Back to Archived Journals » Biosimilars » Volume 6
Development, safety, and efficacy of biosimilar adalimumab: the data so far
Authors Gabbani T, Deiana S, Bagnoli S, Annese V
Received 30 July 2015
Accepted for publication 18 November 2015
Published 29 January 2016 Volume 2016:6 Pages 1—7
DOI https://doi.org/10.2147/BS.S55900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Shein-Chung Chow
Tommaso Gabbani, Simona Deiana, Siro Bagnoli, Vito Annese
Emergency Department, Division of Gastroenterology 2, AOU Careggi University Hospital, Florence, Italy
Abstract: Biologics are defined as active substances derived from living cells or organisms with the aid of biotechnology methods. A similar biological medicinal product, commonly referred to as a biosimilar, is a copy version of an approved original biological medicine whose data protection has expired. The main advantage of biosimilars is to lower prices and create financial savings for health care systems. Adalimumab (Humira) is a human recombinant monoclonal antibody against tumor necrosis factor-α, licensed for the treatment of several rheumatologic diseases and inflammatory bowel diseases. The original biologic agent has worldwide sales of approximately 12.5 billion US dollars, but the patent will expire in 2016 for the US and in 2018 for Europe. Adalimumab biosimilar is already currently marketed and used in a clinical setting in India, but many other companies are testing adalimumab biosimilars in order to request authorization from the European Medicines Agency and the US Food and Drug Administration at the time of patent expiry. In this gold rush, the long-term safety and immunogenicity of these drugs are an issue, but the striking rise of pharmaceutical expenses makes these drugs highly attractive for health care systems and pharmaceutical companies.
Keywords: adalimumab, IBD, biosimilar, biologics, Crohn's disease
Market review
Biological drugs have represented a revolution in the treatment of patients with rheumatologic conditions and inflammatory bowel disease (IBD) since their appearance, allowing physicians to set and achieve more ambitious therapeutic targets.
Biological medicinal products (or biologics) are defined as active substances derived from living cells or organisms with the aid of biotechnology methods (recombinant DNA, controlled gene expression, antibody technologies).1 A similar biological medicinal product, commonly referred to as biosimilar, is a copy version of an approved original biological medicine whose data protection has expired.2
In the European Union (EU), biological medicines need to be authorized by the European Medicines Agency (EMA). The EMA has established a specific legal pathway for biosimilars approval in 2005, in which it is recognized that biosimilars cannot be identical to the primary compound, but must be similar to the original EU-approved molecules in terms of quality, safety, and efficacy.3
A total of 19 biosimilar medicines have been evaluated and 14 authorized in the EU currently. All authorized biosimilars can be divided in three product classes: human growth hormones, erythropoietin, and granulocyte colony-stimulating factors.4
Nowadays, use of biological agents is increasing, with new indications and increased patient demand. In 2013, it was already estimated that six of the world’s top ten selling drugs were biological drugs. Sales of biological drugs are expected to continue to grow at least twice the rate of sales of other molecules.5 All biologic drugs are expensive for the health care system, even though cost-efficacy analysis should take into account impact on patient, disease outcomes, and related expenses (ie, hospitalization, sick leaves).6
The main advantage of biosimilars is to create conditions for economic competition, and the latter notoriously means lower prices and financial savings for health care systems and might increase the number of patients in therapy. The size of the financial savings seems to depend on many factors, in particular the size of the market for biological drugs, and the price reduction that will be achieved. Both factors are difficult to estimate: nowadays, the market is not very large, but in the medium term, it could achieve many billions of euros. However, this figure is likely to be overestimated because patients in the meantime might move to new innovative medicines, or part of the savings could be turned into a wider usage. In addition, not for all biologic drugs, a biosimilar could be available. In the area of cost, it is problematic to make forecasts today, but certainly, the complexity and requirements of production for biosimilars (development, comparability exercise, clinical trials, etc) required for their access to the market probably will never permit a price reduction comparable to that of equivalent drugs (generics).
Chemically synthesized drugs are usually small molecules with well-known structures and low immunogenicity potential. Instead, biologic drugs are large molecules with a three-dimensional high complex structure. Biologic agents are engineered and produced by living cells, which define the structure and posttranslational modifications, and it is a major potential source of manufacturing heterogeneity; that is why, the production of biosimilars is associated with several variables. Molecular heterogeneity of biologics results from the complex interplay of the primary, secondary, and higher order structures. In addition, intra- and intermolecular interactions and posttranslational modifications can result in millions of chemical forms. The type and the degree of glycosylation have a strong influence on interaction with receptors, and such changes in the glycosylation pattern of biosimilars could have unpredictable effects on efficacy and safety (ie, immunogenicity).7 Moreover, the final biosimilar product is determined by the manufacturing process, but even after patent expiration, biosimilar companies do not have access to the manufacturing process of the original drug. Manufacture includes several steps: selection of the type of vector and the host cell expression system, the cell expansion procedure, the protein recovery mechanism, the purification process, and the formulation of the target protein into the drug.8
For these reasons, biosimilars are highly unlikely to be identical with their reference products and they require regulatory approval processes. Demonstration of comparability between biosimilars and the initial biologic drug product is therefore necessary.9 In contrast, given that the efficacy of the innovator biologic drug has already been established, the clinical studies required for approval of a biosimilar could be reduced compared with clinical trials required for approval of the first original biologic. In addition, the attraction of developing a biosimilar comes from the opportunity of getting the approval for all indications held by the original drug, the so-called extrapolation. These indications can be achieved with less extensive nonclinical data, and few clinical data related to only one or two indications. EMA, US Food and Drug Administration (FDA), and Health Canada have generally allowed the drug companies for extrapolation, based on a number of comparability exercises without clinical data for all indications.10
Anti-TNF agents in IBD
The most effective and used class of biologic drugs in IBD are anti-tumor necrosis factor (anti-TNF) agents with four biologics currently approved by the EMA and FDA. These are anti-TNF monoclonal antibodies: infliximab (IFX), adalimumab, golimumab, and certolizumab (the latter is approved only by the FDA). Other two biologics are anti-integrin antibodies: natalizumab, which has been authorized by the FDA for Crohn’s disease (CD) in 2008, and the new one, vedolizumab, recently approved in both Europe and US.
Regarding luminal inflammatory CD, a recent meta-analysis of all controlled trials with anti-TNF agents indicated that adalimumab, certolizumab pegol, and IFX are all efficacious for induction of remission.11 Adalimumab was superior to placebo in the induction of remission in CD patients naïve to IFX (Classic I: remission rate at 4 weeks: 36% vs 12% placebo, P<0.05)12 and in CD patients with secondary failure to IFX (GAIN: remission rate at 4 weeks: 21% vs 7% placebo, P<0.05).13 Patients with early CD (<2 years from diagnosis) had higher remission rates compared to those with a longer disease duration (CHARM: 60% vs 40%, P<0.05).
Although adalimumab was found to be superior to placebo in maintenance of clinical remission in CD patients at week 54 (CHARM: relative risk [RR] 3.28, 95% CI 2.13–5.06; CLASSIC 2: RR 1.82, 95% CI 1.06–3.13), in two studies, adalimumab was not much better than placebo in preventing relapse of quiescent luminal CD.14
Adalimumab is also licensed for the treatment of moderate or severely active ulcerative colitis (UC) in Europe from 2012. There are two placebo-controlled trials that have evaluated the efficacy of adalimumab in patients with moderately active UC despite conventional therapy. In one of them, adalimumab has also shown benefit over placebo for the induction of remission in IFX-naïve patients with moderately active UC (remission rate at week 8: 18.5% vs 9.2%, P=0.031).15 In the second trial,16 patients previously exposed to anti-TNF were also included; clinical remission was achieved significantly more with adalimumab than placebo at week 8 (16.5% and 9.3%, P=0.02) and week 52 (17.3% and 8.5%, P=0.01), with an higher clinical remission rate in patients naïve to anti-TNF. Furthermore, there are small case series that reported benefit of adalimumab in patients with active UC previously exposed to IFX.17–19 Finally, adalimumab allows steroid-free remission in 13.3% of patient at week 52.20
IFX biosimilar
In 2012, the first biosimilar agent of IFX CT-P13 (Inflectra – Hospira, Lake Forest, IL, USA; and Remsima – Celltrion, Incheon, South Korea) has been authorized for the treatment of autoimmune diseases and IBD in India and South Korea.21 Data on efficacy and safety come from two trials. In the first (randomized, Phase II, double-blind) study, efficacy and safety of CT-P13 and IFX 5 mg/kg were compared in 250 patients with ankylosing spondylitis. The pharmacokinetic profile of CT-P13 was judged to be equivalent to original IFX; in addition, the two agents also displayed comparable efficacy and safety profiles. Infusion reactions occurred in 3.9% of CT-P13 patients group vs 4.9% in the IFX group, and 27.4% of the CT-P13 patients developed antidrug antibodies vs 22.5% of those on IFX.22
In the second (Phase III, double-blind) trial, 604 patients with rheumatoid arthritis (RA) were randomized to CT-P13 or IFX 3 mg/kg (both given with methotrexate). The two patient groups at the end of trial demonstrated an equivalent rate of responses, drug-related adverse events, and rate of antidrug antibodies.23
On the basis of these data, the EMA approved the biosimilar of IFX for treatment of RA, ankylosing spondylitis, psoriatic arthritis, and IBD in September 2013.
The indications of the originator product IFX include autoimmune arthritis of various etiologies, psoriasis, and IBD. Although in the EU, like in Korea and Japan, all indications of the reference product were licensed, extrapolation to IBD was not granted in Canada.24
Adalimumab biosimilars
Adalimumab (Humira) is a human recombinant human IgG1 monoclonal antibody. It binds and neutralizes TNF-α, preventing it from binding with TNF receptors, which cause activation of the inflammatory reaction cascade. Moreover, it acts on cell surface expressing TNF by inducing apoptosis by a reverse intracellular signal. Adalimumab is licensed for the treatment of RA, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, psoriasis, CD, and UC. The original biologic agent (AbbVie’s Humira) adalimumab was approved by the FDA in December 2002 and by the EMA in September 2003. In 2014, it had worldwide sales of ~12.5 billions of US dollars, but the patent will expire in the US in December 2016, while in Europe, only in April 2018. In Table 1, a summary of adalimumab biosimilars approved or in development is detailed.
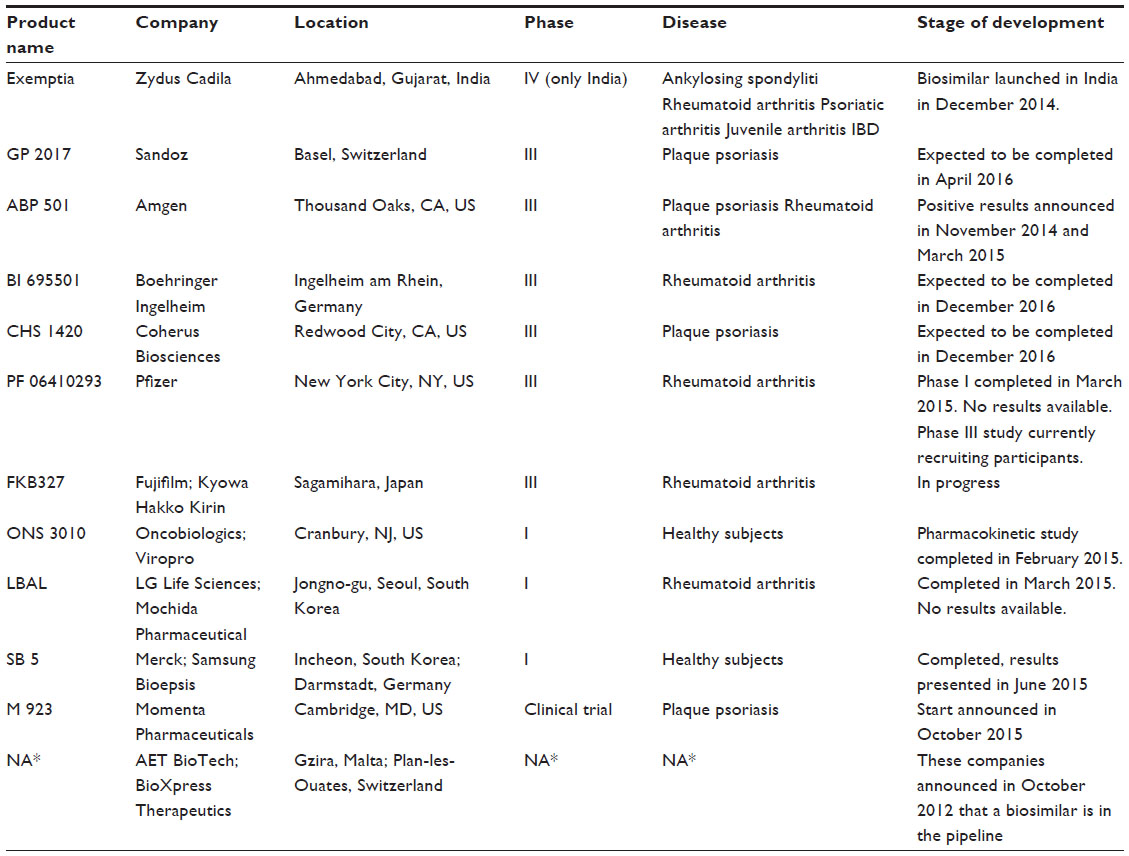 | Table I Biosimilars of adalimumab approved or in development |
The Indian generics maker Zydus Cadila Healthcare Ltd launched its adalimumab biosimilar (ZRC-3197) in India in December 2014 under the brand name Exemptia (adalimumab). The company claims that it is the first company anywhere in the world to launch a biosimilar of adalimumab.25 Biosimilarity has been demonstrated with analytical techniques to characterize the physicochemical and functional properties of ZRC-3197 in comparison with the originator. Indistinguishable primary and secondary structures with similar level of purity and heterogeneity were showed by ZRC-3197 when compared to the original Humira product. Both ZRC-3197 and original adalimumab showed comparable key functional properties, which have been assessed by in vitro cell-based assay and surface plasmon resonance technique. ZRC-3197 exhibited a TNF-α neutralizing activity and a binding affinity for FcγRIIIa receptor similar to Humira.26 A randomized, parallel-group, active-controlled, Phase III trial started in October 2013, conducted by Cadila Healthcare to evaluate efficacy, tolerability, and safety of their biosimilar compared to adalimumab in patients with rheumatoid arthritis. One hundred and twenty Indian subjects were enrolled with age 18–65 years, history of RA (as defined by American College of Rheumatology [ACR] Classification) for at least 6 months, and a moderate-to-severe active seropositive disease. Moreover, patients had a history of treatment with methotrexate, 10–25 mg per week for at least 12 weeks with stable dose in the last 4 weeks. Patients were randomized to receive 40 mg of adalimumab (AbbVie) or the same dosage of adalimumab biosimilar (Zydus) by subcutaneous injection every other week for 3 months. The primary end point was proportion of patient with an ACR 20 response in both the treatment groups on day 84 as compared to baseline. Secondary end points were change from baseline in Disease Activity Score 28 C-Reactive Protein, proportion of patient with an ACR 50 response in both the treatment groups, proportion of patient with an ACR 70 response in both the treatment groups, and percentage of subjects who develop detectable antidrug antibodies.27 In this trial, ZRC-3197 seems to exhibit similar level of efficacy, safety, and tolerability of the originator adalimumab drug, but full data are not yet published. Based on these findings, Indian market authorization has been granted for ZRC-3197. It should be noted that this biosimilar product approved in India might not have been authorized in the EU due to a more strict regulatory process required by the EMA.28,29
Biotech company Amgen, and pharmaceutical company Sandoz, appear to be in pole position in the race to obtain biosimilar adalimumab approval in Europe and in the US. Both companies are already carrying out Phase III trials with their candidate drugs. Sandoz, the generic drug division of Swiss drug giant Novartis, already sells some biosimilars of somatropin (Omnitrope), epoetin α (Binocrit), and filgrastim (Zarzio) in some countries across Europe and US. It announced on December 2013 the start of a Phase III clinical trial for the biosimilar of adalimumab. The Sandoz trial will evaluate the safety, efficacy, and immunogenicity of the biosimilar adalimumab (GP2017) vs Humira (AbbVie) in patients with moderate or severe plaque-type psoriasis. The trial is a multicenter, double-blind, randomized, active-controlled, parallel-group study. The main objective of the study is to demonstrate equivalent efficacy of GP2017 with respect to Humira (adalimumab) in patients with plaque-type psoriasis. Four hundred and forty-eight patients from Bulgaria, France, Germany, Hungary, India, Poland, Romania, Russia, Slovakia, Spain, and US should be enrolled and randomized to receive GP2017 or Humira (adalimumab) with subcutaneous injections. The initial dose of 80 mg will be followed by 40 mg every other week, starting at week 1 and ending at week 33. The trial is expected to be completed in June 2015; currently, it is still ongoing, but patients recruiting is finished. This trial should be strong enough to support regulatory submissions in the US and EU.30,31 This study is the eighth Phase III trial that Sandoz has initiated across six biosimilar molecules. Sandoz is also currently conducting Phase II and III clinical trials for a biosimilar version of rituximab, filgrastim, and pegfilgrastim.
Amgen completed two multicenter, randomized, double-blind, parallel-group, Phase III studies for its biosimilar adalimumab (ABP501). The first trial evaluated efficacy and safety of ABP501 compared to adalimumab in subjects with moderate-to-severe RA. Five hundred and twenty-six patients were enrolled from the US, Canada, UK, Russia, Poland, Spain, Romania, Mexico, Hungary, Germany, Bulgaria, Argentina, and Czech Republic. A solution for subcutaneous injection in prefilled syringe was administered in a dose of 40 mg every 2 weeks for 22 weeks. The primary efficacy end point was 20% improvement in ACR core set measurements. Key secondary end points were Disease Activity Score 28 and 50% or 70% improvement in ACR core set measurements. In this study, completed in November 2014, the biosimilar ABP501 hits its primary and key secondary end points.32,33 The second study evaluated the efficacy and safety of ABP501 compared to adalimumab in adults with moderate-to-severe plaque psoriasis. Three hundred and fifty patients from Australia, France, Canada, Poland, Germany, and Hungary were enrolled. Solution for subcutaneous injection in prefilled syringe was administered in a dose of 80 mg at week 1/day 1, and 40 mg at week 2 and every 2 weeks until week 14. At week 16, subjects with a good response will remain on study for up to 52 weeks; subjects initially randomized to adalimumab will be re-randomized (1:1) to ABP501 or adalimumab treatment for weeks 16–48. This study was completed in March 2015, but results are not available yet.34
Finally, an open-label, single-arm extension study to evaluate long-term safety and efficacy of ABP501 in subjects with moderate-to-severe RA is still ongoing.35
ONS-3010 is the adalimumab biosimilar developed by Oncobiologics Inc.; the company recently announced that its candidate biosimilar drug met the primary end points in its first clinical study. A three-arm single-dose pharmacokinetic study was performed in healthy volunteers to compare ONS-3010 to Humira. All of the pharmacokinetic end points met the bioequivalence criteria of the geometric mean ratios within 90% confidence interval of 80%–125%. Safety and immunogenicity were similar across the three arms. An exploratory ex vivo pharmacodynamic study also showed good results between ONS-3010 and the reference products on TNF-α blockade and the induction of specific inflammatory responses. This first clinical trial was conducted in the Netherlands, but no results are available.36
The biosimilar BI 695501 (Boehringer Ingelheim Pharmaceuticals, Ingelheim, Germany) was also compared in a clinical trial against adalimumab in healthy subjects. A randomized, open-label, single-dose, parallel-arm, active comparator clinical Phase I study was realized in 2012 in New Zealand. Primary end points were bioequivalence of the area under the concentration–time curve of the analyte in plasma over the time, and maximum measured concentration of the analyte in plasma in comparison with US-Humira and EU-Humira products. This trial is completed, but no data are available yet. Another pharmacokinetic trial with BI 695501 was completed in 2014, but again, data are not available in literature. Currently, a clinical trial of BI 695501 and adalimumab, in patients with active RA, is started and still recruiting patients.37
Conclusion
The availability of biosimilars in the near future will be a challenge for physicians and patients. About the possibility to expand the indication of biosimilars to areas not directly researched, EMA recommendations are inconclusive. EMA leaves a door open to this possibility, for cases based on sound scientific reasoning. Although antecedent cases of biosimilar approved for multiple indications already exist, in other foreign drug agencies, there is nothing to suggest that the EMA should follow this way.
Several scientific rheumatologic and gastroenterological societies have raised concern to extrapolate results from one condition to all the others and have solicitated the promotion and realization of specific trials for each indication. However, given the high cost of biologic therapy and the increasing number of patients in treatment, a rapid reduction in the price represents a priority for each health care system and government. In this complex situation, in the interest of patients and pharmaceutical companies alike, it is essential to maintain high safety standards and adequate testing. No compromise of health safety should be accepted in order to obtain economic advantages. Finally, we believe that the final decision on choice should be in the hands of physicians, without strong impositions and always made on individual basis.
Disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. The authors report no conflicts of interest in this work.
References
Annese V, Vecchi M; on behalf of the Italian Group for the Study of IBD (IG-IBD). Use of biosimilars in inflammatory bowel disease: statements of the Italian Group for Inflammatory Bowel Disease. Progress report. Dig Liver Dis. 2014;46:963–968. | |
Rinaudo-Gaujous M, Paul S, Tedesco ED, Genin C, Roblin X, Peyrin-Biroulet L. Review article: biosimilars are the next generation of drugs for liver and gastrointestinal diseases. Aliment Pharmacol Ther. 2013;38:914–924. | |
European Medicines Agency. Questions and answers on biosimilar medicines (similar biological medicinal products). 2012. Available from: http://www.ema.europa.eu/ema/pages/includes/document/open_document.jsp?webContentId=WC500020062. Accessed January 18, 2013. | |
European Medicines Agency. Human medicines – biosimilars. 2013. Available from: http://www.ema.europa.eu/ema/. Accessed January 18, 2013. | |
McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011;3(2):209–217. | |
McCamish M, Woollett G. The rise of the biosimilar. Expert Rev Clin Pharmacol. 2012;5:597–599. | |
Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. 2009;8:226–234. | |
Misra M. Biosimilars: current perspectives and future implications. Indian J Pharmacol. 2012;44:12–14. | |
Gecse KB, Khanna R, Van den Brink GR, et al. Biosimilars in IBD: hope or expectation? Gut. 2013;62(6):803–807. | |
Feagan BG, Choquette D, Ghosh S, et al. The challenge of indication extrapolation for infliximab biosimilars. Biologicals. 2014;42:177–183. | |
Peyrin-Biroulet L, Deltenre P, de Suray N, et al. Efficacy and safety of anti-tumor necrosis factor agents in Crohn’s disease: a meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol. 2008;6:644–653. | |
Hanauer SB, Sandborn WJ, Rutgeerts P, Fedorak RN, Lukas M, MacIntosh D. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology. 2006;130:323–333. | |
Sandborn WJ, Rutgeerts P, Enns R, et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann Intern Med. 2007;146:829–838. | |
Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology. 2007;132:52–65. | |
Reinisch W, Sandborn WJ, Hommes DW, D’Haens G, Hanauer S, Schreiber S. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011;60:780–787. | |
Sandborn WJ, van Assche G, Reinisch W, Colombel JF, D’Haens G, Wolf DC. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;142:257–265. e1–e3. | |
Peyrin-Biroulet L, Laclotte C, Roblin X, Bigard M-A. Adalimumab induction therapy for ulcerative colitis within tolerance or lost response to infliximab: an open-label study. World J Gastroenterol. 2007;13:2328–2332. | |
Oussalah A, Laclotte C, Chevaux JB, Bensenane M, Babouri A, Serre AA. Long-term outcome of adalimumab therapy for ulcerative colitis with intolerance or lost response to infliximab: a single-centre experience. Aliment Pharmacol Ther. 2008;28:966–972. | |
Afif W, Leighton JA, Hanauer SB, et al. Open-label study of adalimumab in patients with ulcerative colitis including those with prior loss of response or intolerance to infliximab. Inflamm Bowel Dis. 2009;15:1302–1307. | |
Sandborn WJ, Van Assche G, Reinisch W, et al. Induction and maintenance of clinical remission by adalimumab in patients with moderate-to-severe ulcerative colitis. J Crohns Colitis. 2011;5:68. | |
Evaluate™. KFDA Approves Remsima(TM) (infliximab), the World’s First Antibody Biosimilar [press release]. Celltrion, Johnson & Johnson; 2012 [July 23]. Available from: http://www.evaluategroup.com/Universal/View.aspx?type=Story&id=320129. Accessed December 29, 2015. | |
Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605–1612. | |
Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613–1620. | |
Health Canada. Summary Basis of Decision (SBD) for Inflectra. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_inflectra_159493-eng.php. Accessed October 15, 2014. | |
Cadila Healthcare Ltd. Zydus Launches World’s First Biosimilar of Adalimumab [press release]. Zydus Cadila. Available at: http://exemptia.com/zydus-launches-worlds-first-biosimilar-of-adalimumab-2/. Accessed October 16, 2015. | |
Bandyopadhyay S, Mahajan M, Mehta T, et al. Physicochemical and functional characterization of a biosimilar adalimumab ZRC-3197. Biosimilars. 2015:5:1–18. | |
Clinical Trial Registry India. New Delhi: Indian National Institute of Medical Statistics (NIMS). Available from: http://ctri.nic.in/Clinicaltrials/showallp.php?mid1=7595&EncHid=&userName=adalimumab. Accessed October 16, 2015. | |
CTRI/2013/10/004040. Medicines Agency. London: Medicines Agency. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/about_us/general/general_content_000109.jsp. Accessed October 16, 2015. | |
Central Drugs Standard Control Organization. Director General of Health Services, Ministry of Health and Family Welfare, Government of India. Available from: http://www.cdsco.nic.in/. Accessed October 16, 2015. | |
NCT02016105. ClinicalTrials.gov. Bethesda, MD: US National Institutes of Health. Available from: https://clinicaltrials.gov/ct2/show/NCT02016105?term=GP+2017&rank=1. Accessed October 16, 2015. | |
CT Number: 2013-000747-11. EU Clinical Trials Register Service. London: European Medicines Agency. Available from: https://www.clinicaltrialsregister.eu/ctr-search/search?query=GP2017. Accessed October 16, 2015. | |
NCT01970475. ClinicalTrials.gov. Bethesda, MD: US National Institutes of Health. Available from: https://clinicaltrials. gov/ct2/show/NCT01970475?term=ABP501&rank=3. Accessed October 16, 2015. | |
CT Number: 2013-004654-13. EU Clinical Trials Register Service. London: European Medicines Agency. Available from: https://www.clinicaltrialsregister.eu/ctr-search/search?query=ABP+501. Accessed October 16, 2015. | |
NCT01970488. ClinicalTrials.gov. Bethesda, MD: US National Institutes of Health. Available from: https://clinicaltrials. gov/ct2/show/NCT01970488?term=ABP501&rank=2. Accessed October 16, 2015. | |
NCT02114931. ClinicalTrials.gov. Bethesda, MD: US National Institutes of Health. Available from: https://clinicaltrials.gov/ct2/show/ NCT02114931?term=ABP501&rank=1. Accessed October 16, 2015. | |
Oncobiologics™. ONS-3010 (Humira®/Adalimumab Biosimilar) Meets Primary Endpoints In First Clinical Study [press release]. Cranbury, NJ: Oncobiologics™; 2015 [February 12]. Available from: http://oncobiologics.com/as_press-releases&recordID=18667. Accessed December 29, 2015. | |
NCT02137226. ClinicalTrials.gov. Bethesda, MD: US National Institutes of Health. Available from: https://clinicaltrials.gov/ct2/show/ NCT02137226?term=Boehringer+Ingelheim+adalimumab&rank=3. Accessed December 29, 2015. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.