Back to Journals » Drug Design, Development and Therapy » Volume 9
Development of lovastatin-loaded poly(lactic acid) microspheres for sustained oral delivery: in vitro and ex vivo evaluation
Received 30 October 2014
Accepted for publication 4 December 2014
Published 10 February 2015 Volume 2015:9 Pages 791—798
DOI https://doi.org/10.2147/DDDT.S76676
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Qigang Guan,1 Wei Chen,2 Xianming Hu2
1Department of Cardiology, The First Affiliated Hospital of China Medical University, Shenyang, People’s Republic of China; 2Department of Pharmaceutical, Shenyang Institute of Pharmaceutical Industry, Shenyang, People’s Republic of China
Background: A novel lovastatin (LVT)-loaded poly(lactic acid) microsphere suitable for oral administration was developed in this study, and in vitro and in vivo characteristics were evaluated.
Methods: The designed microspheres were obtained by an improved emulsion-solvent evaporation method. The morphological examination, particle size, encapsulation ratio, drug loading, and in vitro release were characterized. Pharmacokinetics studies were used to show that microspheres possess more advantages than the conventional formulations.
Results: By using the emulsion-solvent evaporation method, it was simple to prepare microspheres and easy to scale up production. The morphology of formed microspheres showed a spherical shape with a smooth surface, without any particle aggregation. Mean size of the microspheres was 2.65±0.69 µm; the encapsulation efficiency was 92.5%±3.6%, and drug loading was 16.7%±2.1%. In vitro release indicated that the LVT microspheres had a well-sustained release efficacy, and ex vivo studies showed that after LVT was loaded to microspheres, the area under the plasma concentration-time curve from zero to the last measurable plasma concentration point and the extrapolation to time infinity increased significantly, which represented 2.63-fold and 2.49-fold increases, respectively, compared to suspensions. The rate of ex vivo clearance was significantly reduced.
Conclusion: This research proved that poly(lactic acid) microspheres can significantly prolong the drug circulation time in vivo and can also significantly increase the relative bioavailability of the drug.
Keywords: lovastatin, microspheres, PLA, in vitro release, pharmacokinetics
Introduction
Lovastatin (LVT; Figure 1), an antihyperlipidemic agent, inhibits the production of cholesterol by the liver.1 It lowers cholesterol levels through reversible and competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase, an enzyme involved in the biosynthesis of cholesterol. Since LVT (with a water solubility of 0.4×10−3 mg/mL) has higher log P-values (4.3) and good oil solubility (38 and 42 mg/mL in carbitol and propylene glycol monocaprylate, respectively), it is considered a desirable intestinal lymphatic transport substrate. In addition, LVT is rapidly metabolized in the intestine and liver, and therefore it has poor oral bioavailability (~5%).2 Cytochrome P450 3A4 metabolizes the lactone form of LVT into hydroxy acid and its metabolites.3,4 As a consequence of extensive hepatic extraction of LVT, availability of the drug to the general circulation is low and variable.5 Moreover, conventional oral dosage leads to fluctuations in plasma drug level, because of the method offers no control over drug delivery. To avoid the effects of individual differences and to prevent rapid clearance of the agent, using a sustained release drug delivery system is a feasible tactic.
 | Figure 1 The structure of lovastatin (A) and simvastatin (B). |
In recent years, drug-loaded microspheres have been used as a gastroretentive system for controlled drug delivery; the advantages are as follows:6–8 1) improvement in dissolution and bioavailability of drugs; 2) sustained and prolonged release of drugs; and 3) targeted drug delivery to a specific site or a particular organ in the body (eg, the colon), including intracellular structures such as lysosomes and the nucleus. Also, magnetically responsive microsphere systems can localize drug carriers and entrap agents at specific in vivo targeting areas. Microspheres can be designed into an optimal drug delivery system through the choice and formulation of various drug–polymer combinations, such as chitosan,9 poly(lactic acid) (PLA),10,11 poly(lactic-co-glycolic acid) (PLGA)12,13 and so on. PLA, a US Food and Drug Administration (FDA)-approved polymer, has been widely used in biodegradable polymer microspheres because of its great advantages of biodegradability and biocompatibility,11 and has gained much attention as a polymer for oral delivery. PLA, composed of lactic acids, would undergo hydrolysis and be metabolized to CO2 and water in the Krebs cycle,14 suggesting good safety with limited toxicity or tissue reaction.
By searching the PubMed database, we found only one article that described LVT microsphere preparation and in vivo and in vitro evaluation, but this study used cyclodextrin as a microsphere material, which has certain limitations.15 Thus, the present work is an effort to improve the bioavailability of LVT utilizing PLA microspheres for gastroretentive drug delivery. LVT-loaded PLA microspheres were prepared via improved emulsion-solvent evaporation method. Physicochemical characteristics were investigated to ensure that LVT-loaded PLA microspheres have good qualities and are suitable for oral delivery. In vitro drug release behavior was also studied for the microspheres as compared with LVT suspensions. In vivo, the pharmacokinetic properties of microspheres in rats were also examined using optimized formulations.
Materials and methods
Materials
LVT and simvastatin (internal standard; Figure 1) were kindly provided by Nanyang Chemical Co, Ltd (Fujian, People’s Republic of China). PLA, Mw 16,000, was obtained from Shandong Institute of Medical Instruments. Polyvinyl alcohol (PVA; MW 30,000) was purchased from Shanghai Sigma Co, Ltd (Shanghai, People’s Republic of China). All other chemical reagents used in this study were of analytical or chromatographical grade.
Preparation of LVT-loaded PLA microspheres
The improved emulsion-solvent evaporation method was used to prepare LVT-loaded PLA microspheres. Briefly, 0.25 mL of organic phase was prepared by completely dissolving both LVT (20 mg) and PLA (100 mg) in dichloromethane. Then, the organic phase was added to 2.5 mL of 2% (w/v) PVA solution dissolved in distilled water by stirring at a constant rate, then stirred for 1 minute at a high speed to form oil-in-water initial emulsion. Subsequently, the initial emulsion was added to 10 mL of 2% (w/v) PVA aqueous solution used as diluent, and magnetically stirred for 3 hours at room temperature to evaporate the organic solvent. The solidified LVT-loaded PLA microspheres were collected by centrifugation (10 minutes, 4,000 rpm) and washed three times with distilled water. The LVT-loaded PLA microsphere powder was obtained by the freeze-drying method. More specifically, the solidified microspheres were located in penicillin bottles, and then freeze-dried at −80°C for 24 hours in a lyophilizer (Alpha 2–4 LSC, CHRIST, Germany).
Morphology, particle size, and zeta potential
The surface and internal morphology of the microspheres were examined under a scanning electron microscope (6010LV; JEOL, Tokyo, Japan). Before analyzing the samples, the freeze-dried microspheres were gold-sputtered in an argon atmosphere.
The size and zeta potential of solidified as well as the freeze-dried microspheres were measured by laser particle size analyzer (Zetasizer Nano; Malvern Instruments, Malvern, UK). The samples were prepared by dispersing the solidified or freeze-dried microspheres in an appropriate amount of distilled water. The results were given by the apparatus automatically.
Drug loading and entrapment efficiency
Drug loading and encapsulation efficiency measures of LVT-loaded PLA microspheres were obtained by high-performance liquid chromatography (HPLC) using an ultraviolet spectrophotometer (LA-10T; Shimadzu Corp, Kyoto, Japan). Briefly, the freeze-dried microspheres (10 mg) were dissolved in an adequate quantity of phosphate buffer at pH 6.8 and then filtered using Whatman® filter paper. The drug loading and entrapment efficiency were calculated by the following equations:
|
|
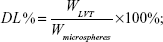
|
|
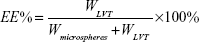
WLVT represents the amount of LVT loaded in the microspheres; Wmicrospheres represents the weight of the LVT microspheres.
Thermal analysis
Differential scanning calorimetry (DSC) was performed on LVT, blank microspheres, a LVT and blank microsphere mix, and LVT-loaded PLA microspheres. For the structural, crystal, and physical state characterization of the drug, DSC studies were performed. The DSC measurements were performed on a Shimadzu DSC (DSC 60), with thermal analyzer. Accurately weighed samples (about 3 mg) were placed in a sealed aluminum pan before heating under nitrogen flow (50 mL/minute) at a scanning rate of 10°C per minute from 100°C to 400°C An empty aluminum pan was used as a reference.17
Powder X-ray diffraction
The crystallinities of LVT and LVT-loaded PLA microspheres were evaluated by X-ray diffraction measurement recorded for LVT, blank microspheres, a physical mixture of LVT and blank microspheres, and LVT-loaded PLA microspheres, using an X-ray diffractometer (D8 Advance; Bruker Optik GmbH, Ettlingen, Germany). Scanning was performed up to 2θ range between 2° and 90° using an Ni-filter.18
In vitro release
Microsphere samples (150 mg) were placed into dialysis bags and suspended in 350 mL of release media of two kinds. One was 0.1 N hydrochloric acid solution (pH 1.2, containing 2% sodium dodecyl sulfate (SDS) and 5,000 unit/mL pectinase enzyme solution) to simulate gastric fluid. The other was phosphate-buffered saline (PBS) (pH 7.0, containing 2% SDS). Both release media had previously been equilibrated to 37°C±0.1°C and stirred at 100 rpm using the USP paddle method. At predetermined times of 2, 4, 8, and 12 hours, and 1, 2, 4, 8, 12, 16, 20, and 30 days, samples (2 mL) were withdrawn with a syringe filter (0.45 μm pore size) from the release media and replaced with an equal volume of the corresponding fresh media to maintain a constant volume. The test solution was analyzed by HPLC method as described in the HPLC analysis section. Triplicates were conducted and the results averaged.
Stability
The stability study program was based on the revised the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidelines. The microspheres were stored at 25°C±2°C and 60%±5% relative humidity (RH) for a period of 6 months and at 5°C±3°C for a period of 12 months, which is the accelerated storage temperature and long-term storage temperature, respectively, for products that are supposed to be refrigerated. During the stability study, the evaluation index of samples was investigated, including drug content, particle size distribution, and any physical changes. The drug content was determined by HPLC method. Acceleration test sampling times were at 0, 2, 4, and 6 months, and long-term storage sampling time trials were performed at 0, 3, 6, 9, and 12 months after preparation of formulation.
Pharmacokinetics
Twelve rats were used for pharmacokinetic experiments, mainly to observe changes in pharmacokinetic parameters of LVT after orally taken microspheres. All rats were randomly divided into two groups and oral administrated suspensions of LVT and LVT-loaded microspheres, respectively, by intragastric administration (10 mg/kg dose).
LVT suspensions (2 mg/mL) were prepared with 0.5% hydroxypropyl methylcellulose solution before administration. Blood samples (0.5 mL) were collected into heparinized tubes from the caudal vein at 0, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 16, and 24 hours after intragastric administration. Blood was immediately processed for plasma by centrifugation at 4000× g for 10 minutes. Plasma samples were collected at −70°C and kept frozen until analysis. The pharmacokinetic parameters were calculated by the drug concentration–time curve. The elimination half-life (T1/2) was determined by linear regression of the terminal portion of the plasma concentration–time data. The area under the plasma concentration-time curve from zero to the last measurable plasma concentration point (AUC0–t) was calculated by the linear trapezoidal method. Extrapolation to time infinity (AUC0–∞) was calculated as follows:
AUC0–∞ = AUC0–t + Ct/ke, | (3) |
where Ct was the last measurable plasma concentration and ke was the terminal elimination rate constant.
HPLC analysis
The amount of LVT in each sample was determined by HPLC (LC-10A; Shimadzu Corp) equipped with an ultraviolet spectrophotometer. Chromatographic separation was achieved using a Dikma Diamonsil® TM C18 column (5 μm, 200 mm ×4.6 mm; Dikma Technologies Inc, Lake Forest, CA, USA) and a precolumn (Nova-Pak, 10 μm, C18; Waters Corp, Milford, MA, USA). Phosphoric acid (0.01%):acetonitrile at a 39:61 ratio (v/v) was used as mobile phase at a flow rate of 1.0 mL/minute. The temperature of the column was maintained at ambient temperature (30°C). The detector wavelength was operated at 238 nm.
Each 100 μL aliquot of plasma was spiked with 10 μL of internal standard working solution (simvastatin, 0.5 mg/mL). Then, 250 μL of dichloromethane was added to the sample. The mixture was vortex mixed for 5 minutes and centrifuged at 4000× g for 5 minutes. The resulting supernatant was transferred to a glass tube and evaporated to dryness under a nitrogen stream at 45°C. The residue was reconstituted with 100 μL of mobile phase, and vortex mixed for 1 minute. The final samples were placed in amber auto sampler vials for HPLC analysis.
Statistical analysis
The data were statistically analyzed by unpaired Student’s t-test. A P-value of less than 0.05 was considered significant.
Results and discussion
Preparation of LVT-loaded PLA microspheres
Different drug delivery systems such as polymeric nanoparticles, microspheres, nano self-microemulsions, and solid lipid nanoparticles are intended to be used in LVT development.19,2,20,21 In these methods, biodegradable microspheres have been widely used for decades. PLA is a type of polymer approved by the FDA, safety biocompatible and biodegradable polymer materials, and has been partially used in medical instruments. After being absorbed by the body, PLA becomes a biodegradable oligomer or a non-toxic acid hydrolysis of the monomer. Because of its high safety, it has been widely used in oral delivery systems.22 Based on this understanding, this study used PLA as a drug carrier. Because of the difference include preparation methods and parameters, microspheres were prepared with different surface characteristics and drug loading capacity. In order to determine the optimal microsphere formulation and preparation process parameters, we first studied the relationship between drugs and PLA proportions. Results of drug loading and encapsulation efficiency indicated that 1:5 (LVT:PLA) was the optimal ratio. Then, we used orthogonal design to optimize the concentrations of PLA and PVA, as well as the ratio of the organic phase and water phase. The results indicated that the optimal formulation was PVT:PLA =1:5, 4% PLA, and an organic phase:water phase ratio of 1:10.
Physicochemical characteristics
Microspheres prepared using the optimal experimental conditions were globular in appearance and dispersed well. Scanning electron microscopy was used to visualize the particle diameter, and the structural and surface morphology of the emulsion-solvent evaporation microspheres (Figure 2). As seen from Figure 2, the surface morphology of the microspheres was smooth and discrete, with a regular spherical to near-spherical shape, which may be due to the use of PVA solution to stabilize emulsion droplets and hence resulted in well-formed microspheres. The size of the microspheres after freeze-drying was 2.65±0.69 μm, and well distributed. The zeta potential of microspheres was −13.1±3.1 mV. The average drug loading and the average encapsulation efficiency were 16.7%±2.1% and 92.5%±3.6%, respectively.
 | Figure 2 Scanning electron micrograph of LVT-loaded PLA microspheres. Magnification ×5,000 (A); and ×500 (B). |
Thermal analysis
Thermal analysis DSC was performed on LVT, blank microspheres, LVT and blank microsphere mix, and LVT-loaded PLA microspheres. For the structural, crystal, and physical state characterization of the drug, DSC studies were performed. Figure 3 is of a DSC thermogram, which showed LVT had a particular peak at about 165°C, which also appeared in the physical mixture of blank microspheres and LVT, indicating that LVT existed as a crystal in its natural state and did not have chemical interaction with the blank microspheres. In contrast, the disappearance of the particular peak in LVT-loaded PLA microspheres revealed that LVT existed in the PLA microspheres in an uncrystalized form, rather than in a crystallized form, just as in the physical mixture of blank microspheres and LVT.
 | Figure 3 DSC analyses of the samples. (A) LVT; (B) blank microspheres; (C) physical mixture of LVT and blank microspheres; and (D) LVT-loaded PLA microspheres. |
Powder XRD
The XRD spectra for pure drug, blank microspheres, the physical mixture of LVT and the blank microspheres, and the LVT-loaded PLA microspheres are depicted in Figure 4. LVT shows characteristic intense peaks between 2θ of 8 and 20, but in the cases of blank microspheres and drug-loaded microspheres, the intensity of peaks was decreased, indicating the amorphous nature of the drug after entrapment into PLA microspheres by freeze-drying.
 | Figure 4 X-ray diffraction spectra of (A) LVT; (B) blank microspheres; (C) physical mixture of LVT and blank microspheres; (D) LVT-loaded PLA microspheres. |
In vitro release
The drug release profiles from LVT-loaded PLA microspheres and LVT suspensions are shown in Figure 5. In comparison with LVT suspensions, LVT release from microspheres was biphasic, consisting of a relative fast phase (up to 24 hours), followed by a sustained release (1–30 days). Of the total LVT in the PLA microspheres, 27.6% (simulated gastric fluid) and 21.5% (PBS media) was released in the first 24 hours, which may reflect the significant amount of LVT adsorbed on, or incorporated near, the surface of the microspheres. In clinical practice, the early release of LVT microspheres would bring about fast effects on patients. After 24 hours, the fast release in the first 24 hours, LVT release from PLA microspheres was very slow. After 30 days, 79.1% of LVT was released from microspheres under the condition of PBS media, whereas only 62.6% of LVT was released from microspheres under the condition of simulated gastric fluid. In contrast, the release rate of LVT from suspension was very quick. Within 6 hours, more than 87.4% of the drug was released. After 24 hours, the cumulative release rate was about 98% (simulated gastric fluid). The in vitro release was kinetically analyzed according to zero-order, first-order, Higuchi, and Weibull models. The relatively high correlation coefficient values obtained from the analysis of the amount of the drug released versus the square root of time demonstrated that the release followed the Higuchi model, as shown in Table 1. These results indicated that LVT microspheres had a good controlled release effect. In our previous preliminary experiments, PBS was used as the release medium. However, since LVT was almost insoluble in water,23 it was difficult during experiments to observe measured concentrations of the drug being released; thus, in this study we added 2% SDS in order to satisfy the sink condition.
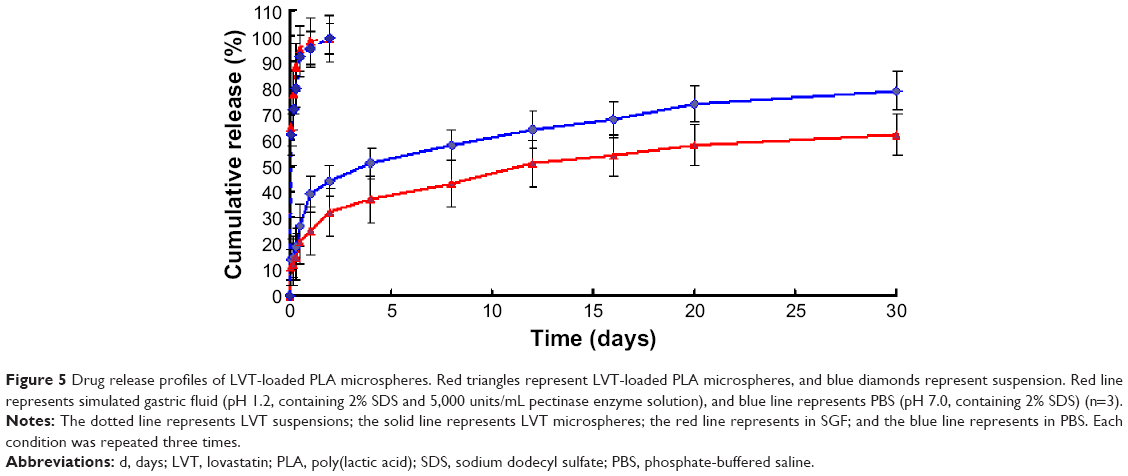 | Figure 5 Drug release profiles of LVT-loaded PLA microspheres. Red triangles represent LVT-loaded PLA microspheres, and blue diamonds represent suspension. Red line represents simulated gastric fluid (pH 1.2, containing 2% SDS and 5,000 units/mL pectinase enzyme solution), and blue line represents PBS (pH 7.0, containing 2% SDS) (n=3). |
 | Table 1 Correlation coefficients for kinetic analysis of release data for LVT-microspheres |
Stability
Long-term stability tests showed that, throughout the 12 months of observation, LVT microspheres underwent little change overall, with only 2.1% decrease in the initial drug concentration. Accelerated stability tests also showed that at 25°C±2°C and 60%±5% RH, the microspheres remained stable during the observation period, and RH fell by only 6.3%. There was no significant change in average particle size; changes in physical and chemical properties were negligible, and did not have an impact on the quality of the microspheres.
Pharmacokinetics
Figure 6 shows the mean plasma concentration–time profiles of LVT after oral administration of a single 10 mg/kg dose of suspensions and microspheres to rats. Based on the analysis of the models and parameters, it was concluded that the in vivo pharmacokinetics of microspheres in plasma could be described by the two-compartment model of under oral administration. The pharmacokinetic parameters are reported in Table 1. It can be seen from Table 2 that in comparison with LVT suspensions, LVT-loaded PLA microspheres altered the distribution of LVT in vivo, and that T1/2 values after oral administration of LVT microspheres (t1/2 =5.48 hours) were prolonged remarkably over T1/2 values (t1/2 =1.32 hours) after oral administration of LVT suspensions. These results indicated that the LVT microspheres had sustained release efficacy. In addition, LVT exhibited poor oral bioavailability because of rapid metabolism in the gut and liver. After the LVT was made to microspheres, AUC0–t and AUC0–∞ increased significantly; values were 2.63-fold and 2.49-fold higher, respectively, compared with suspensions. The rate ex vivo clearance was significantly reduced. This reduction proved that PLA microspheres can significantly prolong the drug circulation time in vivo and significantly increase the relative bioavailability of the drug.
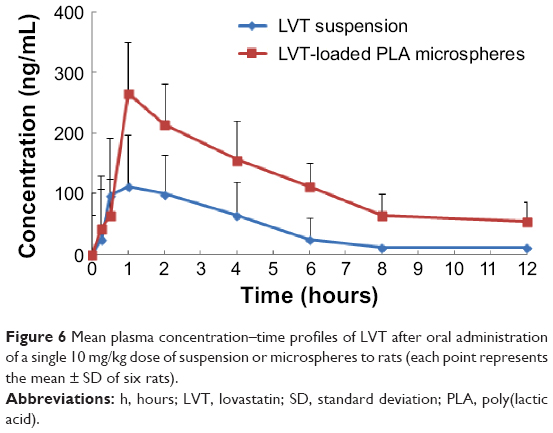 | Figure 6 Mean plasma concentration–time profiles of LVT after oral administration |
 | Table 2 Pharmacokinetic parameters of the two formulations |
Conclusion
The improved emulsion-solvent evaporation method was successfully used to prepare LVT-loaded PLA microspheres. The surface morphology of the microspheres was smooth and discrete, with a regular spherical to near-spherical shape. The size of the microspheres after freeze-drying was 2.65±0.69 μm, and the zeta potential was −13.1±3.1 mV. The average drug loading and the average encapsulation efficiency were 16.7%±2.1% and 92.5%±3.6%, respectively. DSC revealed that LVT existed in the PLA microspheres in an uncrystalized form, rather than in a crystal form. In vitro and in vivo studies indicated that LVT-loaded PLA microspheres had a well-controlled release efficacy.
Disclosure
The authors report no conflicts of interest in this work.
References
Jenkins DJ, Kendall CW, Marchie A, et al. Effects of a dietary portfolio of cholesterol-lowering foods vs lovastatin on serum lipids and C-reactive protein. JAMA. 2003;290(4):502–510. | ||
Rao S, Tan A, Boyd BJ, Prestidge CA. Synergistic role of self-emulsifying lipids and nanostructured porous silica particles in optimizing the oral delivery of lovastatin. Nanomedicine (Lond). Epub 2014 Jun 18. | ||
Lennernäs H, Fager G. Pharmacodynamics and pharmacokinetics of the HMG-CoA reductase inhibitors. Similarities and differences. Clin Pharmacokinet. 1997;32(5):403–425. | ||
Chen CH, Uang YS, Wang ST, et al. Interaction between red yeast rice and CYP450 enzymes/P-glycoprotein and Its implication for the clinical pharmacokinetics of lovastatin. Evid Based Complement Alternat Med. 2012;2012:127043. | ||
Sun JX, Niecestro R, Phillips G, Shen J, Lukacsko P, Friedhoff L. Comparative pharmacokinetics of lovastatin extended-release tablets and lovastatin immediate-release tablets in humans. J Clin Pharmacol. 2002;42(2):198–204. | ||
Thombre NA, Gide PS. Floating-bioadhesive gastroretentive Caesalpinia pulcherrima-based beads of amoxicillin trihydrate for Helicobacter pylori eradication. Drug Deliv. Epub 2014 May 28. | ||
Dube TS, Ranpise NS, Ranade AN. Formulation and evaluation of gastroretentive microballoons containing baclofen for a floating oral controlled drug delivery system. Curr Drug Deliv. 2014;11(6):805–816. | ||
Goswami N, Joshi G, Sawant K. Floating microspheres of valacyclovir HCl: formulation, optimization, characterization, in vitro and in vivo floatability studies. J Pharm Bioallied Sci. 2012;4(Suppl 1):S8–S9. | ||
Sun Y, Cui F, Shi K, Wang J, Niu M, Ma R. The effect of chitosan molecular weight on the characteristics of spray-dried methotrexate-loaded chitosan microspheres for nasal administration. Drug Dev Ind Pharm. 2009;35(3):379–386. | ||
Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28(1):5–24. | ||
Ruan G, Feng SS. Preparation and characterization of poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) (PLA-PEG-PLA) microspheres for controlled release of paclitaxel. Biomaterials. 2003;24(27):5037–5044. | ||
Tai IC, Fu YC, Wang CK, et al. Local delivery of controlled-release simvastatin/PLGA/HAp microspheres enhances bone repair. Int J Nanomedicine. 2013;8:3895–3904. | ||
Nath SD, Son S, Sadiasa A, Min YK, Lee BT. Preparation and characterization of PLGA microspheres by the electrospraying method for delivering simvastatin for bone regeneration. Int J Pharm. 2013;443(1–2):87–94. | ||
Tamber H, Johansen P, Merkle HP, Gander B. Formulation aspects of biodegradable polymeric microspheres for antigen delivery. Adv Drug Deliv Rev. 2005;57(3):357–376. | ||
Kumar S, Nagpal K, Singh S, Mishra D. Improved bioavailability through floating microspheres of lovastatin. Daru. 2011;19(1):57–64. | ||
Obeidat WM, Price JC. Preparation and evaluation of Eudragit S 100 microspheres as pH-sensitive release preparations for piroxicam and theophylline using the emulsion-solvent evaporation method. J Microencapsul. 2006;23(2):195–202. | ||
Tadwee I, Shahi SR, Thube MW. Spray dried nasal mucoadhesive microspheres of carbamazepine: preparation and invitro/ex-vivo evaluation. Int J Sci Publ Res Pharm. 2011;2:23–32. | ||
Mahajan HS, Gattani SG. Nasal administration of ondansetron using a novel microspheres delivery system Part II: ex vivo and in vivo. Pharm Dev Technol. 2010;15(6):653–657. | ||
Ho MH, Chiang CP, Liu YF, et al. Highly efficient release of lovastatin from poly(lactic-co-glycolic acid) nanoparticles enhances bone repair in rats. J Orthop Res. 2011;29(10):1504–1510. | ||
Beg S, Sandhu PS, Batra RS, Khurana RK, Singh B . QbD-based systematic development of novel optimized solid self-nanoemulsifying drug delivery systems (SNEDDS) of lovastatin with enhanced biopharmaceutical performance. Drug Deliv. Epub 2014 Mar 27. | ||
Suresh G, Manjunath K, Venkateswarlu V, Satyanarayana V. Preparation, characterization, and in vitro and in vivo evaluation of lovastatin solid lipid nanoparticles. AAPS Pharm Sci Tech. 2007;8(1):24. | ||
Mohamed F, van der Walle CF. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J Pharm Sci. 2008;97(1):71–78. | ||
Frishman WH, Rapier RC. Lovastatin: an HMG-CoA reductase inhibitor for lowering cholesterol. Med Clin North Am. 1989;73(2):437–448. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.