Back to Journals » International Journal of Nanomedicine » Volume 14
Development of a stable single-vial liposomal formulation for vincristine
Authors Mao W, Wu F, Lee RJ, Lu W, Wang J ![]()
Received 13 February 2019
Accepted for publication 8 May 2019
Published 21 June 2019 Volume 2019:14 Pages 4461—4474
DOI https://doi.org/10.2147/IJN.S205276
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Wenxue Mao,1 Fan Wu,2 Robert J Lee,3 Weigen Lu,4 Jianxin Wang1
1Department of Pharmaceutics, School of Pharmacy, Fudan University & Key Laboratory of Smart Drug Delivery, Ministry of Education, Shanghai 201203, People’s Republic of China; 2College of Medicine, Des Moines University, Des Moines, IA 50312, USA; 3Division of Pharmaceutics and Pharmaceutical Chemistry, Ohio State University, Columbus, OH 43210, USA; 4China State Institute of Pharmaceutical Industry, Shanghai 201203, People’s Republic of China
Background: Vincristine is a potent therapeutic agent with well-defined activity against hematologic malignancies and solid tumors. It is a cell-cycle specific drug with concentration and exposure duration dependent activity. When used by liposomal delivery, it exhibits enhanced anti-tumor activity. However, vincristine liposome formulation in the clinic is supplied as a 3-vial-kit due to lacking sufficient stability. So it has to be prepared in situ prior to use through a multi-step process.
Purpose: The purpose here is to develop a more stable and ready-to-use liposomal formulation for vincritstine in one vial.
Patients and methods: A series of preparations were investigated based on sphingomyelin/cholesterol/PEG2000-DSPE lipid composition, with different drug/lipid (D/L) ratios (1/10, 1/5, 1/2), using an active sucrose octasulfate triethylamine salt gradient loading method. In this work, compared to generic vincristine sulfate liposome injection (GVM), the stability both in vivo and in vitro and efficacy in vivo of novel vincristine liposomes were investigated.
Results: It was shown that the degradation of vincristine during 2–8°C storage was significantly decreased from 8.2% in 1 month (GVM) to 2.9% in 12 months (D/L ratio 1/5). The half-time for sphingomyelin/cholesterol/PEG2000-DSPE liposomes in vivo could be adjusted from 17.4 h (D/L ratio 1/10) to 22.7 h (D/L ratio 1/2) in rats, while the half-time for GVM was only 11.1 h. The increase in drug retention contributed to the lower in vivo toxicity. The antitumor efficacy was evaluated using a human melanoma tumor model and showed remarkable improvement compared to GVM.
Conclusion: The study demonstrates that the new formulation with the drug/lipid ratio of 1/5 owns a higher encapsulation efficiency, better stability, lower toxicity and superior antitumor efficacy, which is screened out for further development.
Keywords: vincristine sulfate, liposome, TEA-SOS, storage stability, and anti-tumor efficacy
Introduction
Vincristine (VCR) is a cytotoxic alkaloid that possesses extensive antitumor activity.1 As a microtubule inhibitor, with an M-phase cell-cycle specific antitumor mechanism, its efficacy is concentration and exposure duration dependent.2 However, the pharmacokinetic (PK) profile shows a rapid clearance rate and a large volume of bio-distribution in body.3 These sub-optimal PK properties and also the dose-related neurotoxicity prevent its full potential.4
Liposomal formulations have been shown to prolong plasma half-life and increase drug accumulation in tumor tissue by enhancing permeability and retention (EPR) effects while mitigating drug toxicity.5–8 In 2012, FDA approved vincristine sulfate liposome injection (VSLI; Marqibo®, Spectrum Pharmaceuticals, Inc., Henderson, NV, USA), which is a sphingomyelin (SM)/cholesterol (Chol) liposome by loading the drug with pH gradient method. It was confirmed to have good therapeutic effect on patients with Philadelphia chromosome-negative (Ph-) acute lymphoblastic leukemia (ALL).9 However, this VCR liposome formulation faces several challenges. One issue is its tedious preparation process. Due to its long-term stability limitation after VCR encapsulation, in order to achieve a nominally stable product, VSLI is supplied as a 3-vial-kit and its preparation requires an on-site, multi-step drug loading process and it must be in a biological safety cabinet or by established pharmacy safety procedures.10
A big challenge for stable liposomal VCR preparation is the chemical instability of VCR. Stability studies for VSLI showed that after VCR-loading, degradation occurred within 24 h at room temperature.11 The typical degradation route for VCR is oxidation and hydrolysis.12 Yunning et al13 attempted to weaken the oxidation of VCR by adding antioxidants into VCR preparations. The result exhibited a positive impact, yet far from enough to long-term storage. The main degradation route for VCR in an aqueous environment is hydrolysis.11 It seems that VCR is most stable in solid state or in an insulated oxygen environment. Freeze-dried VCR liposome formulation has been developed.14 But the lyophilization process means higher cost and longer manufacture time, usually one to several days. Meanwhile, the encapsulation efficiency (EE) of VCR would also be decreased during the rehydration process, and the increase of free VCR would strengthen the safety concern of drugs. Beside freeze–drying technology, different drug loading methods can also influence the formulation stability through changing the existential form of encapsulated VCR. By generating a transmembrane pH gradient, VSLI successfully detains VCR in a solubilization state inside the liposomes, but that does not prevent the drug hydrolysis.11,15,16 Ion gradient is another active loading method often used for amphipathic weak bases. Two of the successful cases are ammonium sulfate gradient and TEA-SOS gradient. Doxil® (doxorubicin HCl liposome injection [DLI], Janssen, Raritan, NJ, USA) was the first FDA approved nano-drug in 1995.17 By utilizing an ammonium sulfate gradient, DLI traps doxorubicin as nanocrystals within the liposomes and has a shelf life of approximately 18 months in a pre-loading formulation.18 The sucrose octasulfate triethylamine salt (TEA-SOS) gradient was first reported for irinotecan liposomes.19 TEA-SOS could form electrostatically stabilized complexes with the amphipathic drugs, such as irinotecan, which improves both the encapsulation efficiency and the in vitro stability of the formulation. This method has been successfully applied in irinotecan liposome injection (Onivyde®, Merrimack Pharmaceuticals, Inc., Cambridge, MA, USA). Other than ammonium sulfate liposome, the inside of the TEA-SOS liposome is composed of polyanion and a substituted ammonium. This combination gives liposomes higher loading efficiency, and more stable inner drug form with less drug release,20 which may be feasible for VCR encapsulation with a purpose of preventing VCR degradation in liposomes and prolonging in vivo circulation time.
Further improvements that can be made for drug release kinetics is altering the lipid composition. This will yield higher drug retention within the liposome during systemic circulation and decrease plasma clearance rate.17 By modifying the amount of Chol in the membrane, the saturation and length of the fatty acid chains will influence the membrane permeability.21,22 In previous VCR liposomal development, the composition varied from egg phosphatidylcholine (EPC)/Chol to distearoyl phosphocholine (DSPC)/Chol and finally to SM/Chol, which ultimately increased therapeutic activity after i.v. administration.23 To reduce plasma clearance, integration of the steric stabilizing lipid PEG-distearoyl phosphatidylethanolamine (DSPE) at 5 mol% into the SM/Chol composition significantly increased the circulation longevity of the SM/Chol liposomes. Conversely, the antitumor effect was not improved due to increased VCR leakage from the PEG-containing liposomes.24 However, considering for a stable VCR intention, it is still a meaningful effort using PEG-modified liposome aiming at its potency enhancement.
This study seeks to optimize the VCR liposome delivery system to extend its application potential by developing a stable and ready-to-use formulation utilizing TEA-SOS intraliposomal stabilization strategy and altering SM/Chol/PEG-lipid composition. In addition, three different drug-to-lipid ratios were investigated in terms of in vitro and in vivo stability, pharmacokinetics, toxicities and antitumor efficacies in animals. In all the studies, VSLI was used as a reference formulation. Finally, this study provides a single vial solution for novel commercial development of VCR liposome formulation.
Materials and methods
Materials
SM, Chol and 1, 2-distearoyl-Sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (PEG2000-DSPE, Mw=2805) were purchased from Lipoid GmbH (Ludwigshafen, Germany). TEA-SOS was donated from Shanghai DDSome Laboratories CO., Ltd. (Shanghai, People's Republic of China). Vincristine sulfate was obtained from Guangzhou Hanfang Pharmaceutical Co., Ltd. (Guangzhou, People's Republic of China). Histidine was provided by Shanghai Ajinomoto Amino Acid Co., Ltd. (Shanghai, People's Republic of China). All other chemicals were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, People's Republic of China). Human melanoma cell line A375 was purchased from Shanghai Institute of Pharmaceutical Industry (Shanghai, People's Republic of China). All chemicals used were of analytical reagent grade or above.
Animals
Male ICR mice (7–8 weeks old, 28±3 g weight), male Wistar rats (8–9 weeks old, 235±10 g weight) and BALB/c nude mouse (7–8 weeks old, 28±3 g weight) were all purchased from SLAC Laboratory Animal (Shanghai, People's Republic of China). All animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the National Research Council the Animal Management Rules of the Ministry of Health of the People’s Republic of China. The protocol was approved by the Institutional Animal Care and Use Committee of Fudan University. Animals were given a commercial diet and water ad libitum.
Liposome preparation and drug loading
Preparation of stable and ready-to-use liposomal VCR (SRLVs)
Required amounts of SM, Chol and PEG2000-DSPE (79:20:1, w/w) were dissolved in appropriate amount of ethanol (phospholipid/ethanol, 1:10, w/v). The resulting organic phase was instilled by a syringe into a defined volume of TEA-SOS (500 mM, 1:9, v/v) under magnetic stirring at 65°C. Then, the formulation was stirred for 30 min and extruded through an 80 nm polycarbonate membrane (Whatman International Ltd, Maidstone, UK) at 65°C. The extrusion step was repeated until a homogeneous batch of liposomes with mean particle size of 95–105 nm was obtained. Followed by cross-flow ultrafiltration, free TEA-SOS and ethanol were removed and blank liposomes were produced. An appropriate amount of vincristine sulfate was dissolved with glucose and histidine to prepare a VCR solution. The lipid concentration of the blank liposome was measured by HPLC and mixed with the vincristine sulfate solution to a final lipid concentration of 10 mg/mL (SRLV-1), 5 mg/mL (SRLV-2), or 2 mg/mL (SRLV-3), and a final drug concentration of 1 mg/mL. VCR-liposomes were obtained by water bath the mixture at 65°C for 30 min.
Preparation of positive control drug
The formulation and preparation method of the control drug were referred from labels and patents11,15,16 of VSLI. Required amounts of SM and Chol (73.5:29.5, w/w) were dissolved in an appropriate amount of CHCl3 (phospholipid: CHCl3, 1:10, w/v) and then the solvent was removed under negative pressure at 65°C. Spontaneously, liposome formation occurred thereafter and the dried film was hydrated with prescribed amounts of citrate buffer (33.6 mg/mL citric acid and 35.4 mg/mL sodium citrate) at 65°C. The suspension was then extruded through an 80 nm polycarbonate membrane as above to a final liposome mean particle size 95–105 nm. VCR was loaded into the liposomes as described in the label of VSLI.
Methods of VCR quantification
VCR quantification was determined by HPLC (2998 PDA Detector, Waters, USA) at 297 nm and 30°C, using a C8 column (250×4.6 mm), 20 μL injection volume, methanol: diethylamine buffer (70:30, v/v) as mobile phase, and 1.0 mL/min flow rate.
Particle size and zeta potential analysis
Both the particle size and zeta potential analysis of the liposomes were measured by Nano-ZS90 Laser Particle Size (Malvern Panalytical, Malvern, UK). The samples for particle size determination were diluted in saline and measured 3 times at 25°C. The zeta potential analysis was tested on the original liquids. All measurements were performed in triplicate.
Cryogenic transmission electron microscopy (Cryo-TEM)
Cryo-TEM was performed using a Talos F200C CryoTwin Transmission Electron Microscope (Thermo Fisher Scientific, Waltham, MA, USA) operated at an acceleration voltage 200 kV in TEM uP SA Zoom Image mode. Images were recorded on the BM-Ceta camera (Thermo Fisher Scientific).
First, the grids with holey carbon film (Quantifoil R 1.2/1.3; Quantifoil Micro Tools GmbH, Großlöbichau, Germany) were activated for 20 s at 8 mA using a Femto plasma cleaner (Diener Electronic, Ebhausen, Germany). Sample preparations were performed by applying 5 μL of the solution on the grid. Excess liquid was removed with filter paper and the samples were vitrified immediately after blotting by plunging the grid into liquid ethane held at approximately −183°C. Samples were kept under liquid nitrogen until TEM analysis. Images at 45,000× were captured for each sample.
Determination of encapsulation efficiency
Encapsulation efficiency (EE) was determined by Sepharose Column method using a Sepharose CL-4B column (10×100 mm). A 200 μL VCR-liposome was loaded on the column which was pre-equilibrated with saline and then eluted with the same solution. Liposome-encapsulated and free VCR were collected respectively. Both encapsulated VCR (CLoaded) and free VCR (CFree) collected from the Sepharose CL-4B column were determined by HPLC after diluted with methanol.
The EE was calculated according to the following equation:
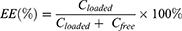
Stability
Storage stability
The stability of the four formulations after drug-loading was investigated by the changes in encapsulation efficiency and vincristine sulfate concentration. After drug loading (as described in the Preparation of stable and ready-to-use liposomal VCR and Preparation of positive control drug sections), both EEs and VCR concentrations of the four VCR liposomes were tested at time 0. For long-term studies, all four samples were deposited in a freeze at 5±3°C (2–8°C) for 12 months and tested in the time of 1st month, 3rd month, 6th month and 12th month. Under accelerated storage conditions, the samples were stored in a constant temperature humidity chamber at 25±2°C/60% ±5% RH for 3 months. The accelerated stability test results were measured in the time of 1st month, 2nd month and 3rd month.
In vitro release
Two different in vitro release systems have been tested in this study. In test A, 1 mL VCR-loaded liposomes were diluted in 4 mL (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)/NaCl serum solution (HEPES/NaCl 2.38 g: 4.21 g: 1 L in 50% serum w/w, pH 7.2). The diluted liposomal drug was then placed into a constant temperature chamber at 37°C
. In test B, 1 mL VCR-loaded liposomes were diluted in 4 mL, C6H15NHCl/histidine solution (50 mM: 200 mM, pH 6.5). This test was performed at 37°C and 42°C respectively. EE was tested by the sepharose column method as described in the Determination of encapsulation efficiency section at 0, 0.5, 2, 4, 8, 12, and 24 h after dilution. The drug release ratio (RR) was calculated as below:
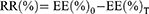
where EE(%)0 is the EE(%) of the samples at time 0 (immediately after dilution) and EE(%)T is the EE(%) at a certain time T.
Animal experiments
Pharmacokinetics studies
Forty healthy Wistar rats (235±20 g) were randomly assigned into four groups of 10. The animals were kept in a temperature-controlled laboratory (25±2°C) with natural lighting, free diet, and were fasted overnight prior to the experiment. All animals were given 1 mg/kg VCR liposome intravenously. After administration, 300–400 μL blood samples were collected from the retro-orbital plexus at designated time points (0.03, 0.5, 1, 2, 4, 8, 12, 24, and 48 h post-injection). The blood samples were placed in heparin sodium anticoagulant tube and centrifuged at 1,600 g for 10 min. The separated plasma was stored at −20°C until analysis.
In vivo toxicity study
Male ICR mice (7–8 weeks old) were housed in a temperature-controlled laboratory (25±2°C) with natural lighting and free diet. After 1 week’s acclimatization, 122 of them were randomized to 17 groups with 6–10 per group. Bodyweights (BW) were recorded once a day during the experiment.
In the single-dose study, 12 groups (n=6) were administered with four different VCR liposomal formulations at doses of 3, 4, or 5 mg/kg, separately, by i.v. injection once and sacrificed after 15 days. In the repeated dose study, four groups of mice (n=10) were administered with four different VCR liposomal formulations (1 mg/kg) by i.v. injection for five consecutive days and sacrificed 15 days after the last dose. The control group (n=10) were given saline (5 mL/kg) once by i.v. injection. The death rate (DR) was calculated as below at the end of the experiment:
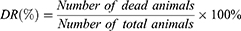
In vivo anticancer efficacy
Tumors were established by subcutaneous flank injections in mice with 2×105 human melanoma A375 cells in 0.1 mL of phosphate buffered balanced salt solution (PBS). Eleven days later, mice (with mean tumor volume, 250±100 mm3) were randomized into five treatment groups of 6 animals per group. Then, the treated animals received three tail vein injections at a dose of 2 mg/kg at day 1, day 5 and day 9. Group 5 were given 25 mL/kg saline solution i.v. on the same day. Tumor size was measured by vernier caliper twice a week, and tumor volume was calculated by the following formula:
Tumor Volume = (Length x width2)/2.
On day 14 post-administration, all mice were sacrificed by cervical dislocation to investigate the antitumor efficacy based on both body weight and the tumor growth inhibition (TGI).
<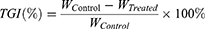
WTreated: average tumor weight in treatment group
WControl: average tumor weight in blank control group
Statistical analysis
All data were presented as the mean ± SD. Statistical analysis was conducted by Student’s t-test or ANOVA analysis. Probability values <0.05 were considered significant.
Results
Preparation and characterization of VCR liposomes
This study attempted to develop a stable nano-vehicle for VCR composed of SM, Chol and PEG2000-DSPE (79:20:1, w/w) with high drug loading efficiency. The drug loading methods used for VCR liposomes are shown in Figure 1. TEA-SOS gradient was chosen for drug encapsulation of SRLVs (Figure 1A), while pH-gradient was used for GVM preparation (Figure 1B). When SRLVs were preparing, the blank liposome was produced by the ethanol injection method, with a size of 100.9 nm, and then VCR was loaded actively into the liposome by TEA-SOS gradient (Figure 1A). After drug loading, three different formulations using one blank liposome suspension were obtained with final drug/lipid ratios of 1/10, 1/5 and 1/2, and the resulting products were named SRLV-1, SRLV-2, and SRLV-3, respectively. As shown in Table 1, the size distributions of three formulations were similar, indicating that different VCR/lipid ratios did not have a significant effect on particle size. The GVM formulation, referring to VSLI, as a positive control drug, was made by film dispersion method, and a pH-gradient was used for VCR-loading (Figure 1B), with mean size of 103.1 nm. PdIs (polydispersity index) of the four formulations were all less than 0.1, demonstrating relatively narrow size distribution of the nanoparticles. There was no significant particle size difference between inter-preparations, conforming to the quality standard of VSLI (100±5 nm). All size distributions meet the study request for the following experiment evaluations.
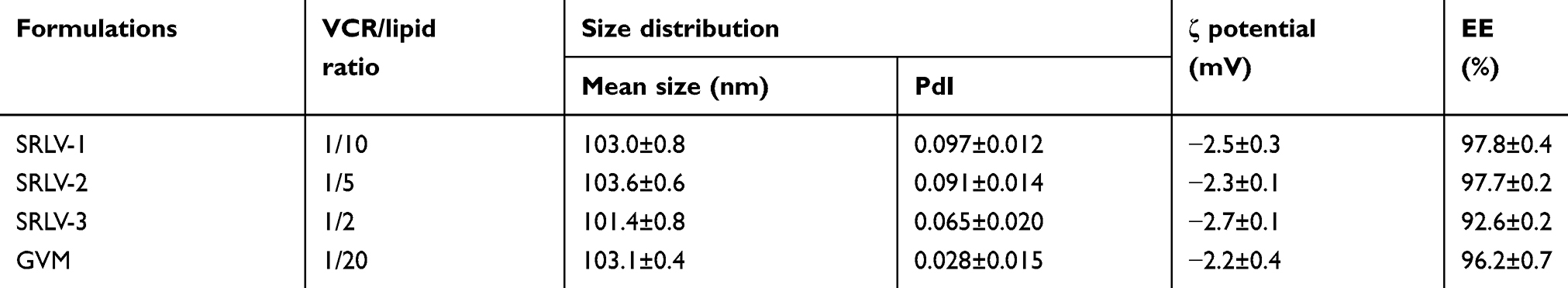 | Table 1 Characterization of SRLV-1, SRLV-2, SRLV-3 and GVM |
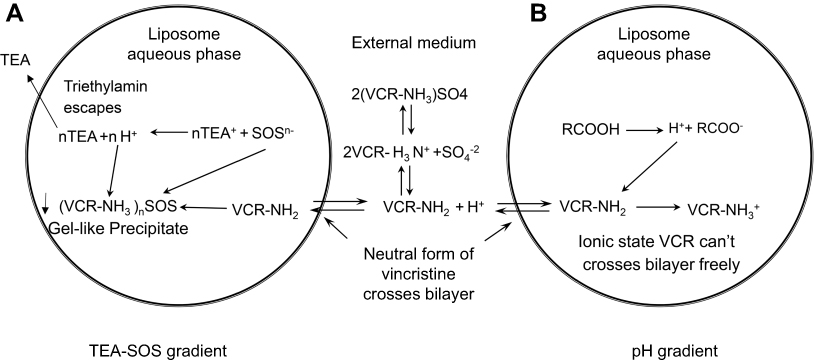 | Figure 1 Mechanisms of VCR drug loading. (A) Schematic of the active loading method of vincristine sulfate into preformed liposomes using a TEA-SOS gradient. VCR forms a gel-like precipitate due to the presence of SOS- inside the liposome. (B) Schematic of the active loading method of vincristine sulfate into performed liposomes using a pH gradient. Detailed descriptions of the method and processes are given in the text. |
Zeta potential and EE of VCR liposomes are presented in Table 1. Both SRLVs and GVM were composed with neutral phospholipids, so the surface charges of four liposomes showed no significant difference, from −2.15 to −2.66 mV. By the advantage of active loading methods, the EEs of all preparations were satisfied, with a minimum of 92.6%.
Figure 2 presents the photos of four formulations taken by Cryo-TEM. Differences in inner liposomal morphology between SRLVs and GVM were detected. For SRLVs, a progressive increase in inner liposome shadow density was observed as the drug/lipid ratio increased from 1/10 to 1/2 (Figure 2A–C). Figure 2D shows the inner structure of GVM, with no obvious evidence for drug precipitates.
 | Figure 2 Cryo-EM micrographs of VCR liposome formulations. (A–C) SM/Chol/mPEG-DSPE (79/20/1, w/w) liposomes using a TEA-SOS gradient for VCR loading at different drug/lipid ratios of 1/10 (A), 1/5 (B) and 1/2 (C). (D) SM/Chol (73.5:29.5, w/w) liposome using a pH gradient for VCR loading. |
Storage stability of VCR liposomes
The storage stability of VCR liposomes was evaluated by the changes of EE and vincristine sulfate concentration in suspension. In order to achieve a stable ready-to-use formulation, the following experiment was performed. After drug loading and filling into the final vials, all four products were stored at 2–8°C for long-term (12 months) study and at 25°C for accelerated (3 months) study for the three SRLVs. As we can see in Figure 3A and B, the vincristine sulfate concentration in suspension of GVM decreased by 10.5% after 1-month storage at 2–8°C, which already failed to meet the product quality standards (90–110%, refer to US Pharmacopeia). In comparison, VCR concentrations of SRLV-1 and SRLV-2 only decreased by 4.3% and 2.9% separately after 12 months storage. For SRLV-3, on the other hand, due to its lower initial EE (about 5% lower than those of SRLV-1 and SRLV-2), the vincristine sulfate concentration quickly reduced by 8.2% after a 1-month refrigeration, and it was maintained unchanged until the end of the experiment, with a total 9.7% decline in 12 months of storage. In the 25°C accelerated stability experiment (Figure 3C and D), the VCR degradation became faster, with degradation ratios of 31.9% for SRLV-1 and 5.5% for SRLV-2 in 3 months. Same performance as at 2–8°C, the VCR concentration of SRLV-3 decreased fast initially followed by a much slower decline, with 9.1% in the first month and ending up with a total decrement of 10.6%. It is clear that compared to GVM, the storage stabilities of SRLV-1, SRLV-2 and SRLV-3 have been improved.
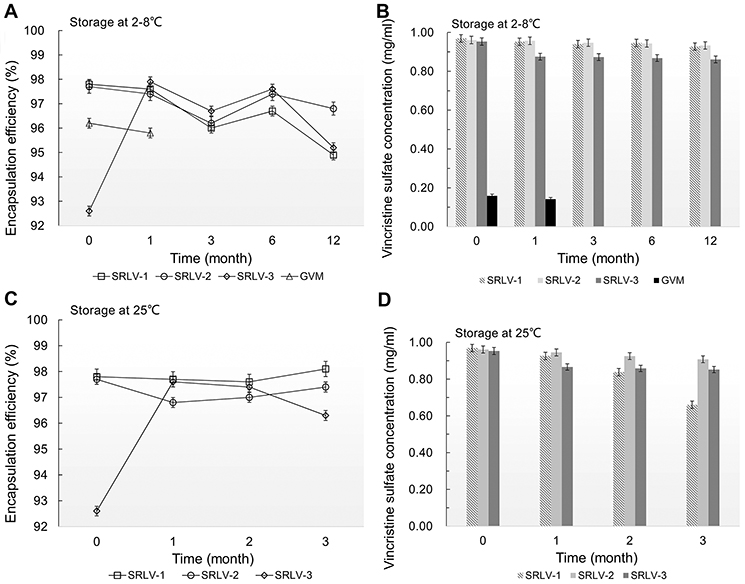 | Figure 3 The changes of encapsulation efficiency (A and C) and vincristine sulfate concentration (B and D) for VCR liposome formulations. The formulations were stored in the time of 0, 1, 3, 6 and 12 months at 2–8°C (A and B) and in the time of 0, 1, 2 and 3 months at 25±2°C/60% ±5% RH (C and D). |
In vitro release
In this study, two in vitro release experiments were conducted by using two different release media for better understanding the release behaviors of four formulations. Figure 4A shows the in vitro release rates in the HEPES/NaCl serum solution of all VCR liposomes after 0.5, 2, 4, 8, 12, and 24 h at 37°C. As can be seen, the drug release of GVM was much faster than that of the other three samples, with a total release ratio of 20.7% at 24 h. The three TEA-SOS loaded liposomes presented VCR/lipid ratio-dependent release performances; the higher the ratio, the slower the release. As a result, the final drug release ratios were 8.4%, 4.9%, and 2.3% for SRLV-1, SRLV-2, and SRLV-3, respectively.
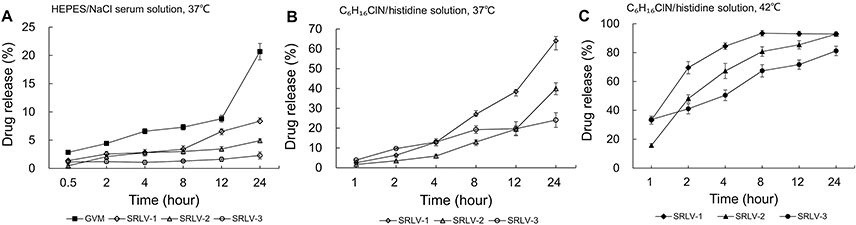 | Figure 4 In vitro release of VCR liposome formulations in medium. (A) In vitro release of SRLV-1, SRLV-2, SRLV-3, and GVM in HEPES/NaCl serum solution (pH 7.2) at 37°C after 0.5, 2, 4, 8, 12, and 24 h incubation. (B and C) In vitro release of SRLV-1, SRLV-2 and SRLV-3 in triethylamine chloride/histidine solution (pH 6.5), at 37°C (B) or 42°C (C) after 0.5, 2, 4, 8, 12, and 24 h incubation. |
To expedite drug leakage from SRLVs, the second test was designed. TEA was added into VCR-loaded TEA-SOS liposomes, and the release behaviors of VCR from SRLV-1, SRLV-2, and SRLV-3 were explored. In our previous studies (unpublished), it has been shown that the presence of NH4+ in the external of liposomes would greatly accelerate the VCR release by enhancing its dissolution in the internal phase liposomes, and inducing the VCR transfer from inside to outside of the lipid bilayer. The tests were measured under two temperatures, 37°C, close to human physiological temperature (Figure 4A), and 42°C, 1°C above the liquid-disordered phase transition temperature (Figure 4B), to find out the difference of the in vitro release regulations of the three formulations under both gel and liquid-disordered phases. It is obvious that the release of three preparations happened much quicker than that in the first test (Figure 4A). In the 37°C study, the release ratios at 12 h were 38.4% (SRLV-1), 19.5% (SRLV-2), and 19.7% (SRLV-3), respectively (Figure 4A). When the temperature goes up from 37°C to 42°C, the lipid membrane transforms from the gel phase to the liquid-disordered phase, and the membrane permeability is significantly augmented. The release ratios at 12 h were greatly increased to 93.1% (SRLV-1), 95.4% (SRLV-2), and 71.7% (SRLV-3), respectively (Figure 4B). In general, it can be concluded that SRLV showed better drug retention with a sustained drug release at higher drug/lipid ratio.
Pharmacokinetics study
Pharmacokinetics of drug-loaded liposomes may provide clues for toxicity and efficacy. We further evaluated the PK behaviors of VCR liposomes in Wistar rats. The total VCR concentrations were examined in serum at 0.03, 0.5, 1, 2, 4, 8, 12, 24, and 48 h after intravenous injection with 1 mg/kg VCR liposomes. The plasma concentration–time curves of GVM, SRLV-1, SRLV-2, and SRLV-3 are shown in Figure 5. The corresponding pharmacokinetic parameters are presented in Table 2. As it is shown, the blood half-lives (t1/2Z) for GVM, SRLV-1, SRLV-2, and SRLV-3 were 11.1, 17.4, 19.2, and 22.7 h, respectively, and the corresponding systemic exposure levels (AUC0-t) were 138.5, 267.1, 366.0, and 366.1 mg/L h. For GVM, the higher leakage rate resulted in shorter blood circulation time and lower systemic exposure. In SRLVs groups, when the VCR/lipid ratio went up, the blood circulation time extended and the systemic exposure increased. For SRLV-3, once again, a high clearance rate was shown in the first hour and then the drug clearance slowed down at the end of the experiment, which makes it consistent with in vitro drug release. In general, both the in vivo blood circulation times and systemic exposures of the three TEA-SOS liposomes, SRLV-1, SRLV-2 and SRLV-3, are significantly improved compared to those of the pH liposome.
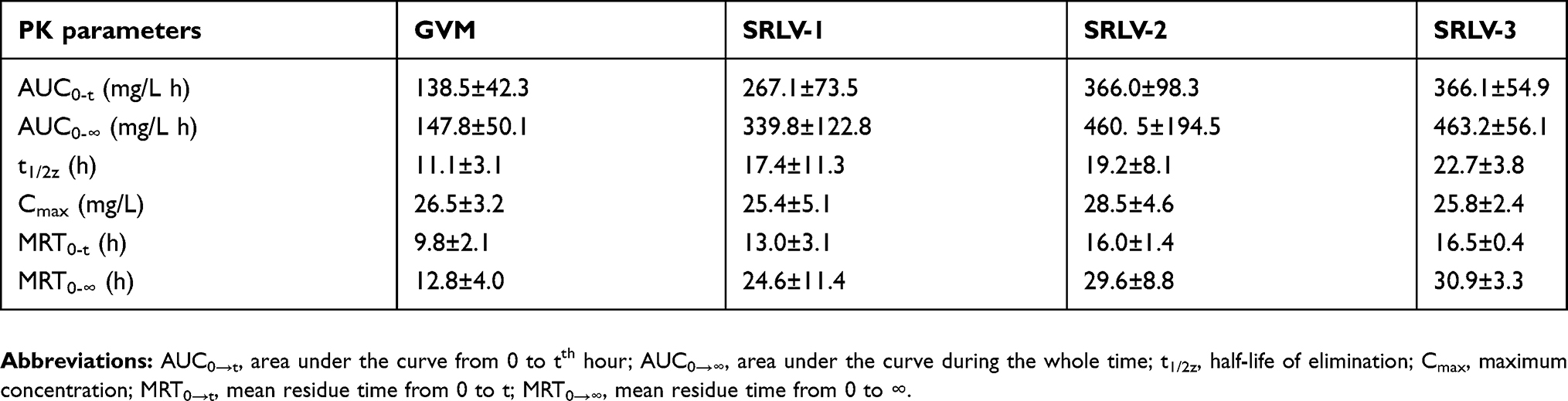 | Table 2 Pharmacokinetics parameters of VCR liposome formulations in rats |
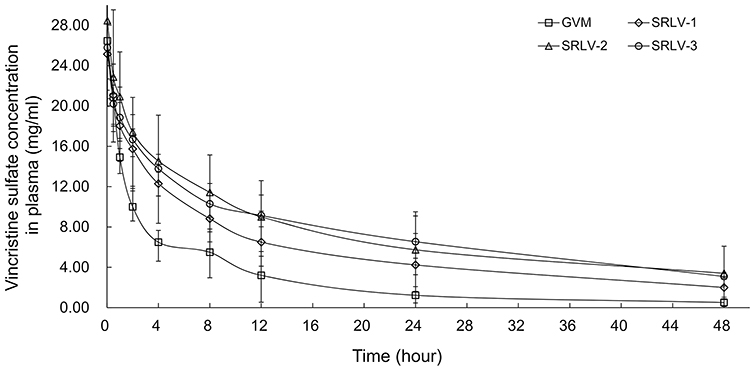 | Figure 5 The plasma concentration of vincristine of VCR liposome formulations in rats after a single dose of 1 mg/kg intravenous injection. Blood samples were taken from retro-orbital plexus at 0.03, 0.5, 1, 2, 4, 8, 12, 24, and 48 h after injection. Data are expressed as mean ± SD (n=10). |
In vivo toxicity study
As previously described, the advantage of liposome as a carrier technology for VCR is not only to prolong in vivo circulation time, but also to reduce toxicity. Here, both single-dose and multiple-dose toxicity studies were set for differentiating formulations. Male ICR mice were injected i.v. with varied doses of VCR liposomes (Table 3). None of the 6 animals died from SRLV-2 at a dose of 3.0 mg/kg, while the death rates of the rest were 4, 3, and 1 of 6 for GVM, SRLV-1, and SRLV-3, respectively. When the dose was raised up to 4.0 mg/kg, all mice that were given GVM or SRLV-1 died. When the dose was raised up to 5.0 mg/kg, all mice died. In the multiple-dose study, 40 animals, 10 per group, received 1.0 mg/kg VCR liposomes daily for 5 consecutive days. All mice that received GVM or SRLV-1 died. One of six experimental animals from GVM group died before the fourth administration. The total death rates of GVM and SRLV-1 were similar in all dose groups, but the death time was delayed when SRLV-1 was given. Among all the four VCR formulations, SRLV-2 had the lowest toxicity with the lowest mortality and the latest death time. Weight loss and physical abnormalities can be seen on all liposomal VCR treated animals. None of the saline group developed any adverse reaction, such as weight loss, behavior disorders or death. On day 14 after the last dose, all living mice started to heal and gained weight for 3 consecutive days. In conclusion, the toxicity of liposomal VCR depends on not only the in vivo drug clearance rate but also the drug encapsulation efficiency.
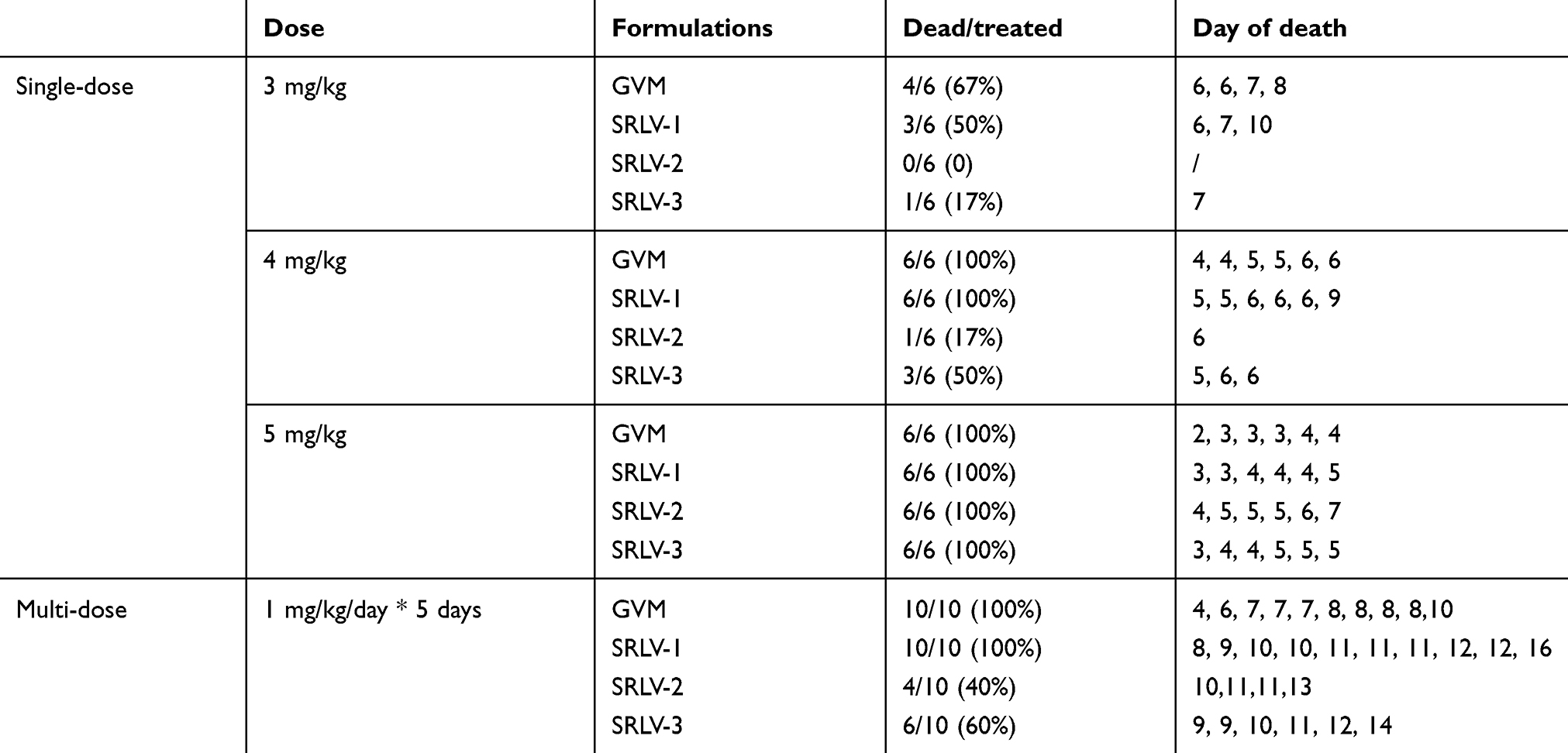 | Table 3 Single dose and multi-dose toxicity studies of VCR liposome formulations in rats |
In vivo anti-tumor efficacy
The in vivo curative efficacy of liposomal VCR was further evaluated using male BALB/c nude mice bearing human melanoma tumor. Mice were treated by 2 mg/kg liposomal VCR or 25 mL/kg saline solution i.v. on day 1, day 5 and day 9. All animals were sacrificed on day 14 and the tumor weights were measured. As shown in Figure 6A, all drug-treated groups exhibited significant therapeutic effect. After the second injection, the tumor size of four treatment group started to reduce, while the tumor size of control group kept rising. Figure 6B showed the final TGIs of four groups. Compared with that of GVM, the TGIs of SRLV-1, SRLV-2 and SRLV-3 were enhanced, and they were presented on trends with drug/lipid ratios. While the drug/lipid ratios increased from 1/10, 1/5 to 1/2, the TGI also elevated from 66.34%, 74.75% to 76.24% respectively. In order to assess the side effects of the treatment, animal behavior and body weight were recorded once a day. The average body weights of GVM, SRLV-1, SRLV-2, SRLV-3, and saline group decreased by 15.7%, 15.0%, 6.3%, 12.1%, and 8.3% on the 14th day, separately. Considering the anti-tumor efficacy and body weight loss, SRLV-2 seems an appropriate candidate for further development.
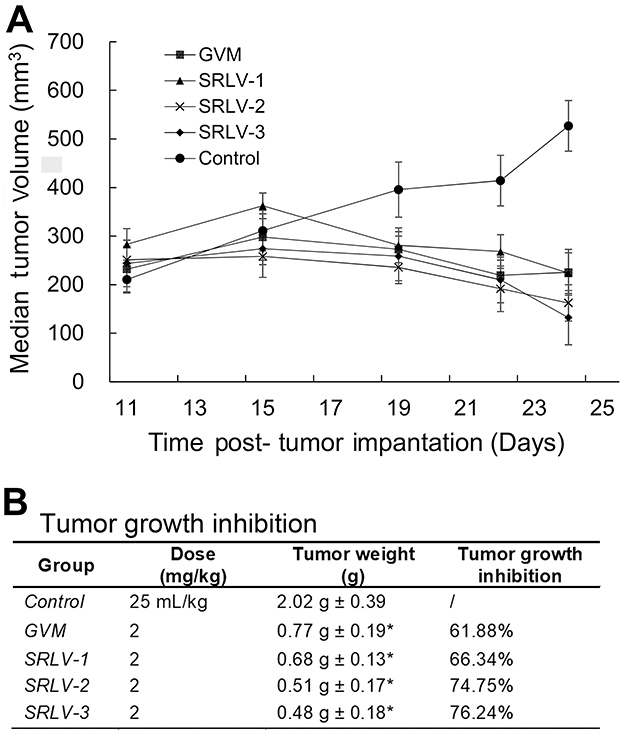 | Figure 6 Antitumor activities of VCR liposome formulations on male BALB/c nude mice bearing human melanoma (NHEM) model. Therapeutic effects are shown as tumor volume change (A) and tumor growth inhibition (B). NHEM tumors were implanted in the dorsal flank of nude mice as described in Materials and methods. Once tumors were appropriately sized (∼250 mm3), mice were treated with VCR liposome formulations at a dose level of 2 mg/kg at day 1, day 5, and day 9. The control group was given 25 mL/kg saline solution i.v. on the same day. All mice began treatment on day 11 after tumor implantation. Tumor weight was recorded at day 14 post-administration. Data represented as mean ± SD (n=6). *p<0.05 as compared to control. |
Discussion
Liposomal delivery has been shown to improve pharmacokinetic profile, reduce the toxicity and widen the therapeutic index of certain anticancer drugs. In this study, we have successfully developed a drug delivery system for VCR. It is composed of a neutral phospholipid SM for liposome formation, Chol for membrane fluidity modification, mPEG-DSPE for avoiding the aggregation of liposomes and TEA-SOS liposome core for drug retention. SM naturally exists in plasma membranes.25 Because of its asymmetric molecular structure and extensive hydrogen-binding capacity,26 compared to other lipid membranes, SM bilayer membranes exhibit a decreased molecule interval and less membrane fluidity, together resulting in a lower drug leakage. Chol is commonly used in liposomal formulations for membrane fluidity modification.27 Ellens et al28 and Kiani et al29 found out that SM with Chol is highly resistant to the destabilization caused by plasma lipoproteins, which can increase the blood circulation time of VCR.
In order to avoid liposome aggregation with the purpose of improving long-term storage stability,30 the surfaces of SM/Chol liposomes with PEG (PEG2000-DSPE) were modified. Liposomes with PEG have been demonstrated to stabilize liposomes both in vitro and in vivo,31 and PEG-modified liposomes have been widely used to prolong in vivo circulation time, so called long-circulating liposomes, such as doxorubicin HCl liposome injection. However, in recent years, it was found that the invisibility of PEG-coated liposomes from the cellular uptake further prevented their rapid escape from endosomes, which led to a subdued efficacy.32 To overcome this so called “PEG dilemma” limitation,33 trace of PEG2000-DSPE (1% of total lipid, w/w) was added. Previous studies (unpublished) showed that small amount of PEG-lipid, such as 1% or less, made no difference on pharmacokinetic characteristics and biodistribution profiles of liposomal drugs, but their stabilities in vitro obviously were improved.
Different drug encapsulating methods can be used to enable drugs loading into liposomes. The simplest way for drug encapsulation is the passive loading method. By dissolving a water-soluble drug in the aqueous buffer for lipid hydration, it is easy to acquire a liposomal formulation with approximately equal drug concentration both inside and outside of liposomes. This method is simple, with a direct preparation procedure and low cost for manufacture, but resulting in low EE. The active loading methods, in contrast, require top-end equipment and high cost in production, but result in high EE (usually above 90%).34 Figure 1A shows the drug-loading principle of SOS-TEA gradient. When uncharged VCR molecules pass the lipid membrane from outside to inside of the liposome, they bind with SOS− inside the liposome to form gel-like precipitates ((VCR-NH3)2SOS) and retain in the liposomes. Meanwhile, when TEA molecules escape from the liposomes, protons would be generated, resulting in a more acidic liposome core. For amphipathic weak bases, such as VCR, an acidic liposome core would enable more drug molecules to transfer into the liposomes. The loading capability of liposome depends on the amount of SOS− inside the liposome. On the other hand, VCR in pH-gradient liposomes exists in a different form. By generating a transmembrane pH gradient between the inside and outside the medium of the blank liposomes, uncharged VCR molecules that cross the lipid bilayer become protonated due to the acidic inner phase, and then the molecules retain within the liposomes (Figure 1B). These two drug loading methods both lead to high EE, but entirely different VCR existence forms in liposomes.
As stated in the introduction, the TEA-SOS gradient was chosen for VCR loading in this research to prevent the degradation of VCR by form precipitates while VCR transfer into liposomes and minimum the VCR leakage from the liposomes. As shown above (Figure 2A–C), increasing internal shadow densities are observed with Cryo-TEM as the drug/lipid ratio increased from 1/10 to 1/2. Unlike linear precipitates observed for doxorubicin liposomes along with great liposomal deformation34–36 or the empty structure for GVM (Figure 2D), the shadow inside liposomes with little deformation could possibly represent an amorphous or gel-like flexible precipitate. It is no surprise that the shadow densities increased with the increasing of amount of VCR encapsulated in individual liposome, indicating a tighter precipitate structure at the higher VCR contents.
Both in vitro and in vivo release studies demonstrate that the release rates of VCR liposomes could be regulated by the drug/lipid ratio. The higher the drug/lipid ratio is, the slower the VCR releases. Meanwhile, the increased VCR/lipid ratio also leads to a better antitumor efficacy. This may be related to higher plasma concentration and prolonged blood circulation time of VCR liposomes. Combined with the Cryo-TEM phenomena, there is reason to speculate that the compactness of precipitate could influence the drug release rates. It is interesting that Noble et al managed to encapsulate VCR into DSPC/Chol/mPEG liposomes at a molar ratio of 3/2/0.015 by TEA-SOS gradient in 2009.37 They similarly investigated the in vivo release behavior of VCR liposomes with different drug/lipid ratios, but resulting in an opposite trend. According to their research, the formulation with the lowest drug/lipid ratio has the longest in vivo retention time (t1/2 31.2 h of 95.1 g drug/mol lipid, t1/2 18.8 h of 192 g drug/mol lipid, t1/2 20.0 h of 375 g drug/mol lipid). The reason for the difference is unknown, but it is worth further investigation.
The change of EE is a classic evaluation indicator for liposomal formulations to assess the storage stability.38 In this experiment, the EEs of the four preparations maintained at high levels (> 95%) under the designed storage conditions. The EE of SRLV-3 after one-month storage increased even from 92.6% to 97.9% at 2–8°C and 97.6% at 25°C (Figure 3A and C). As mentioned before, VCR is chemically instable and easy to hydrolyze. However, after transferring from outside to inside the liposomes, VCR would be protected from hydrolysis by the formation of precipitation. Yet the unencapsulated free VCR remains uncovered in the external phase buffer and will be in degradation. The EE of VCR liposomes is calculated as loaded VCR divide by total VCR (free VCR plus loaded). While free VCR hydrolyzed as time increased, and if the loaded VCR is released very little, the EE showed an illusion of increasing. The reduction of VCR content along with increased EE after 1-month storage provides new evidence for the degradation of free VCR. Therefore, EE is neither sensitive nor specific in stability evaluation of VCR liposomes.
Because of the slower in vivo and in vitro drug release, we predicated a lower toxicity for SRLV-3, but it turned out to be the opposite. Although SRLV-3 performed better than both GVM and SRLV-1 on the in vitro drug release and pharmacokinetics studies, the death rate was higher and the death time was earlier than those of SRLV-2. It should be noted that both treatment effect and toxicity were optimized for SRLV-2 and SRLV-3, compared with GVM. Nevertheless, SRLV-2 demonstrated the best therapeutic effect with the highest TGI and the least body weight loss. One other result to note is that no animal died during the experiment, although the cumulative dose (6.0 mg/kg) of treatment groups was higher than that of toxicological study. In the toxicological study, all mice died after a single dose of 5.0 mg/kg and 30 of 40 died after a multiple dose of 1.0 mg/kg/day for 5 consecutive days (cumulative dose 5.0 mg/kg). This result suggests that enough recovery time can largely improve toxicity tolerance of mice. The reason why SRLV-2 presented the lowest toxicity, or in other words, why SRLV-3 has the slowest VCR release ratio and best TGI but resulted a higher toxicity than SRLV-2 is unknown.
The last point of discussion concerns the contradiction between the decreased drug release ratio and the increased toxicity and antitumor efficiency, as reflected by the study outcomes of SRLV-3 (D/L ratio 1/2) and SRLV-2 (D/L ratio 1/5). A possible explanation for this is the structural damage of liposome caused by high D/L ratio. In one study of doxorubicin liposome, Johnston et al36 found that growing precipitate would lead to physical disruption of the liposome bilayer and resulting in leakage of encapsulated drug or degradation of the ion gradient, giving rise to the pH gradient driving drug loading. According to the experiment, when the D/L ratio increased above the critical value (1/5), growing crystal would cause lipid membrane damage. A similar rule was obtained on pH gradient drug encapsulation VCR liposomes. With the increasing D/L ratio, leakage of inner phase sucrose was observed. Although we utilized a different drug loading gradient in this research and VCR existed in a gel-like state inside the liposomes instead of the linear crystal precipitates of doxorubicin, the results of Johnston et all36 should be particularly instructive. Further work will focus on the influence of VCR precipitates to lipid membrane and the relation between in vivo release and antitumor efficiency leading to optimized therapeutic properties.
Conclusion
Compared to the positive control drug used in this research, the new formulation achieved the following advantages: enhanced stability, improved antitumor efficacy and lowered toxicity. Among the three preparations with varied drug/lipid ratios which have been explored in this study, the formulation with drug/lipid ratio of 1/5 (SRLV-2) performed the best, with a high EE, the lowest degradation rate after 12 months at 2–8°C and 3 months at 25°C storage, the lowest death rate in the toxicity study and approximately highest anti-tumor efficacy on human melanoma tumor model. More importantly, the improved stability of the new liposomal VCR eliminates the need for multi-step preparation at the hospital pharmacy and could provide important advantages in practice in terms of convenience and safety. Therefore, there is potential for clinical translation of this single-vial formulation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Carbone PP, Bono V, Frei E
2. Horton JK, Houghton PJ, Houghton JA. Relationships between tumor responsiveness, vincristine pharmacokinetics and arrest of mitosis in human tumor xenografts. Biochem Pharmacol. 1988;37(20):3995–4000.
3. El-Sayed A, Cordell GA. Catharanthus alkaloids. XXXIV. Catharanthamine, a new antitumor bisindole alkaloid from Catharanthus roseus. J Nat Prod. 1981;44(3):289–293. doi:10.1021/np50015a009
4. Moore A, Pinkerton R. Vincristine: can its therapeutic index be enhanced? Pediatr Blood Cancer. 2009;53(7):1180–1187. doi:10.1002/pbc.22161
5. Webb MS, Harasym TO, Masin D, Bally MB, Mayer LD. Sphingomyelin-cholesterol liposomes significantly enhance the pharmacokinetic and therapeutic properties of vincristine in murine and human tumour models. Br J Cancer. 1995;72:896. doi:10.1038/bjc.1995.430
6. Zhu G, Oto E, Vaage J, et al. The effect of vincristine-polyanion complexes in STEALTH liposomes on pharmacokinetics, toxicity and anti tumor activity. Cancer Chemother Pharmacol. 1996;39(1–2):138–142.
7. Sivakumar PA, Panduranga Rao K. The use of cholesteryl pullulan for the preparation of stable vincristine liposomes. Carbohydr Polym. 2003;51(3):327–332. doi:10.1016/S0144-8617(02)00187-X
8. Boehlke L, Winter JN. Sphingomyelin/cholesterol liposomal vincristine: a new formulation for an old drug. Expert Opin Biol Ther. 2006;6(4):409–415. doi:10.1517/14712598.6.4.409
9. Silverman JA, Deitcher SR. Marqibo(R) (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71(3):555–564. doi:10.1007/s00280-012-2042-4
10. Harrison TS, Lyseng-Williamson KA. Vincristine sulfate liposome injection: a guide to its use in refractory or relapsed acute lymphoblastic leukemia. BioDrugs. 2013;27(1):69–74. doi:10.1007/s40259-012-0002-5
11. Monte WT, Abra RM, Luo B, Zhang Y A Ready-to-use Formulation for Vincristine Sulfate Liposome Injection.
12. Webb MS, Sarris AH, Cabanillas F, et al. Clinical and preclinical pharmacology of liposomal vincristine. J Liposome Res. 2000;10(4):501–512. doi:10.3109/08982100009031114
13. Yang Y, Guo Y, Tan X, et al. Vincristine-loaded liposomes prepared by ion-paring techniques: effect of lipid, pH and antioxidant on chemical stability. Eur J Pharm Sci. 2018;111:104–112. doi:10.1016/j.ejps.2017.09.045
14. Yan Z, Zhu ZL, Qian ZZ, et al. Pharmacokinetic characteristics of vincristine sulfate liposomes in patients with advanced solid tumors. Acta Pharmacol Sin. 2012;33(6):852–858. doi:10.1038/aps.2012.44
15. Webb MS, Bally MB, Mayer LD, Miller JJ, Tardi PG Sphingosomes for enhanced drug delivery.
16. Sarris AH, Cabanillas F, Logan PM, Burge CT, Goldie JH, Webb MS Compositions and methods for treating lymphoma.
17. Barenholz Y. Doxil(R)–the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi:10.1016/j.jconrel.2012.03.020
18. Jiang W, Lionberger R, Yu LX. In vitro and in vivo characterizations of PEGylated liposomal doxorubicin. Bioanalysis. 2011;3(3):333–344. doi:10.4155/bio.10.204
19. Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66(6):3271–3277. doi:10.1158/0008-5472.CAN-05-4007
20. Hong K, Drummond DC, Kirpotin D Liposomes useful for drug delivery.
21. Pietzyk B, Henschke K. Degradation of phosphatidylcholine in liposomes containing carboplatin in dependence on composition and storage conditions. Int J Pharm. 2000;196(2):215–218.
22. Maurer N, Fenske DB, Cullis PR. Developments in liposomal drug delivery systems. Expert Opin Biol Ther. 2001;1(6):923–947. doi:10.1517/14712598.1.6.923
23. Mayer LD, Krishna R, Webb M, Bally M. Designing liposomal anticancer drug formulations for specific therapeutic applications. J Liposome Res. 2000;10(2–3):99–115. doi:10.3109/08982100009029381
24. Wang X, Song Y, Su Y, et al. Are PEGylated liposomes better than conventional liposomes? A special case for vincristine. Drug Deliv. 2016;23(4):1092–1100. doi:10.3109/10717544.2015.1027015
25. Ahmed SN, Brown DA, London E. On the origin of sphingolipid/cholesterol-rich detergent-insoluble cell membranes: physiological concentrations of cholesterol and sphingolipid induce formation of a detergent-insoluble, liquid-ordered lipid phase in model membranes. Biochemistry. 1997;36(36):10944–10953. doi:10.1021/bi971167g
26. Slotte JP. The importance of hydrogen bonding in sphingomyelin’s membrane interactions with co-lipids. Biochim Biophys Acta. 2016;1858(2):304–310. doi:10.1016/j.bbamem.2015.12.008
27. Ulrich AS. Biophysical aspects of using liposomes as delivery vehicles. Biosci Rep. 2002;22(2):129–150.
28. Ellens H, Morselt H, Scherphof G. In vivo fate of large unilamellar sphingomyelin-cholesterol liposomes after intraperitoneal and intravenous injection into rats. Biochim Biophys Acta. 1981;674(1):10–18.
29. Kiani MF, Yuan H, Chen X, Smith L, Gaber MW, Goetz DJ. Targeting microparticles to select tissue via radiation-induced upregulation of endothelial cell adhesion molecules. Pharm Res. 2002;19(9):1317–1322.
30. Kostarelos K, Tadros T, Luckham P. Physical conjugation of (Tri-) block copolymers to liposomes toward the construction of sterically stabilized vesicle systems. Langmuir. 1999;15(2):369–376.
31. Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–436. doi:10.2165/00003088-200342050-00002
32. Xu H, Paxton JW, Wu Z. Enhanced pH-responsiveness, cellular trafficking, cytotoxicity and long-circulation of PEGylated liposomes with post-insertion technique using gemcitabine as a model drug. Pharm Res. 2015;32(7):2428–2438. doi:10.1007/s11095-015-1635-0
33. Hatakeyama H, Akita H, Harashima H. The polyethyleneglycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 2013;36(6):892–899. doi:10.1248/bpb.b13-00059
34. Li T, Cipolla D, Rades T, Boyd BJ. Drug nanocrystallisation within liposomes. J Control Release. 2018;288:96–110. doi:10.1016/j.jconrel.2018.09.001
35. Peretz Damari S, Shamrakov D, Varenik M, et al. Practical aspects in size and morphology characterization of drug-loaded nano-liposomes. Int J Pharm. 2018;547(1–2):648–655. doi:10.1016/j.ijpharm.2018.06.037
36. Johnston MJ, Edwards K, Karlsson G, Cullis PR. Influence of drug-to-lipid ratio on drug release properties and liposome integrity in liposomal doxorubicin formulations. J Liposome Res. 2008;18(2):145–157. doi:10.1080/08982100802129372
37. Drummond DC, Noble CO, Guo Z, et al. Improved pharmacokinetics and efficacy of a highly stable nanoliposomal vinorelbine. J Pharmacol Exp Ther. 2009;328(1):321–330. doi:10.1124/jpet.108.141200
38. Celik B, Sagiroglu AA, Ozdemir S. Design, optimization and characterization of coenzyme Q10- and D-panthenyl triacetate-loaded liposomes. Int J Nanomedicine. 2017;12:4869–4878. doi:10.2147/IJN.S140835
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.