Back to Journals » International Journal of Nanomedicine » Volume 15
Development of Rifapentine-Loaded PLGA-Based Nanoparticles: In vitro Characterisation and in vivo Study in Mice
Authors Liang Q, Xiang H, Li X, Luo C, Ma X, Zhao W, Chen J, Tian Z, Li X, Song X
Received 10 April 2020
Accepted for publication 26 August 2020
Published 6 October 2020 Volume 2020:15 Pages 7491—7507
DOI https://doi.org/10.2147/IJN.S257758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Qiuzhen Liang1 ,* Haibin Xiang1 ,* Xinyu Li,2 Chunxia Luo,2 Xuehong Ma,2 Wenhui Zhao,2 Jiangtao Chen,1 Zheng Tian,1 Xinxia Li,2 Xinghua Song3,4
1Department of Orthopaedics, The First Affiliated Hospital of Xinjiang Medical University, Urumqi 830054, People’s Republic of China; 2School of Pharmacy, Xinjiang Medical University, Urumqi 830011, People’s Republic of China; 3Department of Orthopaedic, The First Affiliated Hospital of Jinan University, Guangzhou, Guangdong Province 510630, People’s Republic of China; 4Department of Orthopaedic, The Affiliated Shunde Hospital of Jinan University, Foshan, Guangdong Province 528303, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xinghua Song
Department of Orthopaedic, The First Affiliated Hospital of Jinan University, Guangzhou, Guangdong Province 510630, People’s Republic of China
Email [email protected]
Xinxia Li
School of Pharmacy, Xinjiang Medical University, No. 393, Xinyi Road, Urumqi 830011, People’s Republic of China
Email [email protected]
Background: Tuberculosis (TB) is a leading cause of death amongst infectious diseases. The poor response to antitubercular agents necessitates the long-term use of high drug doses, resulting in low patient compliance, which is the main reason for chemotherapy failure and contributes to the development of multidrug-resistant TB. Patient non-compliance has been a major obstacle in the successful management of TB. The aim of this work was to develop and characterise rifapentine (RPT)-loaded PLGA-based nanoparticles (NPs) for reducing dosing frequency.
Methods: RPT-loaded PLGA and PLGA–PEG NPs were prepared using premix membrane homogenisation combined with solvent evaporation method. The resulting NPs were characterised in terms of physicochemical characteristics, toxicity, cellular uptake and antitubercular activity. NPs were further evaluated for pharmacokinetic and biodistribution studies in mice.
Results: The resulting NPs showed suitable and safe physicochemical characteristics and could be taken up by macrophages. RPT-loaded NPs were more effective against Mycobacterium tuberculosis than free RPT. In vivo studies revealed that NPs could improve pharmacokinetic parameters, particularly for RPT/PLGA–PEG NPs. Moreover, both formulations had no toxicity to the organs of mice and could reduce hepatotoxicity.
Conclusion: The application of PLGA-based NPs as sustained-release delivery vehicles for RPT could prolong drug release, modify pharmacokinetics, increase antitubercular activity and diminish toxicity, thereby allowing low dosage and frequency.
Keywords: tuberculosis, rifapentine, drug delivery system, nanoparticles
Introduction
Tuberculosis (TB) is an infectious bacterial disease caused by Mycobacterium tuberculosis (M. tuberculosis). The increasing morbidity and mortality caused by TB continues to be a global health problem. According to the 2019 World Health Organization report, TB has surpassed HIV as the most lethal infectious disease in the world. Moreover, without proper treatment, up to two-thirds of people infected with TB will die.1
Antimicrobial therapy of TB is a challenge due to poor response to drug therapy, and the low permeation of antimycobacterial agents necessitates the long-term use of high drug doses.2 Furthermore, the degradation of drugs may occur before they reach target tissues. Conventional treatments for TB include daily therapy with high doses of drugs for at least 6 months and even more than 1 year for osteoarticular TB.3,4 Patients usually complain about severe side effects after a short while of starting therapy, such as hepatotoxicity, which may even progress into hepatitis. Consequently, patients drop out early therapy, resulting in low patient compliance and the emergency of multidrug-resistant TB.5 Therefore, patient non-compliance has been a major obstacle in the successful management of TB and has compelled researchers to develop sustained-release drug formulations so that the dosing frequency may be reduced.6
Although the development of new antimycobacterial drugs is an obvious and necessary strategy to fight TB, mechanisms to improve the efficacy of existing drugs could represent a faster approach.3 The current strategy for enhancing the therapeutic activity of currently available drugs is to entrap drugs within a delivery system. Amongst various drug delivery systems, nanoparticles (NPs) represent a very promising approach to improve the solubility of hydrophobic drugs.7,8 This formulation offers potential advantages over free drug, including prolonging drug release or availability of drug into the systemic circulation, increasing therapeutic efficacy and diminishing toxicity, thereby allowing less frequent dosing.2 Another feature of NPs, which makes them potentially useful for the treatment of intracellular infections, including M. tuberculosis, is their ability to accumulate in macrophages.9
Polymeric NPs can be degraded in vivo, either enzymatically or by hydrolysis or both, to produce biocompatible, toxicologically safe by-products that are further eliminated by the normal metabolic pathways.10,11 PLGA (poly-d,l-lactide-co-glycolide) has been approved from the Food and Drug Administration (FDA) and European Medicine Agency for drug delivery system, and has been widely used as an implantable drug carrier.12 Importantly, they offer the chance for further structural modification particularly on the surface of NPs to enhance their therapeutic performance.13 For example, PEG (poly ethylene glycol) is a hydrophilic and inert polymer that provides a steric barrier on the surface of the NPs. Adding PEG is useful to prolong NPs circulation and to decrease premature drug release.14 Consequently, several copolymers of PLGA with PEG have been synthesised, encapsulating a wide variety of therapeutic drugs. Different production techniques such as nanoprecipitation, emulsion/solvent diffusion, spray drying and salting-out have been reported for the synthesis of PLGA–PEG NPs.11 Amongst them, membrane emulsification is highly attractive due to the low energy input required and narrow particle size distribution.15
In the present study, rifapentine (RPT)-loaded PLGA and PLGA–PEG NPs were prepared using premix membrane homogenisation combined with solvent evaporation method. RPT was chosen as a model drug for the following reasons: (i) Although rifampicin is the first-line anti-TB drug amongst rifamycin antibiotics, there were half a million new cases of rifampicin-resistant TB in 2018,1 which may limit its clinical application. RPT is a rifamycin derivative, and RPT-based combination therapy could prevent the emergence of rifampicin-resistant M. tuberculosis.16 (ii) Compared with rifampicin, RPT is several times more active against M. tuberculosis with MIC of 0.02–0.06 μg/mL,17 which is beneficial for reducing the dosage. (iii) Importantly, the major advantage of RPT is that it has a much longer half-life than rifampicin,18 which makes it an ideal rifamycin antibiotic for reducing the dosing frequency. However, RPT also suffers from some drawbacks such as poor water solubility, elevation in serum aminotransferase and clinically apparent acute liver injury.19,20 In addition, the fact that M. tuberculosis is an intra-macrophage pathogen limits the use of conventional RPT, and if they even reach the target site, they have inefficient intracellular penetration. Therefore, we encapsulated RPT into PLGA or PLGA–PEG NPs as PLGA-based nanostructures will offer a very suitable medium for enhancing the advantages whilst reducing the disadvantages of RPT described above. To the best of our knowledge, polymeric nano-formulations of RPT have not yet been reported in the literature, and this is the first study that investigated the effect of RPT-loaded PLGA-based NPs on their cellular uptake, antitubercular activity, pharmacokinetics and biodistribution after oral administration to mice.
Materials and Methods
Materials
All chemicals and reagents were purchased from Sigma Chemical (St. Louis, Missouri, USA) unless otherwise stated. RPT (content: 97%) was obtained from Sichuan Med-Shine Pharmaceutical Co.,Ltd. (Sichuan, China). PLGA (Mw: 17000; lactide: glycolide = 50:50) and PLGA–PEG (Mw: 19000; lactide: glycolide = 50:50) were purchased from Shandong Institute of Medical Instrument (Jinan, China). Shirasu porous glass (SPG) membranes were purchased from SPG Technology Co., Ltd. (Miyazaki, Japan). Murine macrophages (J774 A.1 cell line) were purchased from Shanghai Honsun Biological Technology Co., Ltd. All chemicals and solvents used in this study were of analytical/HPLC grade. Ultrapure water was collected from the Milli-Q water purifier unit and was used for aqueous solution preparation.
Preparation of RPT-Loaded NPs
RPT-loaded NPs were prepared using the SPG premix membrane homogenisation combined with modified solvent evaporation method. In brief, 20 mg of RPT was dissolved in 10 mL of the prepared solution of PLGA (200 mg) or PLGA–PEG (250 mg) in dichloromethane (DCM). This oil phase was further emulsified to an aqueous phase (PVA, 3% w/v) through ultrasonication (S-450D Digital Sonifier, Branson, USA) at 200 W for 30 s to form coarse emulsions. Then, the coarse emulsion was subjected to five homogenisation cycles in an open system under positive transmembrane pressure using an external pressure type micro-kit emulsification device (SPG Technology Co., Ltd, Sadowaracho, Japan) equipped with an SPG membrane (0.1 μm pore size, SPG Technology). The obtained emulsion was poured quickly into 100 mL of 1% (w/v) PVA solution and left under magnetic stirring (5×g) for 6 h at 40°C to allow DCM evaporation. NPs were collected and washed three times with cold distilled water at 21,000×g for 20 min, and finally freeze dried. To make coumarin 6-labelled NPs for the cell internalisation studies, coumarin 6 was added to the DCM lipid phase, as the only change to the procedure described above.
Physicochemical Characterisation
Particle Size, Polydispersity Index (PDI), and Zeta Potential (ZP)
Particle size and PDI were obtained by dynamic light scattering technique, whilst ZP was determined by measuring the electrophoretic mobility. Each sample was diluted in purified water at 25°C. Determinations were performed in triplicate using a ZetaSizer Nano-ZS (Malvern Instruments Ltd., Malvern, UK).
Morphological Characterisation
The morphology of RPT-loaded NPs was observed using a transmission electron microscopy (TEM, JEM-1230, Jeol Ltd., Japan). NPs were diluted suitably, mounted on copper grid and added with 2% w/v solution of phosphotungstic acid. The sample was air-dried at room temperature before microscopic analysis.
Determination of Drug Entrapment Efficiency (%EE) and Drug Loading (%DL)
The indirect method for the determination of %EE and %DL of RPT in PLGA or PLGA–PEG NPs was followed. The emulsion was centrifuged at 20,000×g for 15 min to obtain a transparent supernatant. The nonencapsulated drug in the supernatant was measured by High-performance liquid chromatography (HPLC). The HPLC analysis was performed using a C18 column (250 mm×4.6 mm, 5 µm; Waters Corporation, Milford, MA, USA) with the mobile phase of methanol–acetonitrile (5:4 v/v). The temperature was set at 35°C, the flow rate was 1 mL/min, and the wavelength was 336 nm (the HPLC conditions were provided by our National Invention Patent, No ZL201210281222.6).
The %EE and %DL were calculated using the following equation:
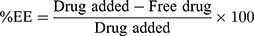

Stability Tests
The freshly prepared freeze-dried NPs were stored in a stable chamber at 25°C for a period of 3 months. Periodically, a sample was collected to measure particle size, PDI and %EE. Moreover, to investigate the stability in gastrointestinal fluids, the NPs were incubated in simulated gastric fluid (SGF, pH 1.2, pepsin 0.32%, w/v) and simulated intestinal fluid (SIF, pH 6.8, pancreatin 1%, w/v) at a concentration of 10 mg/mL for 2 h. The overall effect was evaluated by measuring the effect on particle size, PDI and %EE compared with the initial amount of encapsulated RPT. Evaluation was performed in triplicate for each analysis.
Drug Release Studies
In vitro RPT release test was assessed by direct dialysis method. Free RPT, RPT/PLGA NPs and RPT/PLGA–PEG NPs (containing 20 mg equivalent of drug) were placed in a 3000 Da dialysis bag and incubated in 50 mL of phosphate-buffered saline medium (pH 7.4). The system was maintained at 37 ± 2°C and stirred at 3×g. Samples were withdrawn at pre-determined intervals and replaced by the same volume of fresh buffer solution. The concentrations of RPT in the samples were analysed by HPLC as described above.
In vitro Cytotoxicity Assay
NP cytotoxicity was evaluated by MTT assay. RPT solution, RPT/PLGA NPs and RPT/PLGA–PEG NPs were incubated with murine macrophages (J774 A.1 cell line) at 37°C for 24 h at final RPT concentrations of 0, 5, 25 and 50 μg/mL. For the positive control group, complete DMEM containing 0.64% phenol was added. Afterwards, 15 μL of MTT was added to each well and incubated further for 4 h at 37°C. The media were removed and 200 μL of DMSO was added into each well to dissolve MTT formazan. After shaking for 20 min, the absorbance of formazan at 570 nm was measured with a microplate reader. Using the same method described above, the macrophages were further treated with various concentrations of blank NPs (ranging from 0 mg/mL to 1.2 mg/mL). The cell viability (%) in relation to negative control cells was calculated using the following equation:

Cellular Uptake
The phagocytic uptake of RPT-loaded NPs by macrophages was evaluated using murine macrophages (J774 A.1 cell line). Briefly, the cells were cultured in a six-well plate at the density of 3×104/well. Different concentrations of coumarin 6 labelled RPT/PLGA NPs and RPT/PLGA–PEG NPs were suspended in the culture medium. The untreated cells and cells treated with a solution of coumarin 6 were used as positive and negative controls, respectively.21 After incubation for 1 h and 4 h, the cells were washed three times with PBS to remove the possible NPs that adhered on the cell membrane, fixed with 5% paraformaldehyde in PBS and stained using 5 μg/mL of DAPI for 15 min for visualisation of the nuclei. The results were photographed using a fluorescent microscope (Leica, Germany). Coumarin 6 and DAPI showed green and blue colourations, respectively.
Haemolytic Toxicity
Haemolytic study in terms of percentage haemolysis was conducted to evaluate the compatibility of RPT-loaded NPs with RBCs (erythrocytes). In this study, whole human blood was collected from healthy human volunteers, which was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of The Xinjiang Medical University (No 20180223–13), and all volunteers signed their informed consent forms. Briefly, human blood was centrifuged, and RBCs were separated and resuspended in 0.9% w/v normal saline. Furthermore, various concentrations (ranging from 0.5 mg/mL to 3.5 mg/mL) of RPT/PLGA NPs and RPT/PEG–PLGA NPs were mixed with normal saline for interaction with RBC suspension. After 1 h, the samples were centrifuged, and plasma was separated from the settled RBCs. A UV–visible spectrophotometer at 545 nm was used for absorbance measurements. RBCs in saline were prepared as a negative control while water was used as a positive control, whilst water was used as a positive control. The haemolytic rate of the sample was calculated by using the following equation:
Haemolytic rate (%) = [(As −Anc)/(Apc−Anc)] × 100%
As absorbance of sample
Anc absorbance of negative control
Apc absorbance of positive control
In vitro Anti-Tubercular Studies
The antimycobacterial activity of RPT NPs against M. tuberculosis was evaluated by microplate Alamar blue assay (MABA) following a previously reported protocol with slight modifications.8 In outer peripheral wells of sterile 96-well plates, sterile deionised water (200 μL) was added to minimise the loss of medium during incubation. Briefly, 100 μL of 7H9GC broth (autoclaved at 15 lbs, 121°C for 12 min, and cooled at 30°C; then, Middlebrook OADC was added and mixed) was added in each well. Approximately 100 μL of RPT solution was added from B to G rows to investigate the minimum inhibitory concentration (MIC) of free drug. Serial dilutions (1:2) were performed in columns 3 to 10, and the excess medium (100 μL) from wells (column 10) was simultaneously discarded to obtain a final RPT concentration range of 1.6 μg/mL to 0.006 μg/mL in each 96-well plate. Afterwards, 100 μL of M. tuberculosis inocula were added in rows B to G (columns 2 to 11). Column 11 was considered as a control (inoculum-only). Parafilm was used to seal the plates and placed for incubation at 37°C for 5 days. On the fifth day, 50 μL of a freshly prepared 1:1 mixture of 10× Alamar Blue reagent and 10% Tween 80 was added to well B11. Afterwards, the plates were allowed to re-incubate for 24 h. All the wells would receive the Alamar dye if well B11 turned pink. The same procedure was applied for the determination of MIC of the RPT-loaded NPs. The final concentration range was as follows: 0.5 μg/mL to 0.002 μg/mL for PLGA NPs and 1.6 μg/mL to 0.006 μg/mL for PLGA–PEG NPs in each well of plates. The results were analysed by means of colour changing in each well: when the colour turned from blue to pink, this indicates bacterial growth, and the MIC of the NPs was calculated based on the lowest concentration at which there is no visible growth of bacteria observed.
In vivo Pharmacokinetics Study
BALB/c male mice (2–3 months, body weight 26±2 g) were obtained from the Animal Center of Xinjiang Medical University (Xinjiang, China). Animal experiments and welfare were complied with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (No 8023), and all experiments were approved by the Ethics Committee of The Xinjiang Medical University (No 20180223–13). The animals were kept under standard hygiene conditions, fed commercial chow and given water ad libitum. For a single-dose drug pharmacokinetics study, the mice were divided into three groups: free RPT, RPT/PLGA NPs and RPT/PLGA–PEG NPs. The mice were given a single dose of 10 mg/kg orally (equivalent to a dose of 10 mg/kg of RPT in each group). Then, blood samples (400μL each) were taken from the retro-orbital plexus of each animal per time point. Plasma was separated by centrifuging the blood samples at 15,000×g for 15 min and stored at −20°C until analysed. Acetonitrile was added to precipitate the plasma proteins. Thereafter, the samples were vortexed and centrifuged at 15,000×g for 20 min. The supernatants were removed, filtered through a 0.2 μm syringe filter and injected on the HPLC column to estimate the amount of RPT. The maximum RPT concentration in plasma (Cmax), the time to reach peak plasma concentration (Tmax), the half-life (t1/2), the area under the curve (AUC0–∞), and the mean residence time (MRT) were determined by blood concentration versus time data.
Tissue Distribution Studies
For a single-dose drug disposition study, the mice were given a single dose of 10 mg/kg orally (equivalent to a dose of 10 mg/kg of RPT in each group). The mice were divided into four groups: control group (normal saline), free RPT, RPT/PLGA NPs and RPT/PLGA–PEG NPs. The animals were sacrificed at a specified time interval. The tissues (lung, liver, spleen and kidney) were excised and washed quickly with normal saline to remove surface blood. They were wrapped with foil and stored at −20°C. Thawed tissues were accurately weighed, and drug levels were estimated in 20% (w/v) tissue homogenates prepared in normal saline. The samples were prepared by protein precipitation and then centrifuged at 21,000×g for 10 min at 4°C. The supernatants were removed, filtered through a 0.2 μm syringe filter and injected on the HPLC column to estimate the amount of RPT in each tissue. The results were expressed as microgram per gram of tissue. To observe the effect of different formulations of RPT on mouse organs, the animals were sacrificed at 24 h in case of normal saline and free RPT, and at 4 and 6 days in case of RPT/PLGA NPs and RPT/PLGA–PEG NPs, respectively. The lung, liver, spleen and kidney tissues of mice were fixed with 4% paraformaldehyde and then paraffin embedded for haematoxylin and eosin (H&E) staining and histological examination. Moreover, before the mice were euthanised, blood samples were drawn from the eye socket for blood biochemistry testing.
Statistical Analysis
All experiments were performed in triplicate at least three times. Results are expressed as mean ± SD. Statistical analyses were performed using the SPSS 17.0 software. One-way ANOVA with Tukey’s test was employed for the comparisons among groups. A value of P<0.05 was considered statistically significant.
Results and Discussion
Physicochemical Characterisation
Physicochemical characterisation (Table 1) shows the most important properties of RPT/PLGA NPs and RPT/PLGA–PEG NPs. The particle size plays an important role in the peculiar properties of NPs, and because of that the precise determination of size population is relevant and crucial for drug delivery systems.5,22 A size range of <10 nm results in the removal of NPs by renal filtration,23,24 whereas size range of >200 nm demonstrates a higher rate of plasma clearance compared with a lower size range.25 In the present study, NPs demonstrated size ranges that were considered to be suitable for administration for long-circulating sustained-release properties. Liu et al26 also reported that NPs of this size not only can be internalised by endocytosis but also are large enough to be maintained in circulation for a long period. The average size of nano-formulations was found to be around 150 nm for RPT/PLGA NPs and 136 nm for RPT/PLGA–PEG NPs. This is explained by the unique proprieties of PEGylated polymers. As an amphiphilic polymer, PEG–PLGA modifies the association of polymer molecules during nanoparticulate formation, eventually resulting in the formation of smaller NPs.27,28 Sheng et al29 found that the size of NPs decreased when their PEG content increased, possibly because of the amphiphilic nature of these copolymers, which reduced the interfacial tension between the aqueous and organic phases.
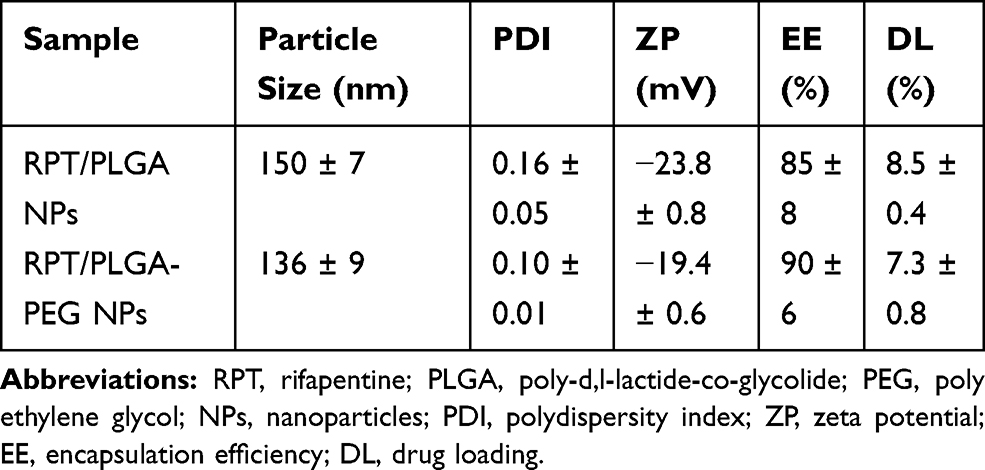 |
Table 1 Physicochemical Characterization of RPT-Loaded NPs |
The surface characteristics also affect the fate of NPs in systemic circulation. Positively charged formulations have a higher rate of plasma clearance compared with neutral and negatively charged formulations. Furthermore, the presence of negatively charged serum proteins can lead to the formation of aggregates after administration of positively charged NPs, resulting in embolism in blood capillaries.30,31 In addition, the highly negative surface charge indicates a strong electrostatic repulsion between particles, representing a great indicator of stability.5 Haggag et al32 reported that PLGA NPs exhibited negative zeta potential, which was significantly higher than that of the PEGylated PLGA types. In our study, the zeta potential of both types of NPs was also negative due to the carboxyl end groups of PLGA chains, and the slight reduction of zeta potential in RPT/PLGA–PEG NPs might be attributed to the hydrophilic ability and the surface charge-shielding effects of hydrophilic PEG outer layer. PEG is known to be amphiphilic, decreasing measurable zeta potential by shielding the native PLGA surface charge.33 A similar phenomenon was also observed on the PLA copolymer. When PEG chains were introduced into the negative PLA segment, the neutral-charged PEG segments occupied the negative site on the surface of NPs.34
The high %EE demonstrated that the method we used to prepare these formulations was properly selected to favour RPT solubility, which may avoid its expulsion from the nanocarrier during preparation. Furthermore, compared with RPT/PLGA NPs, RPT/PLGA–PEG NPs showed higher %EE with lower %DL. This phenomenon might be ascribed to the following reasons: (i) PEGylated polymers can form NPs more effectively than PLGA because of their well-defined lipophilic portion remaining inside the NPs and their hydrophilic component projected to the outside.27 This creates a more suitable environment for encapsulating hydrophobic RPT. (ii) PEGylated polymers with low solubility in DCM will solidify quicker during the solidification stage.32 (iii) RPT is more soluble in DCM containing PLGA alone, which may give rise to higher drug loading. (iv) The concentration of PEGylated polymer was higher than that of the PLGA polymer during nanoparticulate formation.
At present, several types of nanocarriers loaded with anti-TB drugs have been reported including NPs, liposome, polymeric micelles, polymersomes and niosomes.35 However, studies on RPT nano-formulation are few. Recently, Magalhães et al36 reported RPT-loaded lipid NPs with particle size of 242 nm, PDI of 0.17, ZP of −22 mV, %EE of 86% and %DL of 2.9%. By contrast, the results in our study seem to be superior to theirs in terms of these characterisations described above. The smaller particle size was considered to be more suitable for increasing the systemic circulation. Lower PDI and higher negative ZP suggest that NPs are more homogeneous and stable. Higher %EE and %DL may contribute to a better application. However, no further results in terms of in vitro release, anti-TB activity, cellular uptake or in vivo pharmacokinetics could be compared. In other previous studies, some non-RPT-loaded nano-formulations were reported, especially for rifampicin (the most common encapsulated drug). The particle size, PDI, ZP, EE% and DL% were also different from each other, mainly because the materials and methods used to prepare these formulations were different. Compared with these studies,37–41 the NPs presented in our study may have a relatively smaller particle size, lower PDI and higher ZP and %EE, which may be ascribed to the combined method used in the present study and the inherent property of RPT. Esmaeili et al42 fabricated PLGA NPs loaded with rifampicin using the combined method of homogenisation and solvent evaporation. These NPs had a particle size of ~200–260 nm and PDI of 0.11–0.26; however, the %EE was less than 1% possibly due to the rapid diffusion of acetone into the aqueous phase when using a high-speed homogeniser at 24,000 rpm. In our study, the combined method refers to the combination of premix SPG membrane homogenisation and solvent evaporation method. Membrane emulsification is attractive due to the low energy consumption and better control of droplet size and droplet size distribution. The emulsions are produced by forcing a primary emulsion through an SPG microporous membrane into a continuous aqueous phase, resulting in much less shear than in conventional emulsification processes, so that the droplets are intact and high %EE and monodispersity could be achieved.43 The premix membrane emulsification is a method that permits the preparation of monodispersed emulsions with productivities of several orders of magnitude higher than those of direct membrane emulsification.44,45 In the present study, pre-emulsion was forced through the pores of an SPG membrane yielding smaller droplets, and to further obtain more uniform particles, the emulsified dispersion was repeatedly extruded through the membrane for five times. Uniform-sized NPs would not only enhance drug delivery but also the decrease side effects of the delivery drugs.46
By contrast, some studies showed better results in terms of these properties described above. Singh et al47 developed solid lipid NPs using a novel microemulsification method to overcome the poor oral bioavailability of rifampicin, with a much higher %DL (50%) and lower particle size (~130 nm). However, the in vivo release time of their NPs was not as long as ours, indicating that the property of RPT and its PEGylation could further prolong the circulation time more effectively. In addition, owing to the enhanced anti-TB activity of RPT-loaded NPs compared with free RPT, a lower %DL may benefit in reducing the side effects of RPT and lowering in vivo burst release of encapsulated RPT.
In stability study, the overall results showed that both NPs were stable at least 3 months given that only slight variations occurred (P>0.05) in the particle size, PDI and EE% (Table 2). Moreover, the gastrointestinal stability of the formulation is one of the major concerns in developing oral drug delivery systems. Thus, the stability of the developed NPs was determined by incubating them in simulated gastric and intestinal fluids, followed by determination of particle size, PDI and %EE. The results (Table 3) revealed that the particles size and PDI of RPT/PLGA NPs increased significantly after 2 h of incubation in SGF, which may be ascribed to the partial aggregation. Acidic environment is responsible for the protonation of the carboxyl groups of PLGA and the aggregation of the NPs.26 Compared with RPT/PLGA NPs, RPT/PLGA–PEG NPs were found to be highly stable in both physiological fluids (SGF and SIF) with an insignificant change in size, PDI and %EE. This stability may be attributed to the presence of PEG chains on the surface, which interfered with the enzymes to react with the NPs.48
 |
Table 2 Stability Evaluation Within Three Months |
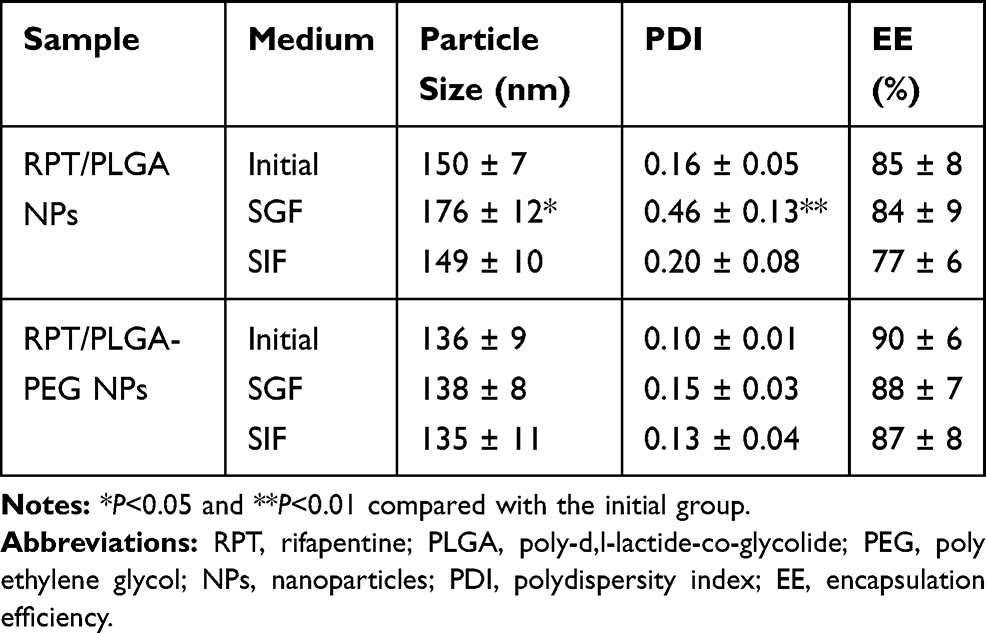 |
Table 3 Stability of NPs in Simulated Gastric and Intestinal Fluids |
Morphology by TEM
The surface morphology of the fabricated RPT-loaded PLGA was investigated by TEM (Figure 1). The image revealed that both types of NPs were dispersed as individual particles with a well-defined spherical shape. The resultant structure of RPT/PLGA–PEG NPs was uniform, and a core surrounded by an outer layer (core–shell structure) at high magnification indicated the encapsulation of drug inside the polymer matrix (Figure 1B).
 |
Figure 1 TEM images of NPs. |
In vitro Release Profiles
The release pattern of RPT from PLGA and PLGA–PEG NPs are shown in Figure 2. The release profile was characterised by an initial burst and subsequent controlled release. The initial drug release from the matrix occurs by diffusion of the drug from the polymer matrix, whereas during the later phases, the release is mediated through the diffusion of the drug and degradation of the polymer matrix itself.49 Within 12 h, the cumulative release was approximately 35% and 17% from RPT/PLGA NPs and RPT/PLGA–PEG NPs, respectively. The hydrophilic PEG outer layer of RPT/PLGA–PEG NPs decreases adsorption of lipophilic PRT on the surface of NPs, thereby decreasing the initial burst drug release. On the contrary, higher amounts of hydrophilic drug were associated with the surface of PEGylated NPs compared with PLGA NPs, causing increasing initial burst release.50 In addition, hydrophilic PEG chains allowed the faster release of the hydrophilic drug through enhanced polymer degradation rate because water was taken up more readily compared with PLGA NPs. Interestingly, Hagaga et al50 found that the burst release decreased following the addition of PVA as an internal aqueous phase stabiliser for hydrophilic drug. In our study, the drug encapsulated in NPs was hydrophobic, which facilitated drug stabilisation in the core of particles even without any stabiliser, thereby contributing to the decreased initial burst release. However, RPT/PLGA–PEG NPs demonstrate higher and faster drug release after the initial burst release compare with RPT/PLGA NPs. This has resulted in an overall 72% and 91% drug depletion from PLGA and PLGA–PEG NPs, respectively. This difference could be explained by the presence of PEGylated surface, causing increased diffusion of water into the core of NPs after the initial burst release. By contrast, free RPT was released in a burst manner within 2 h initially and approximately 97% in 9 h. Overall, this controlled release profile is a relevant and desirable property during TB treatment once the therapeutic concentration can be maintained over a longer period of time, which reduces the number of doses administered and consequently adverse effects.
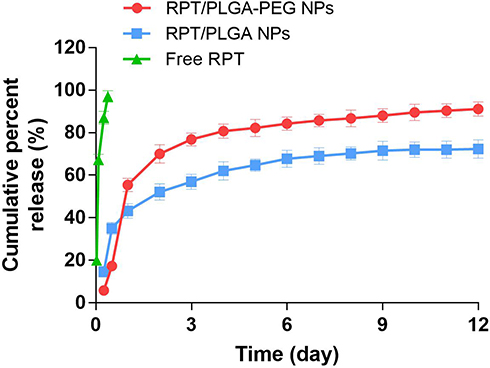 |
Figure 2 In vitro cumulative release curves of free RPT, RPT/PLGA NPs and RPT/PLGA-PEG NPs. |
In vitro Cytotoxicity Assay
Cytotoxicity was evaluated on J744A.1 cells. The results exhibited in Figure 3A showed that both NPs were non-toxic for macrophages, because the cell viability was over 90% in all concentrations tested, whilst it was only 19.5% in the positive control group. Compared with the negative control, both drug-loaded NPs did not present a significant difference at the concentration of 5 and 25 μg/mL (P>0.05); however, RPT/PLGA NPs were considerably more toxic at a concentration of 50 μg/mL (P<0.01). By contrast, free RPT presented a cell viability of less than 80% at concentrations of 25 and 50 μg/mL, demonstrating that the toxicity of RPT was remarkably reduced after encapsulation. To further assess the effect of blank NPs on cell viability, the cells were exposed to NPs at different concentrations for 24 h. The results revealed that cell viability was still above 90% at a concentration of 0.6 mg/mL; however, for the highest concentration used (1.2 mg/mL), the cell viability was less than 60% (Figure 3B).
 |
Figure 3 Cytotoxicity studies on macrophages. (A) Cytotoxicity of RPT and RPT-loaded NPs. (B) Cytotoxicity of Blank NPs. |
Cellular Uptake
In order to observe the uptake of NPs into macrophages, the macrophages were incubated in the presence of coumarin 6-loaded NPs. As shown in Figure 4, a green fluorescence inside the cells could be observed after 1 h of incubation, and the fluorescence intensity increased in a time- and concentration-dependent manner, which suggested that both NPs could be taken up by macrophages. When the cells were incubated with coumarin 6 solution, a negligible fluorescence was observed in the cells. As M. tuberculosis lives inside the macrophage, RPT-loaded NPs endocytosis is favourable within these infected cells.
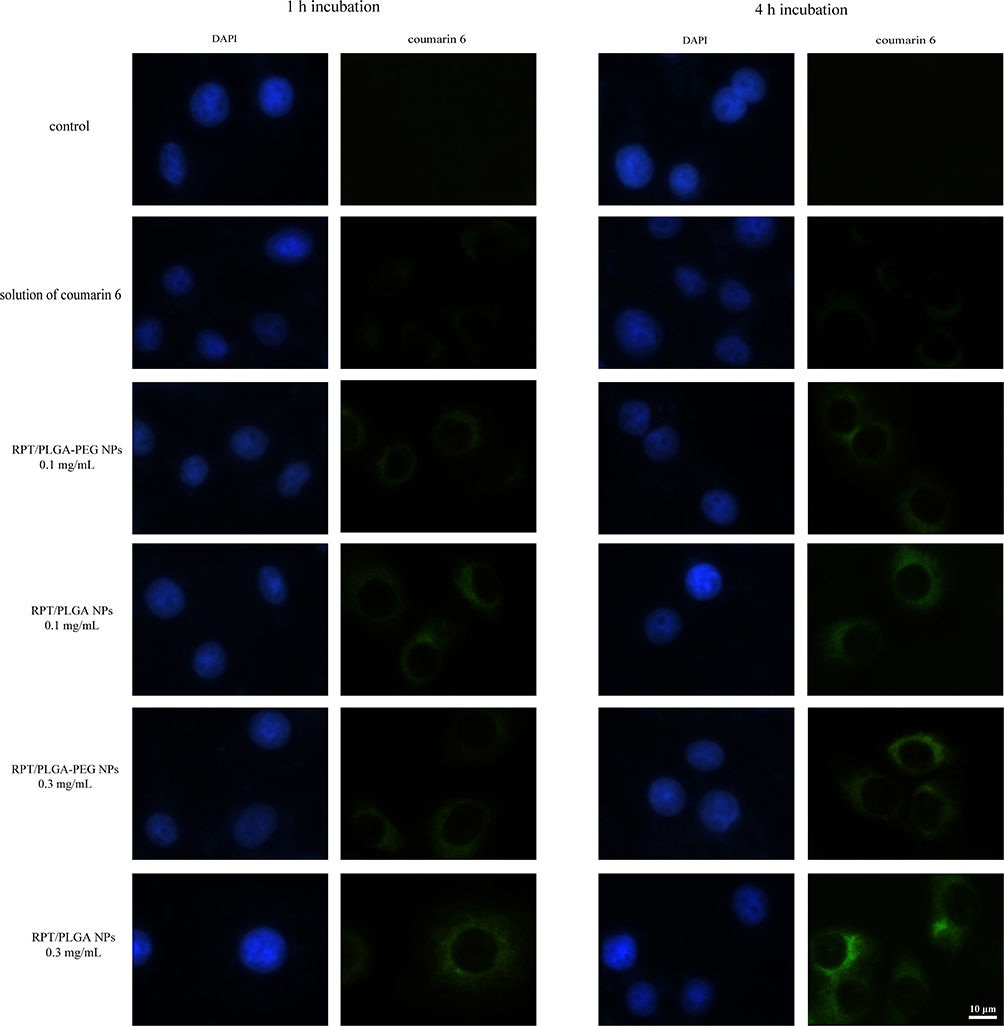 |
Figure 4 Fluorescent microscopy images of macrophages after incubation with NPs. |
Moreover, RPT/PLGA–PEG NPs were less internalised by macrophages than RPT/PLGA NPs. This might be due to a hydrophilic and inert PEG, which provides a steric barrier on the surface of NPs. Similar phenomena were reported in other studies, proposing that PEG will repel nanocarriers from the cell surface, especially for macrophages.29,51 Shan et al34 investigated the effect of PEG-grafted degree on the in vitro macrophage uptake. The results showed that PEGylation could reduce recognition by phagocytic cells, and the higher the PEG content in the copolymer, the more phagocytic cells are repelled. Sheng et al29 reported that when the PEG content increased from 5wt% to 10 wt%, the phagocytic uptake percentage was drastically reduced by 55%. The effect of PEG on the cellular uptake of NPs is widely discussed in the literature. PEGylation can significantly affect physicochemical properties such as particle size and surface charge, which are known to influence NP uptake and biological activity in organ and tissue structures.27 Haggag et al21 investigated the efficiency of the cellular uptake of PLGA and PEGylated PLGA NP by cancer cells. The results showed that in vitro uptake was enhanced with increasing PEG content. In their study, higher PEG content was associated with lower negative zeta potential and particle size, which might give rise to a high uptake. However, in our study, the difference in particle size or surface charge between the two types of NPs was relatively small, which may not afford a definitive statement about the size or surface charge dependence of cellular uptake. Thus, the less internalisation in the PLGA–PEG group may be ascribed to the intrinsic properties of the PEG layer and its interaction with proteins in serum-containing media. These effects were often explained by postulating that PEGylation prevents the formation of a protein corona on the NP surface. However, Pelaz et al52 further provided evidence that PEGylated NPs in serum-containing media are not devoid of a protein corona and that the adsorbed proteins penetrate and deform the PEG layer on the NP surface, thereby reducing NP uptake.
Haemolytic Toxicity
Haemolytic activity is an important factor to investigate the quality of NPs for administration. Excessive haemolysis activity could be seriously life-threatening.53 As shown in Figure 5, both types of RPT-loaded NPs developed in the present study were found to possess negligible haemolysis of erythrocytes. Even at the highest concentrations, the haemolytic rate was less than 5%. Therefore, RPT-loaded NPs were potentially feasible for systemic drug delivery through the blood circulation.
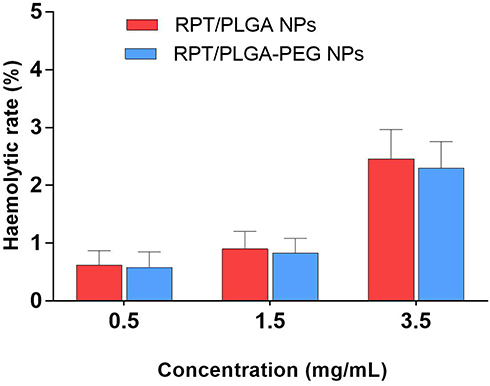 |
Figure 5 Haemolytic toxicity of various concentrations of NPs. |
In vitro Anti-Tubercular Studies
Microplate Based Alamar Blue Assay (MABA) is one of the robust methods to determine the inhibitory concentration of test formulations against M. tuberculosis bacterial strain.8 MIC was investigated to precisely determine the lowest concentration of NPs and free RPT needed to inhibit M. tuberculosis growth in vitro. The results (Figure 6) showed that the MIC of free PRT was 0.2 μg/mL, whereas those of PLGA NPs and PLGA–PEG NPs were 0.031 and 0.025 μg/mL (approximately 6.4- and 8-fold lower) (P<0.001), respectively, proving that both types of NPs were more effective than free drug. As NPs reached the same efficacy after employing lower doses, they improved the antimycobacterial activity of the RPT probably due to an enhanced penetration through the mycobacterial cell wall given that NPs have a higher contact surface and better delivery of RPT to its site of action. Moreover, PLGA–PEG NPs were more effective than PLGA NPs, which might be due to the following reasons: (i) PLGA–PEG NPs had a smaller size (ie a higher contact surface) than PLGA NPs. (ii) Poly-L-lactic acid/polyethylene glycol itself has shown antimicrobial and antibacterial activities on different microbes such as bacteria.54,55
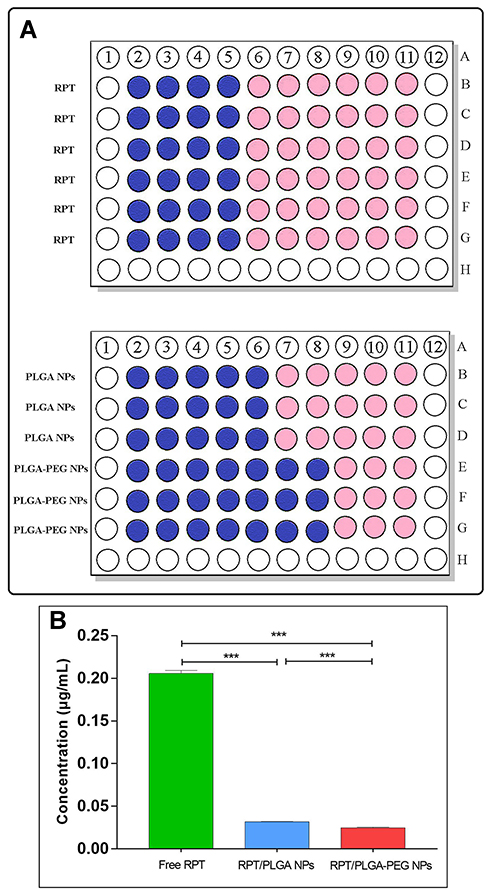 |
Figure 6 In vitro anti-tubercular studies for determination of MIC value. |
Pharmacokinetics Studies
The RPT serum concentration versus time curve for NPs and free drug after oral administration is shown in Figure 7. Drug-loaded NPs showed a prolonged drug release profile for up to 10 days (PLGA NPs) and 14 days (PLGA–PEG NPs) compared with only 3 days for free RPT. This is attributed to the role of NPs as long-circulating sustained-release drug delivery vehicles. The major pharmacokinetic parameters are shown in Table 4. Compared with free drug, both NPs resulted in a decrease in the elimination rate of the drug from serum, which was reflected in their half-lives (3.4- and 5.5-fold increases for PLGA and PLGA–PEG NPs, respectively). This difference between the two kinds of NPs was attributed to the presence of PEG on the surface of PLGA NPs and its role in evading the CI mechanisms of the body.22,56 Importantly, the bactericidal activity of RPT is exposure-dependent and related to the area under the concentration–time curve.17 The MRTs exhibited 3.3- and 5.2-fold increases for PLGA (85.28 h) and PLGA–PEG (135.82 h) NPs, respectively, compared with free RPT (26.18 h). As a result, AUC0–∞ was increased in case of PLGA–PEG NPs followed by PLGA NPs. On the other hand, no drug was detectable in the plasma beyond 3 days after administration of the free drug. The concept of slow and sustained release from a biodegradable particle is a crucial aspect of NP delivery. According to literature, PLGA-based nanostructure has gained significant attention due to its salient features including (i) biocompatibility and biodegradability, (ii) feasibility to design sustained release, (iii) practicable surface modifications for provision of stealth and effective biological interactions, (iv) protection of drug from degradation, (v) well-described preparations and synthesis approaches in accordance with various types of drugs and (vi) possibility to target specific organs or cells.57
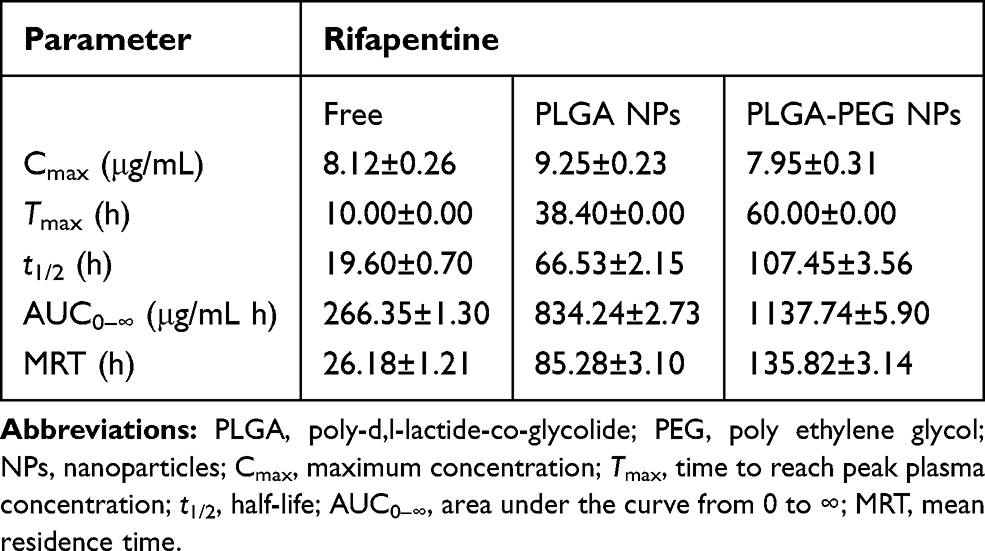 |
Table 4 Pharmacokinetic Parameters in Different Groups |
 |
Figure 7 Plasma concentration versus time after administration of different drug formulations. |
When freely administered RPT was given, the drug reached a high concentration in the plasma in hours but was then rapidly degraded. By contrast, when the same dose of RPT was delivered through a single administration of NPs, the plasma levels remained above the MIC at all time points measured. This finding means that PLGA NPs and especially PLGA–PEG NPs increased the average transit time of RPT molecules throughout the body. The surface decoration of NPs with PEG forms a barrier-like layer that sterically hinders the surface of the original NPs. Other than steric hindrance, it confers a hydrophilic neutral attribute to the surface of NPs that works as a layer opposing the interaction of the NPs surface and other materials.22,58 This provides efficient protection to NPs and gives long-circulation properties to RPT/PLGA–PEG NPs.
Tissue Distribution of RPT
The tissue distribution of RPT in different groups is shown in Figure 8. In the case of free RPT, the drug concentration in the liver was significantly higher than that of its NPs within the first 24 h, indicating the possibility of hepatotoxicity. Later, with further recirculation of blood in the liver, the RPT concentration tended to decrease gradually. The efficient localisation of PLGA NPs in the lungs might be due to the following reasons: (i) PLGA NPs can become physically trapped in the lung after recirculation being a result of lung capillary filtration effects. (ii) PLGA NPs were removed from the blood stream through phagocytosis by the cell of pulmonary reticuloendothelial system. M. tuberculosis mainly resides within the lung macrophages, and thus, delivery of RPT into the lungs at bactericidal concentrations is necessary for treating TB.59 Hence, it may be utilised as a potential tool for the delivery of RPT to the lung tissues through oral administration. In the case of RPT/PLGA–PEG NPs, the biodistribution in the liver, spleen, lung and kidney was elevated gradually and decreased afterward. The drug concentrations of the lung and liver in the PLGA–PEG group were much lower than those in the PLGA group within the first 48 h, which indicated that PLGA–PEG NPs could escape rapid uptake by phagocytic cells in mice. The fate of NPs in the body has been correlated to the surface properties.60 PLGA–PEG NPs have significantly reduced systemic clearance compared with similar particles without PEG, which is especially important for systemic circulation. Moreover, PLGA–PEG NPs had smaller sizes compared with PLGA NPs, which also contributed to escaping phagocytosis.
 |
Figure 8 In vivo organ distribution in mice. |
The toxicity of normal organs has always been a common limiting factor in the clinical use of antitubercular agents. One thing that needs to be noted is that the particles mainly accumulate in the liver following phagocytosis, and liver toxicity is one of the side effects of RPT in clinical application. Thus, the major organs of mice were sectioned and stained with H&E for morphological analysis. The results showed that there were no obvious abnormalities in the spleen, lung or kidney of the four groups, suggesting that free drug and its nano-formulations had no obvious acute toxicity in the spleen, lung or kidney. Moreover, histopathological evaluation of liver tissue of RPT/PLGA and RPT/PLGA–PEG groups showed no necrosis compared with the control group, whereas that of the free PRT group showed local necrosis (Figure 9). Blood biochemistry analysis was also conducted to evaluate the toxicity of RPT-loaded NPs. Most of the NPs were taken up and eliminated by ERS organs including the liver and kidneys.61 Therefore, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) for hepatic function and blood urea nitrogen (BUN) for kidney function were measured (Figure 10). The results showed that the AST levels of free RPT were higher (P<0.05) than those of other groups. The increase in AST levels in serum after the administration of free RPT appeared to be response due to the short-term accumulation of large doses (without slow-release effect) of the free drug in the liver. The amounts of ALT and BUN in all groups remained low and did not present significant differences compared with the control group (P>0.05).
 |
Figure 9 H&E staining of the organs of mice. |
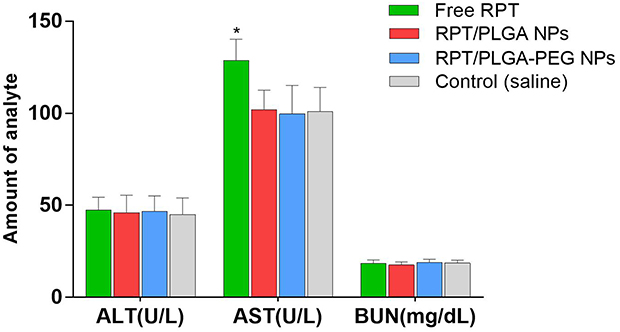 |
Figure 10 Serum biochemistry data on ALT, AST and BUN. |
Conclusion and Future Prospective
RPT-loaded PLGA and PLGA–PEG NPs were prepared using premix membrane homogenisation combined with solvent evaporation method. The resulting NPs showed suitable physicochemical characterisation without cellular or haemolytic toxicity and could be taken up by macrophages. In contrast to free drug, RPT-loaded NPs were more effective against M. tuberculosis in vitro. Moreover, the formulations were found to be well tolerated in mice and reduced hepatotoxicity, and the PEGylated formulations could provide sustained release of RPT above the MIC for at least 14 days in plasma and 16 days and longer in tissues. Hence, the formulations may be superior to existing ones not only because they have greater bactericidal activity but also because they have the potential to reduce the dosing frequency. Hence, daily conventional free drug treatment can be replaced with intermittent doses of NP-based drugs, which can lead to greater patient compliance and thus decrease the chances of chemotherapy failure and development of drug resistance.
Although the features of the developed formulations are promising, the results presented in this preliminary phase of the study are not sufficient to predict their effectiveness in human TB treatment, and translation from bench to bedside is challenged by their own toxicity concerns. To draw a principle on their toxicity, further in-depth investigations need to be conducted in various areas such as metabolism and biointeraction, long-term toxicity, oral capsule or tablet and effectiveness on infected cells and animal models. In pre-clinical phase, the animal studies will be further involved to demonstrate long-term efficacy and to identify appropriate dose ranges. Furthermore, the use of membrane emulsification method at the industrial scale is still hindered by the lack of a robust technique able to translate the results from laboratory scale to mass production. Thus, new technologies are required to overcome these challenges and significantly accelerate the clinical translation of NPs. However, nanomedicine may be the long-sought solution for improving patient compliance in TB chemotherapy. In the future, it will not only be achievable but also worthwhile in the treatment of TB.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (81860394).
Disclosure
The authors report no conflicts of interest in this study.
References
1. World Health Organization. Global tuberculosis report; 2019. Available from: https://www.who.int/tb/publications/global_report/en/.
2. Sukhithasri V, Vinod V, Varma S, Biswas R. Mycobacterium tuberculosis treatment modalities and recent insights. Curr Drug Deliv. 2014;11(6):744–752. doi:10.2174/1567201811666140619121728
3. Gaspar MM, Cruz A, Penha AF, et al. Rifabutin encapsulated in liposomes exhibits increased therapeutic activity in a model of disseminated tuberculosis. Int J Antimicrob Agents. 2008;31(1):37–45. doi:10.1016/j.ijantimicag.2007.08.008
4. Li L, Chou K, Deng J, et al. Two-stage total hip arthroplasty for patients with advanced active tuberculosis of the hip. J Orthop Surg Res. 2016;11(1):38. doi:10.1186/s13018-016-0364-3
5. Carneiro SP, Carvalho KV, de Oliveira Aguiar Soares RD. Functionalized rifampicin-loaded nanostructured lipid carriers enhance macrophages uptake and antimycobacterial activity. Colloids Surf B Biointerfaces. 2019;175:306–313. doi:10.1016/j.colsurfb.2018.12.003
6. Pandey R, Sharma A, Zahoor A, Sharma S, Khuller GK, Prasad BJ. Poly (DL-lactide-co-glycolide) nanoparticle-based inhalable sustained drug delivery system for experimental tuberculosis. Antimicrob Chemother. 2003;52(6):981–986. doi:10.1093/jac/dkg477
7. Saraogi GK, Gupta P, Gupta UD, Jain NK, Agrawal GP. Gelatin nanocarriers as potential vectors for effective management of tuberculosis. Int J Pharm. 2010;385(1–2):143–149.
8. Rani S, Gothwal A, Pandey PK, et al. HPMA-PLGA based nanoparticles for effective in vitro delivery of rifampicin. Pharm Res. 2018;36(1):19. doi:10.1007/s11095-018-2543-x
9. Shivangi MLS, Meena LS. A novel approach in treatment of tuberculosis by targeting drugs to infected macrophages using biodegradable nanoparticles. Appl Biochem Biotechnol. 2018;185(3):815–821. doi:10.1007/s12010-018-2695-5
10. Luque-Michel E, Imbuluzqueta E, Sebastián V, Blanco-Prieto MJ. Clinical advances of nanocarrier-based cancer therapy and diagnostics. Expert Opin Drug Deliv. 2017;14(1):75–92. doi:10.1080/17425247.2016.1205585
11. Albisa A, Piacentini E, Sebastian V, Arruebo M, Santamaria J, Giorno L. Preparation of drug-loaded PLGA-PEG nanoparticles by membrane-assisted nanoprecipitation. Pharm Res. 2017;34(6):1296–1308. doi:10.1007/s11095-017-2146-y
12. Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymer. 2011;3(3):1377–1397. doi:10.3390/polym3031377
13. Rafiei P, Haddadi A. Pharmacokinetic consequences of PLGA nanoparticles in docetaxel drug delivery. Pharm Nanotechnol. 2017;5(1):3–23. doi:10.2174/2211738505666161230110108
14. Xu Q, Ensign LM, Boylan NJ, et al. Impact of surface polyethylene glycol (PEG) density on biodegradable nanoparticle transport in mucus ex vivo and distribution in vivo. ACS Nano. 2015;9(9):9217–9227. doi:10.1021/acsnano.5b03876
15. Joseph S, Bunjes H. Evaluation of shirasu porous glass (SPG) membrane emulsification for the preparation of colloidal lipid drug carrier dispersions. Eur J Pharm Biopharm. 2014;87(1):178–186. doi:10.1016/j.ejpb.2013.11.010
16. Jarvis B, Lamb HM. Rifapentine. Drugs. 1998;56(4):607–617. doi:10.2165/00003495-199856040-00008
17. Chan JG, Bai X, Traini D. An update on the use of rifapentine for tuberculosis therapy. Expert Opin Drug Deliv. 2014;11(3):421–431. doi:10.1517/17425247.2014.877886
18. Zumla A, Chakaya J, Centis R, et al. Tuberculosis treatment and management-an update on treatment regimens, trials, new drugs, and adjunct therapies. Lancet Respir Med. 2015;3(3):220–234. doi:10.1016/S2213-2600(15)00063-6
19. Burman WJ, Gallicano K, Peloquin C. Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibacterials. Clin Pharmacokinet. 2001;40(5):327–341. doi:10.2165/00003088-200140050-00002
20. LiverTox. Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012.
21. Haggag YA, Matchett KB, Dakir E-H. Nano-encapsulation of a novel anti-Ran-GTPase peptide for blockade of regulator of chromosome condensation 1 (RCC1) function in MDA-MB-231 breast cancer cells. Int J Pharm. 2017;521(1–2):40–53. doi:10.1016/j.ijpharm.2017.02.006
22. Rafiei P, Haddadi A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: pharmacokinetics and biodistribution profile. Int J Nanomedicine. 2017;12:935–947. doi:10.2147/IJN.S121881
23. Rao J. Shedding light on tumors using nanoparticles. ACS Nano. 2008;2(10):1984–1986. doi:10.1021/nn800669n
24. Kircher MF, Mahmood U, King RS, Weissleder R, Josephson L. A multimodal nanoparticle for preoperative magnetic resonance imaging and intraoperative optical brain tumor delineation. Cancer Res. 2003;63(23):8122–8125.
25. Owens DE
26. Liu Y, Wu X, Mi Y, et al. PLGA nanoparticles for the oral delivery of nuciferine: preparation, physicochemical characterization and in vitro/in vivo studies. Drug Deliv. 2017;24(1):443–451. doi:10.1080/10717544.2016.1261381
27. Haggag YA, Matchett KB, Falconer RA, et al. Novel ran-RCC1 inhibitory peptide-loaded nanoparticles have anti-cancer efficacy in vitro and in vivo. Cancers (Basel). 2019;11(2):222. doi:10.3390/cancers11020222
28. Haggag YA, Faheem AM, Tambuwala MM, et al. Effect of poly(ethylene glycol) content and formulation parameters on particulate properties and intraperitoneal delivery of insulin from PLGA nanoparticles prepared using the double-emulsion evaporation procedure. Pharm Dev Technol. 2018;23(4):370–381. doi:10.1080/10837450.2017.1295066
29. Sheng Y, Yuan Y, Liu C, Tao X, Shan X, Xu F. In vitro macrophage uptake and in vivo biodistribution of PLA-PEG nanoparticles loaded with hemoglobin as blood substitutes: effect of PEG content. J Mater Sci Mater Med. 2009;20(9):1881–1891. doi:10.1007/s10856-009-3746-9
30. Yoo JW, Chambers E, Mitragotri S. Factors that control the circulation time of nanoparticles in blood: challenges, solutions and future prospects. Curr Pharm Des. 2010;16(21):2298–2307. doi:10.2174/138161210791920496
31. Wang J, Sui M, Fan W. Nanoparticles for tumor targeted therapies and their pharmacokinetics. Curr Drug Metab. 2010;11(2):129–141. doi:10.2174/138920010791110827
32. Haggag Y, Abdel-Wahab Y, Ojo O, et al. Preparation and in vivo evaluation of insulin-loaded biodegradable nanoparticles prepared from diblock copolymers of PLGA and PEG. Int J Pharm. 2016;499(1–2):236–246. doi:10.1016/j.ijpharm.2015.12.063
33. Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385(1–2):113–142. doi:10.1016/j.ijpharm.2009.10.018
34. Shan X, Liu C, Yuan Y, et al. In vitro macrophage uptake and in vivo biodistribution of long-circulation nanoparticles with poly(ethylene-glycol)-modified PLA (BAB type) triblock copolymer. Colloids Surf B Biointerfaces. 2009;72(2):303–311. doi:10.1016/j.colsurfb.2009.04.017
35. Grotz E, Tateosian N, Amiano N, et al. Nanotechnology in tuberculosis: state of the art and the challenges ahead. Pharm Res. 2018;35(11):213.
36. Magalhães J, Chaves L, C. Vieira A, G. Santos S, Pinheiro M, Reis S. Optimization of rifapentine-loaded lipid nanoparticles using a quality-by-design strategy. Pharmaceutics. 2020;12(1):75. doi:10.3390/pharmaceutics12010075
37. Chokshi NV, Khatri HN, Patel MM. Formulation, optimization, and characterization of rifampicin-loaded solid lipid nanoparticles for the treatment of tuberculosis. Drug Dev Ind Pharm. 2018;44(12):1975–1989. doi:10.1080/03639045.2018.1506472
38. Kumar G, Sharma S, Shafiq N, Khuller GK, Malhotra S. Optimization, in vitro-in vivo evaluation, and short-term tolerability of novel levofloxacin-loaded PLGA nanoparticle formulation. J Pharm Sci. 2012;101(6):2165–2176. doi:10.1002/jps.23087
39. Changsan N, Chan HK, Separovic F, Srichana T. Physicochemical characterization and stability of rifampicin liposome dry powder formulations for inhalation. J Pharm Sci. 2009;98(2):628–639. doi:10.1002/jps.21441
40. Vieira ACC, Chaves LL, Pinheiro S, et al. Mucoadhesive chitosan-coated solid lipid nanoparticles for better management of tuberculosis. Int J Pharm. 2018;536(1):478–485. doi:10.1016/j.ijpharm.2017.11.071
41. Tukulula M, Gouveia L, Paixao P, Hayeshi R, Naicker B, Dube A. Functionalization of PLGA nanoparticles with 1,3-β-glucan enhances the intracellular pharmacokinetics of rifampicin in macrophages. Pharm Res. 2018;35(6):111.
42. Esmaeili F, Hosseini-Nasr M, Rad-Malekshahi M, Samadi N, Atyabi F, Dinarvand R. Preparation and antibacterial activity evaluation of rifampicin-loaded poly lactide-co-glycolide nanoparticles. Nanomedicine. 2007;3(2):161–167. doi:10.1016/j.nano.2007.03.003
43. Pradhan R, Kim YI, Jeong JH, Choi HG, Yong CS, Kim JO. Fabrication, characterization and pharmacokinetic evaluation of doxorubicin-loaded water-in-oil-in-water microemulsions using a membrane emulsification technique. Chem Pharm Bull (Tokyo). 2014;62(9):875–882. doi:10.1248/cpb.c14-00231
44. Doan TV, Couet W, Olivier JC. Formulation and in vitro characterization of inhalable rifampicin-loaded PLGA microspheres for sustained lung delivery. Int J Pharm. 2011;414(1–2):112–117. doi:10.1016/j.ijpharm.2011.05.007
45. Coran T. Preparation of microemulsions and nanoemulsions by membrane emulsification. Colloids Surf A. 2019;579:1–14.
46. Sun G, Qi F, Wu J, Ma G, Ngai T. Preparation of uniform particle-stabilized emulsions using SPG membrane emulsification. Langmuir. 2014;30(24):7052–7056. doi:10.1021/la500701a
47. Singh H, Jindal S, Singh M, Sharma G, Kaur IP. Nano-formulation of rifampicin with enhanced bioavailability: development, characterization and in-vivo safety. Int J Pharm. 2015;485(1–2):138–151. doi:10.1016/j.ijpharm.2015.02.050
48. Jain AK, Goyal AK, Mishra N, Vaidya B, Mangal S, Vyas SP. PEG-PLA-PEG block copolymeric nanoparticles for oral immunization against hepatitis B. Int J Pharm. 2010;387(1–2):253–262. doi:10.1016/j.ijpharm.2009.12.013
49. Sahana DK, Mittal G, Bhardwaj V, Ravi Kumar MNV. PLGA nanoparticles for oral delivery of hydrophobic drugs: influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J Pharm Sci. 2008;97(4):1530–1542. doi:10.1002/jps.21158
50. Haggag YA, Osman MA, El-Gizawy SA, et al. Polymeric nano-encapsulation of 5-fluorouracil enhances anti-cancer activity and ameliorates side effects in solid ehrlich carcinoma-bearing mice. Biomed Pharmacother. 2018;105:215–224. doi:10.1016/j.biopha.2018.05.124
51. Griffiths G, Nyström B, Sable SB, Khuller GK. Nanobead-based interventions for the treatment and prevention of tuberculosis. Nat Rev Microbiol. 2010;8(11):827–834.
52. Pelaz B, Del Pino P, Maffre P, et al. Surface functionalization of nanoparticles with polyethylene glycol: effects on protein adsorption and cellular uptake. ACS Nano. 2015;9(7):6996–7008.
53. Wei YM, Liang J, Zheng XL, et al. Lung-targeting drug delivery system of baicalin-loaded nanoliposomes: development, biodistribution in rabbits, and pharmacodynamics in nude mice bearing orthotopic human lung cancer. Int J Nanomedicine. 2017;12:251–261. doi:10.2147/IJN.S119895
54. Rani S, Gothwal A, Khan I, et al. Smartly engineered PEGylated di-block nanopolymeric micelles: duo delivery of isoniazid and rifampicin against mycobacterium tuberculosis. AAPS PharmSciTech. 2018;19(7):3237–3248. doi:10.1208/s12249-018-1151-8
55. Sahan Y, Gurbuz O, Goncagu G, Kara A, Ozakin C. Antimicrobial effect of PEG–PLA on food-spoilage microorganisms. Food Sci Biotechnol. 2017;26(4):1123–1128. doi:10.1007/s10068-017-0138-7
56. Jain S, Spandana G, Agrawal AK, Kushwah V, Thanki K. Enhanced antitumor efficacy and reduced toxicity of docetaxel loaded estradiol functionalized stealth polymeric nanoparticles. Mol Pharm. 2015;12(11):3871–3884. doi:10.1021/acs.molpharmaceut.5b00281
57. Mir M, Ahmed N, Rehman AU. Recent applications of PLGA based nanostructures in drug delivery. Colloids Surf B Biointerfaces. 2017;159:217–231. doi:10.1016/j.colsurfb.2017.07.038
58. Patel B, Gupta N, Ahsan F. Particle engineering to enhance or lessen particle uptake by alveolar macrophages and to influence the therapeutic outcome. Eur J Pharm Biopharm. 2015;89:163–174. doi:10.1016/j.ejpb.2014.12.001
59. Liu Z, Li X, Xiu B, et al. A novel and simple preparative method for uniform-sized PLGA microspheres: preliminary application in antitubercular drug delivery. Colloids Surf B Biointerfaces. 2016;145:679–687. doi:10.1016/j.colsurfb.2016.05.085
60. Guo J, Gao X, Su L, et al. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials. 2011;32(31):8010–8020. doi:10.1016/j.biomaterials.2011.07.004
61. Ganipineni LP, Ucakar B, Joudiou N, et al. Magnetic targeting of paclitaxel-loaded poly(lactic-co-glycolic acid)-based nanoparticles for the treatment of glioblastoma. Int J Nanomedicine. 2018;13:4509–4521. doi:10.2147/IJN.S165184
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.