP242, an active site adenosine triphosphate-competitive inhibitor of both mTORC1 and mTORC2, as well as low doses of rapamycin (an inhibitor of mTORC1), we were able to distinguish between the rapamycin-sensitive and rapamycin-insensitive effects80–83 of mTORC1 signaling in order to dissect the critical pathway components. PP242 inhibited phosphorylation of both S6 ribosomal protein and 4E-BP1, whereas rapamycin only inhibited phosphorylation of the S6 ribosomal protein. These studies revealed that the proapoptotic effects of PI3K/AKT/mTOR inhibitors correlated with an inhibition of 4E-BP1/eIF4E function, suggesting that inhibition of cap-dependent protein translation mediates the apoptotic effect in BL cells. (It is worth noting here that higher doses of rapamycin can inhibit both S6K and 4E-BP1 and, therefore, do not always discriminate between different mTOR functions in every study reported.) It is possible that activation of the S6-kinase by mTOR could additionally have a positive effect on MYC-mediated transcription through the phosphorylation and degradation of the MYC inhibitor, MAD1,84 but in terms of 4E-BP1 function, it is interesting to note that the activity of eIF4E has been previously reported to rescue cells from MYC-induced apoptosis.85 We found that inhibition of the PI3K/AKT/mTOR signaling pathway in BL cells decreased the expression of both c-MYC and MCL-1 (a prosurvival member of the BCL-2 family). Both proteins, which are short-lived, require continual protein synthesis for the maintenance of expression levels such that the inhibition of cap-dependent translation would rapidly affect their level of expression. In support of these findings, enhanced mTORC1 activity in mice (in the Eμ-MYC/TSC2+/– model) accelerates MYC-driven oncogenesis through lower levels of apoptosis and increased mTORC1-dependent MCL-1 expression.86 In addition, targeting mTORC1 with Everolimus (RAD001; Novartis AG, Basel, Switzerland), an inhibitor that is now being investigated in clinical trials, both protected Eμ-MYC mice from lymphoma development and induced regression of preformed lymphoma.87 Further evidence to support the conclusion that 4E-BP1 activity is important for lymphoma survival has subsequently been reported using the pan PI3K/mTOR inhibitor BEZ235 in the Eμ-MYC murine model. In this case, apoptosis was reported to be dependent on upregulation of the BH3-only protein Bcl-2 modifying factor (BMF), which is induced in response to inhibition of 4E-BP1/eIF4E mediated 5’ cap-dependent protein translation.88,89 Furthermore Eμ-MYC lymphomas have been shown to have elevated levels of 4E-BP1 phosphorylation on mTOR-specific sites (Thr37/46) and the mTOR active site inhibitor MLN0128 was shown to induce apoptosis in the BL Raji cell line.90
A promising outcome of this research then, in terms of identifying potential strategies for the treatment of BL, is that mTOR activity is an attractive target for therapeutic intervention. An important finding was that the combination of PI3K, AKT, or mTOR inhibitors act synergistically to induce apoptosis in combination with the BH3-mimetic compound ABT-737.22 ABT-737 acts like BIM or PUMA to inhibit BCL-XL function and induce intrinsic mitochondrial apoptosis (Figure 2). PI3K inhibition rendered previously resistant BL cells sensitive to the effects of ABT-737, indicating that PI3K can induce chemoresistance to agents that target one of the critical oncogene-induced apoptosis pathways. These data also imply that MCL-1 inhibitors could be effective in combination with BH3 mimetics or activators of wild type p53 as a therapeutic strategy in BL. Such combination therapies may offer significant advantages over current treatment regimens by reducing drug-associated toxicity.
The impact of Epstein–Barr virus on c-MYC and PI3K
EBV persistently infects greater than 90% of the population and remains, in most cases, benign for life. In some individuals, however, EBV infection is clearly associated with the development of BL. It is not entirely clear how EBV infection may contribute to disease pathogenesis but, presumably, EBV infection creates the right environment for cells to undergo, and perhaps more significantly to survive, the process of MYC gene translocation. How EBV may complement the activation of MYC to promote pathogenesis has been reviewed comprehensively elsewhere.91 However, in light of new studies, which have now established that the PI3K/AKT/mTOR pathway is a critical partner in MYC-driven lymphomagenesis, it is worth revisiting some aspects of EBV biology that may impact on these pathways. It is perhaps no coincidence that infection with this tumor-associated virus shows remarkable mimicry of many of the effects of the genomic mutational events recently described in BL.
Following infection, an initial phase of B-cell activation and proliferation is driven by viral genes and orchestrated by the viral TCF Epstein–Barr nuclear antigen (EBNA)-2.92 Significantly, one of the main cellular genes targeted by EBNA-2 for this purpose is c-MYC.93,94 In addition, EBNA-2 and two EBV-encoded membrane proteins activate the PI3K pathway. EBNA-2 appears to induce phospho-AKT via induction of the microRNA, miR-21 (probably through the posttranscriptional processing rather than by the induction of the primary transcript).95 Meanwhile, latent membrane protein (LMP)-2A, a mimic of functional BCRs, is expressed by EBV potentially to inhibit negative selection. LMP-2A constitutively activates PI3K,96,97 thus providing the cell with the “tonic” BCR-like survival signals that are evident following TCF3 and ID3 mutation in BL. Interestingly, LMP-2A is often detected in tumor biopsies of EBV-related malignancies, although LMP-2A is down-regulated during later stages of viral latency and can, therefore, play no further role in promoting cell survival. It seems likely then, that LMP-2A may have a potential role in augmenting the survival of cells98 carrying tumorigenic genetic abnormalities within the GC, possibly by PI3K/AKT-mediated upregulation of BCL-XL.96 A second EBV encoded membrane protein, LMP-1, functionally mimics activated CD40,99 requires no ligand, and is capable of transforming rodent fibroblasts.100 LMP-1 signals via a number of intracellular pathways such as nuclear factor-kappa B,101 mitogen-activated protein kinase,102 Janus kinase,103 but also, through the binding of its cytoplasmic tail to the p85 regulatory subunit, which signals via PI3K to AKT.104,105 Thus, through the expression of EBNA-2, LMP-1, and LMP-2A, EBV provides similar survival and transforming signals that have been shown to cooperatively induce malignant transformations in BL.
There are also numerous viral products that can block the ability of infected cells to mount an effective intrinsic apoptotic response to oncogene-induced stress (reviewed in Spender and Inman).6 EBV blocks this response either by directly interfering with BAX/BAK function (mediated by the viral genes LMP-1, BHRF-1, and BALF-1), inducing expression of other prosurvival BCL-2 family members such as MCL-1106 and BFL-1 (induced by LMP-1),107 or by inhibiting the function or expression of the BH3-only proteins PUMA (BHRF-1, miR-BART5) and BIM (EBNAs-3A and 3C).108,109 Inhibiting the function of BIM and PUMA or inducing MCL-1 are, as discussed previously, also the consequence of promoter methylation, MYC mutation or increased mTORC activity, all described in BL.
In light of the evidence that EBV infection appears to recapitulate the events required for BL pathogenesis (ie, MYC induction, activation of PI3K, and inactivation of the intrinsic apoptosis pathway), one might expect that p53 oncogene-induced stress response would also be targeted. Indeed, two potent viral transcriptional repressors (EBNA-3A and 3C) are implicated in the repression of p14ARF,110 which is required for p53 induction. There are also data reporting that EBV infection deubiquitinates and stabilizes the p53-negative regulator MDM2 (a function of EBNA-3C),111 or mediates p53 degradation independently of MDM2 (BZLF-1),112 thus reducing p53 expression. The formation of EBNA-leader protein (LP)/MDM2/p53 complexes has also been proposed to selectively block p53-mediated transcription (of p21) and provide an explanation as to how rapidly proliferating infected cells tolerate high levels of wild type p53 without succumbing to p53-induced cell cycle arrest.113
One deviation from the hypothesis that EBV infection exactly mimics the changes seen in BL applies to the expression of cyclins. Mutation and expression of cyclin D3 is associated with BL pathogenesis; however, cyclin D3 expression is rarely seen in EBV-infected cells with an activated phenotype.114 The viral TCF EBNA-2 does, however, indirectly induce cyclin D2.94 Cyclin D2 can compensate for loss of cyclin D3, at least in some circumstances,115 so perhaps cyclin D2 upregulation in the context of EBV infection has similar net pro-proliferative effects as cyclin D3 activity within BL.
Taken together, the data suggest that EBV infection may functionally mimic the BL cellular environment, perhaps enabling the genomic aberrations associated with BL to establish themselves during the tumorigenic process. The latent viral gene expression program that persists within BL is very restricted, involving only EBNA-1, which is required for viral genome maintenance, and two small RNA species called Epstein-Barr virus encoded small RNAs (EBER)1 and EBER2, whose potential targets are unknown. Both EBNA-1 and the EBERs, however, also appear to have a role in preventing BL cell apoptosis,116 which could help promote lymphomagenesis. It would also be relevant to study the significance of infection with different EBV subtypes and their association with BL. EBV type 1 (also called type A) differs from type 2 (or type B) predominantly through sequence variation of the EBNA-2 gene. While the type A virus is much more efficient than the type B virus at driving proliferation of EBV-infected lymphoblastoid cell lines in vitro117 (potentially via better induction of LMP-1 expression),118 in some areas of endemic BL, infection with the type 2 strain predominates.119 Differential regulation of certain cell genes does occur in response to type 1 and type 2 EBNA-2 expression,118 but it is presently unclear whether these genes may significantly affect BL pathogenesis. The new models of BL pathogenesis targeting the cell of origin may help to decipher the cooperating roles of mutated and viral genes in BL development – in particular, which EBV genes are actually required, and when they are required, to contribute to BL development. Such studies will also provide excellent model systems in which to test new therapeutic strategies.
Acknowledgments
Research performed in our laboratory described in this review was funded by an Association for International Cancer Research Fellowship and by Cancer Research UK.
Disclosure
The authors report no conflicts of interest in this work.
References
Burkitt D. A sarcoma involving the jaws in African children. Br J Surg. 1958;46(197):218–223. | |
Crawford DH. Biology and disease associations of Epstein-Barr virus. Philos Trans R Soc Lond B Biol Sci. 2001;356(1408):461–473. | |
Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79(24):7824–7827. | |
Taub R, Kirsch I, Morton C, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A. 1982;79(24):7837–7841. | |
Moormann AM, Snider CJ, Chelimo K. The company malaria keeps: how co-infection with Epstein-Barr virus leads to endemic Burkitt lymphoma. Curr Opin Infect Dis. 2011;24(5):435–441. | |
Spender LC, Inman GJ. Inhibition of germinal centre apoptotic programmes by epstein-barr virus. Adv Hematol. 2011;2011:829525. | |
Chang TC, Yu D, Lee YS, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40(1):43–50. | |
O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–843. | |
Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H. Sequence-specific DNA binding by the c-Myc protein. Science. 1990;250(4984):1149–1151. | |
Blackwell TK, Huang J, Ma A, et al. Binding of myc proteins to canonical and noncanonical DNA sequences. Mol Cell Biol. 1993;13(9):5216–5224. | |
Lüscher B. MAD1 and its life as a MYC antagonist: an update. Eur J Cell Biol. 2012;91(6–7):506–514. | |
Taub R, Moulding C, Battey J, et al. Activation and somatic mutation of the translocated c-myc gene in burkitt lymphoma cells. Cell. 1984;36(2):339–348. | |
ar-Rushdi A, Nishikura K, Erikson J, Watt R, Rovera G, Croce CM. Differential expression of the translocated and the untranslocated c-myc oncogene in Burkitt lymphoma. Science. 1983;222(4622):390–393. | |
Hayday AC, Gillies SD, Saito H, et al. Activation of a translocated human c-myc gene by an enhancer in the immunoglobulin heavy-chain locus. Nature. 1984;307(5949):334–340. | |
Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98(6):779–790. | |
Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7(3):295–302. | |
Bouchard C, Stellar P, Eilers M. Control of cell proliferation by Myc. Trends Cell Biol. 1998;8(5):202–206. | |
Park SS, Kim JS, Tessarollo L, et al. Insertion of c-Myc into Igh induces B-cell and plasma-cell neoplasms in mice. Cancer Res. 2005;65(4):1306–1315. | |
Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22(40):6151–6159. | |
Gomez-Curet I, Perkins RS, Bennett R, Feidler KL, Dunn SP, Krueger LJ. c-Myc inhibition negatively impacts lymphoma growth. J Pediatr Surg. 2006;41(1):207–211; discussion 207–211. | |
Sampson VB, Rong NH, Han J, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67(20):9762–9770. | |
Spender LC, Inman GJ. Phosphoinositide 3-kinase/AKT/mTORC1/2 signaling determines sensitivity of Burkitt’s lymphoma cells to BH3 mimetics. Mol Cancer Res. 2012;10(3):347–359. | |
Schuhmacher M, Staege MS, Pajic A, et al. Control of cell growth by c-Myc in the absence of cell division. Curr Biol. 1999;9(21):1255–1258. | |
Schlosser I, Hölzel M, Hoffmann R, et al. Dissection of transcriptional programmes in response to serum and c-Myc in a human B-cell line. Oncogene. 2005;24(3):520–524. | |
Marinkovic D, Marinkovic T, Mahr B, Hess J, Wirth T. Reversible lymphomagenesis in conditionally c-MYC expressing mice. Int J Cancer. 2004;110(3):336–342. | |
Schuhmacher M, Kohlhuber F, Hölzel M, et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 2001;29(2):397–406. | |
Ji H, Wu G, Zhan X, et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One. 2011;6(10):e26057. | |
Lin CY, Lovén J, Rahl PB, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151(1):56–67. | |
Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A. 2003;100(14):8164–8169. | |
Seitz V, Butzhammer P, Hirsch B, et al. Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PLoS One. 2011;6(11):e26837. | |
Fan J, Zeller K, Chen YC, et al. Time-dependent c-Myc transactomes mapped by Array-based nuclear run-on reveal transcriptional modules in human B cells. PLoS One. 2010;5(3):e9691. | |
Love C, Sun Z, Jima D, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44(12):1321–1325. | |
Hann SR, Eisenman RN. Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol Cell Biol. 1984;4(11):2486–2497. | |
Sakamuro D, Prendergast GC. New Myc-interacting proteins: a second Myc network emerges. Oncogene. 1999;18(19):2942–2954. | |
Kuttler F, Amé P, Clark H, et al. c-myc box II mutations in Burkitt’s lymphoma-derived alleles reduce cell-transformation activity and lower response to broad apoptotic stimuli. Oncogene. 2001;20(42):6084–6094. | |
Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14(19):2501–2514. | |
Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18(3):717–726. | |
Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol. 2000;20(7):2423–2435. | |
Bahram F, von der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95(6):2104–2110. | |
Hoang AT, Lutterbach B, Lewis BC, et al. A link between increased transforming activity of lymphoma-derived MYC mutant alleles, their defective regulation by p107, and altered phosphorylation of the c-Myc transactivation domain. Mol Cell Biol. 1995;15(8):4031–4042. | |
Bouchard C, Lee S, Paulus-Hock V, Loddenkemper C, Eilers M, Schmitt CA. FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev. 2007;21(21):2775–2787. | |
Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13(20):2658–2669. | |
Post SM, Quintás-Cardama A, Terzian T, Smith C, Eischen CM, Lozano G. p53-dependent senescence delays Emu-myc-induced B-cell lymphomagenesis. Oncogene. 2010;29(9):1260–1269. | |
Farrell PJ, Allan GJ, Shanahan F, Vousden KH, Crook T. p53 is frequently mutated in Burkitt’s lymphoma cell lines. EMBO J. 1991;10(10):2879–2887. | |
Vousden KH, Crook T, Farrell PJ. Biological activities of p53 mutants in Burkitt’s lymphoma cells. J Gen Virol. 1993;74(Pt 5):803–810. | |
Leventaki V, Rodic V, Tripp SR, et al. TP53 pathway analysis in paediatric Burkitt lymphoma reveals increased MDM4 expression as the only TP53 pathway abnormality detected in a subset of cases. Br J Haematol. 2012;158(6):763–771. | |
Lindström MS, Klangby U, Wiman KG. p14ARF homozygous deletion or MDM2 overexpression in Burkitt lymphoma lines carrying wild type p53. Oncogene. 2001;20(17):2171–2177. | |
Pujals A, Renouf B, Robert A, Chelouah S, Hollville E, Wiels J. Treatment with a BH3 mimetic overcomes the resistance of latency III EBV (+) cells to p53-mediated apoptosis. Cell Death Dis. 2011;2: e184. | |
Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004; 101(16):6164–6169. | |
Dang CV, O’donnell KA, Juopperi T. The great MYC escape in tumorigenesis. Cancer Cell. 2005;8(3):177–178. | |
Hemann MT, Bric A, Teruya-Feldstein J, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436(7052):807–811. | |
Eischen CM, Roussel MF, Korsmeyer SJ, Cleveland JL. Bax loss impairs Myc-induced apoptosis and circumvents the selection of p53 mutations during Myc-mediated lymphomagenesis. Mol Cell Biol. 2001;21(22):7653–7662. | |
Paschos K, Smith P, Anderton E, Middeldorp JM, White RE, Allday MJ. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 2009;5(6):e1000492. | |
Richter-Larrea JA, Robles EF, Fresquet V, et al. Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood. 2010;116(14):2531–2542. | |
Erlacher M, Labi V, Manzl C, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med. 2006;203(13):2939–2951. | |
Garrison SP, Jeffers JR, Yang C, et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol. 2008;28(17):5391–5402. | |
Spender LC, Carter MJ, O’Brien DI, et al. Transforming growth factor-β directly induces p53-up-regulated modulator of apoptosis (PUMA) during the rapid induction of apoptosis in myc-driven B-cell lymphomas. J Biol Chem. 2013;288(7):5198–5209. | |
Spender LC, O’Brien DI, Simpson D, et al. TGF-beta induces apoptosis in human B cells by transcriptional regulation of BIK and BCL-XL. Cell Death Differ. 2009;16(4):593–602. | |
Sander S, Calado DP, Srinivasan L, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22(2):167–179. | |
Rohn JL, Hueber AO, McCarthy NJ, et al. The opposing roles of the Akt and c-Myc signalling pathways in survival from CD95-mediated apoptosis. Oncogene. 1998;17(22):2811–2818. | |
Zhao JJ, Gjoerup OV, Subramanian RR, et al. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3(5):483–495. | |
Kumar A, Marqués M, Carrera AC. Phosphoinositide 3-kinase activation in late G1 is required for c-Myc stabilization and S phase entry. Mol Cell Biol. 2006;26(23):9116–9125. | |
Han SS, Yun H, Son DJ, et al. NF-kappaB/STAT3/PI3K signaling crosstalk in iMyc E mu B lymphoma. Mol Cancer. 2010;9:97. | |
Bouchard C, Marquardt J, Brás A, Medema RH, Eilers M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 2004;23(14):2830–2840. | |
Curnock AP, Knox KA. LY294002-mediated inhibition of phosphatidylinositol 3-kinase activity triggers growth inhibition and apoptosis in CD40-triggered Ramos-Burkitt lymphoma B cells. Cell Immunol. 1998;187(2):77–87. | |
Brennan P, Mehl AM, Jones M, Rowe M. Phosphatidylinositol 3-kinase is essential for the proliferation of lymphoblastoid cells. Oncogene. 2002;21(8):1263–1271. | |
Peled JU, Yu JJ, Venkatesh J, et al. Requirement for cyclin D3 in germinal center formation and function. Cell Res. 2010;20(6):631–646. | |
Cato MH, Chintalapati SK, Yau IW, Omori SA, Rickert RC. Cyclin D3 is selectively required for proliferative expansion of germinal center B cells. Mol Cell Biol. 2011;31(1):127–137. | |
Møller MB, Nielsen O, Pedersen NT. Cyclin D3 expression in non-Hodgkin lymphoma. Correlation with other cell cycle regulators and clinical features. Am J Clin Pathol. 2001;115(3):404–412. | |
Schmitz R, Young RM, Ceribelli M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490(7418):116–120. | |
Goldfarb AN, Flores JP, Lewandowska K. Involvement of the E2A basic helix-loop-helix protein in immunoglobulin heavy chain class switching. Mol Immunol. 1996;33(11–12):947–956. | |
Richter J, Schlesner M, Hoffmann S, et al; ICGC MMML-Seq Project. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012;44(12):1316–1320. | |
Spender LC, Lucchesi W, Bodelon G, et al. Cell target genes of Epstein-Barr virus transcription factor EBNA-2: induction of the p55alpha regulatory subunit of PI3-kinase and its role in survival of EREB2.5 cells. J Gen Virol. 2006;87(Pt 10):2859–2867. | |
Schild C, Wirth M, Reichert M, Schmid RM, Saur D, Schneider G. PI3K signaling maintains c-myc expression to regulate transcription of E2F1 in pancreatic cancer cells. Mol Carcinog. 2009;48(12):1149–1158. | |
van Weeren PC, de Bruyn KM, de Vries-Smits AM, van Lint J, Burgering BM. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB. J Biol Chem. 1998;273(21):13150–13156. | |
Amente S, Zhang J, Lavadera ML, Lania L, Avvedimento EV, Majello B. Myc and PI3K/AKT signaling cooperatively repress FOXO3a-dependent PUMA and GADD45a gene expression. Nucleic Acids Res. 2011;39(22):9498–9507. | |
Ashcroft M, Ludwig RL, Woods DB, et al. Phosphorylation of HDM2 by Akt. Oncogene. 2002;21(13):1955–1962. | |
Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282(5392):1318–1321. | |
Devlin JR, Hannan KM, Ng PY, et al. AKT signalling is required for ribosomal RNA synthesis and progression of Eμ-Myc B-cell lymphoma in vivo. FEBS J. Epub January 19, 2013. | |
Hsieh AC, Costa M, Zollo O, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17(3):249–261. | |
Dowling RJ, Topisirovic I, Alain T, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328(5982):1172–1176. | |
Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7(2):e38. | |
Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009;5(5):725–726. | |
Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A. 2008;105(18):6584–6589. | |
Li S, Takasu T, Perlman DM, et al. Translation factor eIF4E rescues cells from Myc-dependent apoptosis by inhibiting cytochrome c release. J Biol Chem. 2003;278(5):3015–3022. | |
Mills JR, Hippo Y, Robert F, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A. 2008;105(31):10853–10858. | |
Wall M, Poortinga G, Stanley KL, et al. The mTORC1 inhibitor everolimus prevents and treats Eμ-Myc lymphoma by restoring oncogene-induced senescence. Cancer Discov. 2013;3(1):82–95. | |
Grespi F, Soratroi C, Krumschnabel G, et al. BH3-only protein Bmf mediates apoptosis upon inhibition of CAP-dependent protein synthesis. Cell Death Differ. 2010;17(11):1672–1683. | |
Shortt J, Martin BP, Newbold A, et al. Combined inhibition of PI3K-related DNA damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood. 2013;121(15):2964–2974. | |
Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A. 2013;110(29):11988–11993. | |
Allday MJ. How does Epstein-Barr virus (EBV) complement the activation of Myc in the pathogenesis of Burkitt’s lymphoma? Semin Cancer Biol. 2009;19(6):366–376. | |
Sinclair AJ, Palmero I, Peters G, Farrell PJ. EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein-Barr virus. EMBO J. 1994;13(14):3321–3328. | |
Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, Kempkes B. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J Virol. 1999;73(5):4481–4484. | |
Spender LC, Cornish GH, Rowland B, Kempkes B, Farrell PJ. Direct and indirect regulation of cytokine and cell cycle proteins by EBNA-2 during Epstein-Barr virus infection. J Virol. 2001;75(8):3537–3546. | |
Rosato P, Anastasiadou E, Garg N, et al. Differential regulation of miR-21 and miR-146a by Epstein-Barr virus-encoded EBNA2. Leukemia. 2012;26(11):2343–2352. | |
Portis T, Longnecker R. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene. 2004;23(53):8619–8628. | |
Mancao C, Hammerschmidt W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood. 2007;110(10):3715–3721. | |
Swanson-Mungerson MA, Caldwell RG, Bultema R, Longnecker R. Epstein-Barr virus LMP2A alters in vivo and in vitro models of B-cell anergy, but not deletion, in response to autoantigen. J Virol. 2005;79(12):7355–7362. | |
Gires O, Zimber-Strobl U, Gonnella R, et al. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997;16(20):6131–6140. | |
Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43(3 Pt 2):831–840. | |
Izumi KM, Kieff ED. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc Natl Acad Sci U S A. 1997;94(23):12592–12597. | |
Roberts ML, Cooper NR. Activation of a ras-MAPK-dependent pathway by Epstein-Barr virus latent membrane protein 1 is essential for cellular transformation. Virology. 1998;240(1):93–99. | |
Eliopoulos AG, Young LS. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1). Oncogene. 1998;16(13):1731–1742. | |
Dawson CW, Tramountanis G, Eliopoulos AG, Young LS. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J Biol Chem. 2003;278(6):3694–3704. | |
Mainou BA, Everly DN Jr, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene. 2005;24(46):6917–6924. | |
Wang S, Rowe M, Lundgren E. Expression of the Epstein Barr virus transforming protein LMP1 causes a rapid and transient stimulation of the Bcl-2 homologue Mcl-1 levels in B-cell lines. Cancer Res. 1996;56(20):4610–4613. | |
Pegman PM, Smith SM, D’Souza BN, et al. Epstein-Barr virus nuclear antigen 2 trans-activates the cellular antiapoptotic bfl-1 gene by a CBF1/RBPJ kappa-dependent pathway. J Virol. 2006;80(16):8133–8144. | |
Clybouw C, McHichi B, Mouhamad S, et al. EBV infection of human B lymphocytes leads to down-regulation of Bim expression: relationship to resistance to apoptosis. J Immunol. 2005;175(5):2968–2973. | |
Anderton E, Yee J, Smith P, Crook T, White RE, Allday MJ. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt’s lymphoma. Oncogene. 2008;27(4):421–433. | |
Maruo S, Zhao B, Johannsen E, Kieff E, Zou J, Takada K. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc Natl Acad Sci U S A. 2011;108(5):1919–1924. | |
Saha A, Murakami M, Kumar P, Bajaj B, Sims K, Robertson ES. Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2. J Virol. 2009;83(9):4652–4669. | |
Sato Y, Shirata N, Kudoh A, et al. Expression of Epstein-Barr virus BZLF1 immediate-early protein induces p53 degradation independent of MDM2, leading to repression of p53-mediated transcription. Virology. 2009;388(1):204–211. | |
Kashuba E, Yurchenko M, Yenamandra SP, et al. Epstein-Barr virus-encoded EBNA-5 forms trimolecular protein complexes with MDM2 and p53 and inhibits the transactivating function of p53. Int J Cancer. 2011;128(4):817–825. | |
Pokrovskaja K, Ehlin-Henriksson B, Bartkova J, et al. Phenotype-related differences in the expression of D-type cyclins in human B cell-derived lines. Cell Growth Differ. 1996;7(12):1723–1732. | |
Mataraza JM, Tumang JR, Gumina MR, Gurdak SM, Rothstein TL, Chiles TC. Disruption of cyclin D3 blocks proliferation of normal B-1a cells, but loss of cyclin D3 is compensated by cyclin D2 in cyclin D3-deficient mice. J Immunol. 2006;177(2):787–795. | |
Brady G, Macarthur GJ, Farrell PJ. Epstein-Barr virus and Burkitt lymphoma. Postgrad Med J. 2008;84(993):372–377. | |
Rickinson AB, Young LS, Rowe M. Influence of the Epstein-Barr virus nuclear antigen EBNA2 on the growth phenotype of virus-transformed B cells. J Virol. 1987;61(5):1310–1317. | |
Lucchesi W, Brady G, Dittrich-Breiholz O, Kracht M, Russ R, Farrell PJ. Differential gene regulation by Epstein-Barr virus type 1 and type 2 EBNA2. J Virol. 2008;82(15):7456–7466. | |
Aitken C, Sengupta SK, Aedes C, Moss DJ, Sculley TB. Heterogeneity within the Epstein-Barr virus nuclear antigen 2 gene in different strains of Epstein-Barr virus. J Gen Virol. 1994;75(Pt 1):95–100. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.