Back to Journals » International Journal of Nanomedicine » Volume 11
Development of a robust pH-sensitive polyelectrolyte ionomer complex for anticancer nanocarriers
Authors Lim C, Youn YS ![]() , Lee KS, Hoang NH
, Lee KS, Hoang NH ![]() , Sim T, Lee ES, Oh KT
, Sim T, Lee ES, Oh KT ![]()
Received 27 October 2015
Accepted for publication 29 December 2015
Published 22 February 2016 Volume 2016:11 Pages 703—713
DOI https://doi.org/10.2147/IJN.S99271
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Chaemin Lim,1,* Yu Seok Youn,2,* Kyung Soo Lee,1 Ngoc Ha Hoang,1 Taehoon Sim,1 Eun Seong Lee,3 Kyung Taek Oh1
1Department of Pharmaceutical Sciences, College of Pharmacy, Chung-Ang University, Seoul, 2Department of Pharmaceutical Sciences, School of Pharmacy, Sungkyunkwan University, Suwon, 3Division of Biotechnology, The Catholic University of Korea, Gyeonggi-do, South Korea
*These authors contributed equally to this work
Abstract: A polyelectrolyte ionomer complex (PIC) composed of cationic and anionic polymers was developed for nanomedical applications. Here, a poly(ethylene glycol)–poly(lactic acid)–poly(ethylene imine) triblock copolymer (PEG–PLA–PEI) and a poly(aspartic acid) (P[Asp]) homopolymer were synthesized. These polyelectrolytes formed stable aggregates through electrostatic interactions between the cationic PEI and the anionic P(Asp) blocks. In particular, the addition of a hydrophobic PLA and a hydrophilic PEG to triblock copolyelectrolytes provided colloidal aggregation stability by forming a tight hydrophobic core and steric hindrance on the surface of PIC, respectively. The PIC showed different particle sizes and zeta potentials depending on the ratio of cationic PEI and anionic P(Asp) blocks (C/A ratio). The doxorubicin (dox)-loaded PIC, prepared with a C/A ratio of 8, demonstrated pH-dependent behavior by the deprotonation/protonation of polyelectrolyte blocks. The drug release and the cytotoxicity of the dox-loaded PIC (C/A ratio: 8) increased under acidic conditions compared with physiological pH, due to the destabilization of the formation of the electrostatic core. In vivo animal imaging revealed that the prepared PIC accumulated at the targeted tumor site for 24 hours. Therefore, the prepared pH-sensitive PIC could have considerable potential as a nanomedicinal platform for anticancer therapy.
Keywords: polyelectrolyte ionomer complex, PEG–PLA–PEI, nanomedicine, pH-sensitive, animal imaging
Introduction
For decades, various types of drug delivery systems, including polymeric micelles, carbon nanotubes, liposomes, polymer-surfactant nanoparticles, conjugated prodrugs, and nanogels have been developed for anticancer chemotherapy, to achieve increased bioavailability of drugs, minimize side effects, control drug release into specific tissues, and enhance drug activity.1–5 Among these nanosized carrier systems, polyelectrolyte ionomer complexes (PICs) have been extensively investigated for current and potential future applications in drug and gene therapy.6,7 The nanosized PIC can be spontaneously formed in aqueous solution from double hydrophilic block copolymers containing ionic and nonionic blocks, upon electrostatic interaction between the ionic blocks and oppositely charged molecules such as genes, polyions, proteins, or surfactants.8–10 The electrostatically neutralized ionic blocks lead to the formation of a hydrophobic core in aqueous solution, which can incorporate various pharmaceutical drugs through hydrophobic interactions and hydrogen bonding. In addition, hydrophilic and nonionic blocks such as poly(ethylene glycol) (PEG) can provide aqueous stability via steric hindrance on the surface of the particle and extended circulation times by avoiding rapid renal clearance and reticuloendothelial system uptake.11–13
Among the various types of polyions that have been developed for drug or gene delivery, poly(ethylene imine) (PEI) has been intensively studied, since the PEI with the highest cationic charge density has improved the endosomal escape ability, which is directly related to the efficacy of drug or gene therapy.14,15 The PEG–PEI block copolymers exhibited improved solubility even under charge-neutralized conditions. However, the PIC system with this type of double hydrophilic block copolymer and oppositely charged molecules can be dissociated in in vivo conditions by other counterions, and showed a lower cell transfection of the therapeutic agents due to the lack of self-assembling aggregation force.6,8 The incorporation of a hydrophobic moiety into the PIC core could overcome these drawbacks, inducing the formation of a tight core and stabilization of the nanoparticles.
In the present study, we developed a novel PIC system based on a cationic poly(ethylene glycol)–poly(lactic acid)–poly(ethylene imine) triblock copolymer (PEG–PLA–PEI) and anionic poly(aspartic acid) (P[Asp]). The hydrophobic PLA block in the triblock polycation was able to provide increased colloidal stability by localizing to the middle layer of PIC and enhancing cell interactions and tissue permeability of the delivery platform.6,10,16,17 The fact is that PIC complexed with PEG–PLA–PEI and P(Asp) at various ratios of cationic PEI and anionic P(Asp) blocks (C/A ratios) shows the pH sensitivity by the protonation and deprotonation of the carboxyl groups in P(Asp) and the amine groups in the PEI blocks. pH-sensitive nanosystems could be used as a cancer reversal strategy through the exploitation of their favorable properties such as improved stability at physiological pH, reduced toxicity, and the controlled release of therapeutic agents at extracellular tumor pH (pHex =~6.5–7.2) or endosomal pH (pHen ≤6.5).18,19 Here, a PIC based on PEG–PLA–PEI and P(Asp) was evaluated for its pH-sensitive anticancer nanomedicinal potential using doxorubicin (dox) as an anticancer model drug.
Materials and methods
Materials
Poly(ethylene glycol) (PEG) methyl ether, molecular weight [MW] 5,000 Da), L-lactide (3,6-dimethyl-1,4-dioxite-2,5-dione), stannous octoate (Sn[Oct]2, Tin[II]2-ethylhexanoate), 4-(dimethylamino)pyridine (DMAP), pyren, succinic anhydride, pyridine, triethylamine (TEA), N-hydroxysuccinicimide (NHS), N,N′-dicyclohexylcarbodiimide (DCC), anhydrous 1,4-dioxane, N,N-dimethylformamide (DMF), D2O-d6, and CDCl3 were purchased from Sigma-Aldrich (St Louis, MO, USA). Triphosgene and branched PEI (MW 10,000 Da) were purchased from Alfa Aesar® Johnson Matthey Korea (Seoul, South Korea). Dichloromethane (DCM), methanol (MeOH), ethanol (EtOH), and toluene were purchased from Honeywell Burdick & Jackson® (Muskegon, MI, USA). Dox·HCl was purchased from Borung Co. (Seoul, South Korea). All other chemicals used were of analytical grade. For cell culture, human breast cancer MCF7 cells and cervical cancer KB cells were obtained from the Korean Cell Line Bank (KCLB, Seoul, South Korea). RPMI 1640 medium, fetal bovine serum (FBS), penicillin, and streptomycin were purchased from Welgene (Seoul, South Korea). Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular Technologies (Tokyo, Japan). P(Asp) was prepared as previously reported.20 The MW of P(Asp) was ~4,000 Da (degree of polymerization =35).
Synthesis of PEG–PLA–PEI triblock copolymers
The synthetic scheme for the polyelectrolyte triblock copolymers, PEG–PLA–PEI, prepared by multistep synthesis is described in Figure 1A. First, the PLA–PEG diblock copolymer was synthesized by the ring-opening polymerization of L-lactide, initiated by the hydroxyl group of PEG in the presence of Sn[Oct]2 as a catalyst, as described in previous studies.20,21 The MW of the prepared PLA–PEG diblock was ~11,000 Da, determined by the 1H-NMR spectra obtained using a 300 MHz Gemini 2000 NMR instrument (Varian Medical Systems, Palo Alto, CA, USA). For the synthesis of PEG–PLA–PEI copolymer, the terminal hydroxyl group of PLA–PEG (0.18 mmol) was carboxylated with succinic anhydride (0.36 mmol), DMAP (0.18 mmol), TEA (0.18 mmol), and pyridine (0.18 mmol) in DCM (30 mL) at room temperature for 1 day. After the reaction, carboxylated PLA–PEG was obtained following reprecipitation from excess diethyl ether. In order to conjugate the PLA–PEG to PEI, the carboxylated PLA–PEG (0.16 mmol) was activated using NHS (0.2 mmol) and DCC (0.2 mmol) in DCM at room temperature for 1 day. After carrying out the reaction, PEG–PLA–PEI was synthesized by a coupling reaction of PEI in DMF and MeOH (1:1) with the activated PLA–PEG, using simple DCC and NHS chemistry. PEI conjugation was confirmed by the presence of a 1H NMR (D2O-d6 with D2O; Cambridge Isotope Lab. Inc., MA, USA) peak at δ2.7–3.1.22,23
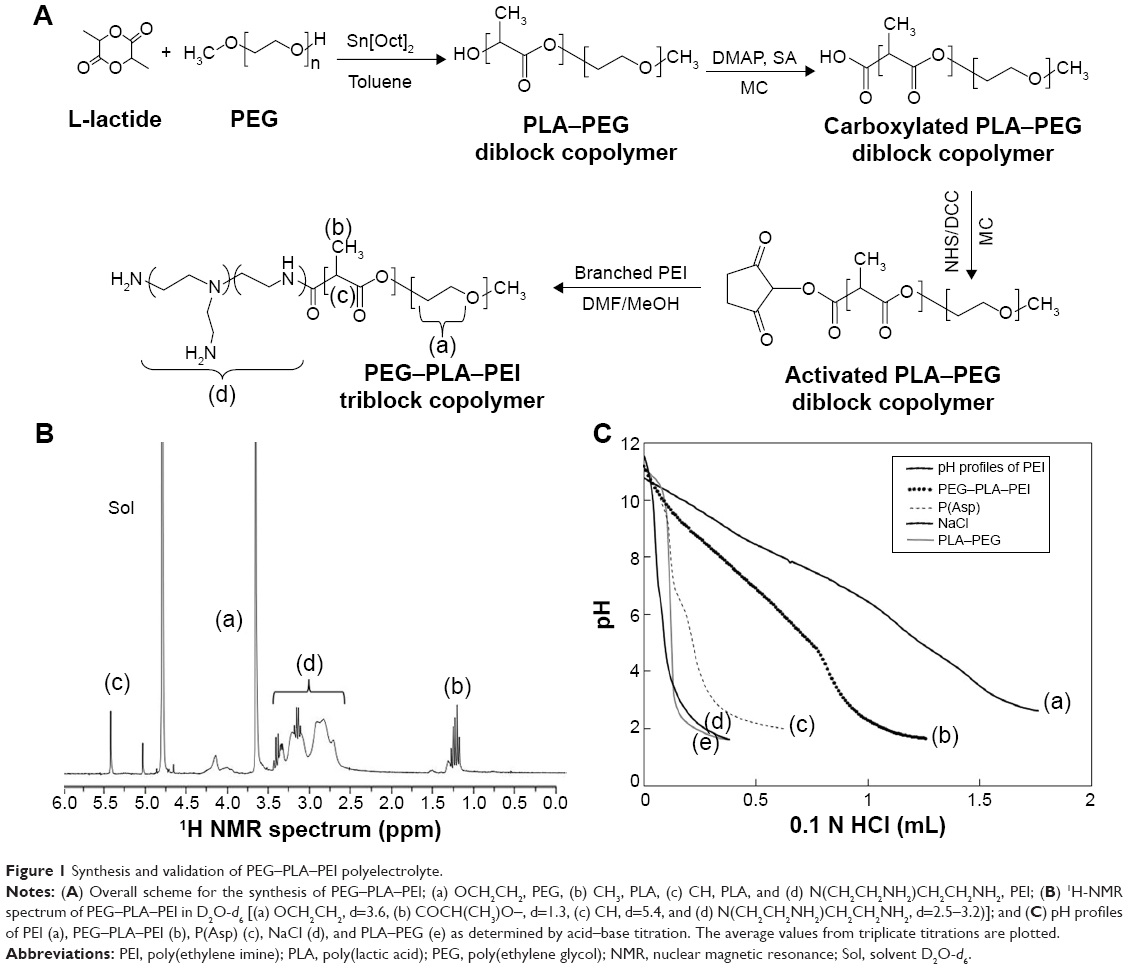 | Figure 1 Synthesis and validation of PEG–PLA–PEI polyelectrolyte. |
Acid–base titration
The titration plots of PLA–PEG, NaCl, P(Asp), PEG–PLA–PEI, and PEI were performed using the potentiometric titration method. The block copolymers (or NaCl as a control) dissolved in deionized water (2 mg/mL) were adjusted to pH 11 with 1 N NaOH. These solutions were titrated by the stepwise addition of 0.1 N HCl to obtain the pH titration profile.24
Preparation of PIC based on PEG–PLA–PEI/P(Asp)
PIC composed of PEG–PLA–PEI and P(Asp) were prepared by mixing 0.1% (wt/vol) aqueous solution of PEG–PLA–PEI with a solution of P(Asp) at various CA ratios (ratio between the nitrogen atom of the cationic polymer and the carboxyl group of P[Asp]), followed by vortexing for 10 seconds and incubation for 20 minutes at room temperature.
Particle size and zeta potential measurement using dynamic light scattering
The effective hydrodynamic diameters (Deff) and zeta potentials of the nanocomplex solution (0.1 mg/mL) were measured by photon correlation spectroscopy using a “Zetasizer Nano-ZS” (Malvern Instruments, Malvern, UK) equipped with the multiangle sizing option BI-MAS (Brookhaven Instrument Corp, NY, USA). The software provided by the manufacturer was used to calculate the Deff and zeta potential values. The average Deff and zeta potential values were calculated from three measurements performed on each sample (n=3).
CAC analysis
Critical association concentration (CAC) was determined by fluorescence measurement using a Scinco FS-2 fluorescence spectrometer (SCINCO Co. Ltd., Seoul, South Korea) as described in previous studies.16 This spectrofluorometer is equipped with polarizers for excitation (334 nm) and emission (372 nm) light beams. Pyrene was used as a fluorescent probe. The various concentrations of nanocomplex sample (from 10−4 g/mL to 10−8 g/mL) were prepared at different ratios using water, mixed with pyrene (at a concentration of 6.0×10−7 M), and stirred overnight at room temperature. CAC values were determined by plotting the ratios of I1 (intensity of the first peak) to I3 (intensity of the third peak) of the emission spectra profiles against the log10 values of the nanocomplex concentration. CAC values were defined as the point of intersection of low polymer concentrations on these plots.
Morphology of PIC
In order to observe the morphology of PIC, a dilute PIC solution (0.1 mg/mL) of the samples was placed onto a glass slide and dried in vacuo. The morphology of PIC was imaged using field emission scanning electron microscopy (FE-SEM, SIGMA, Carl Zeiss Meditec AG, Jena, Germany).
Preparation and characterization of dox-loaded PIC
The dox-loaded PIC system was prepared using the bottom flask method.20,21 Before loading dox into the complex system, dox·HCl was stirred with TEA (2 mol) in DCM overnight to obtain the dox base. PEG–PLA–PEI/P(Asp) (10 mg) and dox (1 mg) were dissolved in 10 mL 50:50 EtOH/DCM solution and transferred into round bottom flasks. For the preparation of the dox-loaded PIC system, the organic phase was removed using a model n-1000 rotary evaporator (EYELA, Tokyo, Japan) to form a thin film in each round bottom flask. Rehydration of the film with a borate buffer solution produced the dox-loaded PIC system, and the pH value of the micelle solution was adjusted with phosphate-buffered saline (PBS) and citric acid buffer. The concentration of dox in the micelles was determined by UV-1200 Spectrophotometer (Labentech, Incheon, South Korea) at λ=481 nm. The drug loading capacity and efficiency were calculated using the following equations:
|
|
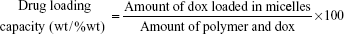
|
|
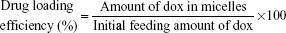
pH-dependent drug release from micelles
For the drug release test, the dox-loaded PIC solution (10%) was transferred into Spectra/Por dialysis membrane tubing (Spectrum Laboratories, Rancho Domingues, CA, USA; MWCO 3500 Da), immersed in a vial containing 10 mL PBS (pH 9.0–4.0), and incubated in a shaking water bath at 37°C and 100 rpm. The released amount of dox from the complex system was measured at predetermined times using UV–vis spectrometry (Genesys 10 UV) at λ=481 nm. After measurement of dox release at specific times, the medium in the vial was replaced with fresh PBS to prevent drug saturation.
Cell viability
MCF-7 cells in growth medium were seeded at a density of 1×104 cells per well of a 96-well plate 24 hours prior to the cytotoxicity test. The dox-loaded complex system in RPMI 1640 medium at pH 6.0 and 7.4, adjusted with 0.1 N HCl, was prepared immediately before use. The medium was removed from the 96-well plate and the preparation was added with different dox concentrations (1–10,000 ng/mL), and incubated for 40 hours. Chemosensitivity was assessed using the CCK assay. Fresh medium (90 μL, according to the pH conditions) containing 10 μL CCK solution was added to each well, and the plate was incubated for an additional 3 hours. The absorbance of each well was read on a Flexstation 3 microplate reader (Molecular Devices, Sunnyvale, CA, USA) at a wavelength of 450 nm.
In vivo fluorescence imaging
In vivo studies were performed using 4- to 6-week-old female nude mice (BALB/c, nu/nu mice; Institute of Medical Science, Tokyo, Japan). The mice were maintained under the guidelines of an approved protocol from the Institutional Animal Care and Use Committee (IACUC) of the Chung-Ang University of Korea. All experiments were performed in compliance with the relevant laws and institutional guidelines. For near-infrared fluorescence real-time tumor imaging, Cy5.5 mono-NHS ester was reacted with the amine of ethylene imine in a solution of PEG–PLA–PEI in DMSO/water for 1 day. The unconjugated Cy5.5 mono-NHS ester was removed by dialysis in water, and Cy5.5-labeled PEG–PLA–PEI was lyophilized by freeze-drying. For the in vivo animal experiments, KB tumor cells were introduced into female nude mice via subcutaneous injection of 1×106 cells suspended in PBS (pH 7.4). When the tumor volume reached 150 mm3, the PIC based on Cy5.5-labeled PEG–PLA–PEI/P(Asp) (C/A ratio: 8) was injected intravenously into tumor-bearing nude mice through the tail vein. A 12-bit CCD camera (Image Station 4000 MM; Kodak, New Haven, CT, USA) was used to take live fluorescence images of the mice. The optical images of Cy5.5-labeled PIC in the mouse model were taken 1, 3, 6, 9, and 24 hours following injection.
Results and discussion
Synthesis of PEG–PLA–PEI
The synthesis was performed using the following steps: 1) synthesis of PEG–PLA using ring-opening polymerization; 2) activation of PLA of the block copolymer; and 3) conjugation of PEI to the PEG–PLA block copolymer using the formation of an amide bond (Figure 1A). The synthesized PLA–PEG block copolymer was analyzed by using 1H NMR and gel permeation chromatography (GPC). In the 1H NMR spectra of the block copolymer dissolved in CDCl3, the characteristic chemical shifts corresponding to both PLA (1.5 and 5.17 ppm) and PEG (3.64 ppm) were observed, and no other peaks were detected (data not shown). The MWs of the PLA blocks were calculated from the integral values of the characteristic peaks of PEG and PLA using the known MW of PEG (5 kDa), and the Mn of PLA blocks was verified by GPC. The MW of PLA was calculated to be 6,000 Da, and in the GPC curve for PLA–PEG, a single sharp peak was shown with 1.15–1.30 polydispersity. These results confirm that the PEG reacted with L-lactide successfully and no homopolymerization of L-lactide occurred during the reaction.
The structure of the triblock copolymer was confirmed by the 1H NMR spectra (Figure 1B). In the 1H NMR spectra of the block copolymer dissolved in D2O, the peak at 3.6 ppm was assigned to protons of the PEG block, and peaks b and c at 1.2 and 5.4 ppm were attributed to the PLA block of CH (δ=5.4) and CH3 (δ=1.2). Peak d at 2.5–3.2 ppm was assigned to protons of PEI. The MW of PLA and PEI in the triblock copolymer can be obtained from the integral ratio of peak a (OCH2CH2, δ=3.6) to peak b (COCH(CH3)O–, δ=1.3) and d (N(CH2CH2NH2)CH2CH2NH2, δ=2.5–3.2). The MW of PEI blocks, calculated from the integral values, was 10,000 Da and the MW of the final triblock copolymer was ~21,000 Da.
Titration of the synthesized PEG–PLA–PEI
The pKa values of the components of the complex system were measured by the potentiometric titration method, and the acid–base titration profiles of the molecules are plotted in Figure 1C. Compared with NaCl and PEG–PLA without charged groups, other polyelectrolytes revealed a buffering zone depending on their structures. The apparent pKa of P(Asp) was ~6.2, with a very narrow buffering zone. However, PEI showed a broad buffering zone due to the cooperation between the high-density amine groups. Interestingly, by grafting PEI to PEG–PLA, the buffering capacity was outstandingly increased, however, slightly lower than PEI alone. This demonstrates that PEI was successfully conjugated to PEG–PLA. The pKb value of PEG–PLA–PEI was similar to that of PEI; however, the titration curve showed a slight change as a result of the hydrophobicity of PLA.25 Therefore, PEG–PLA–PEI has a more similar electron effect to PEI rather than to PEG–PLA, leading to a behavior similar to PEI, which shows the facilitation of osmotic swelling and rupture of endosomes by the proton sponge effect. In addition, drug-loaded PIC could release the drug into the cytosol from PIC.15,26–28
Characterization of PIC
PEG–PLA–PEI itself did not form aggregates due to the hydrophilicity of PEI and PEG compared with the hydrophobicity of PLA, and was soluble in aqueous solution. However, the complexes formed with cationic PEI and anionic P(Asp) by electrostatic interaction possessed a hydrophobic compartment due to neutralization, which could provide a driving force to form micelle-like aggregation. PIC is formed with hydrophobic cores from PLA and complexes with PEI and P(Asp), and hydrophilic coronas from PEG and unreacted PEI.29,30
The particle sizes of PIC at different C/A ratios, measured using dynamic light scattering (DLS), are presented in Figure 2A. PIC particles of different sizes were formed by mixing PEG–PLA–PEI and P(Asp). PIC at a 1:1 C/A ratio formed huge unstable particles ~940 nm in size. As the amount of added P(Asp) increased, the particle sizes of PIC drastically decreased, and the PIC with a C/A ratio of 8 formed ~130 nm particles. In contrast, a decrease in P(Asp) led to a slight decrease in the particle sizes of PIC, which may be due to the repulsive forces as a result of the negative charge. In addition, PIC formation in aqueous solution was examined using a fluorescence technique in the presence of pyrene as a probe. In PIC at a C/A ratio of 2, 4, and 8, the CAC values were 2.3, 6.5, and 10.3 μg/mL, respectively (Table 1). These results suggest that with a low amount of P(Asp), charged PEI and P(Asp) blocks could not provide enough hydrophobicity to form a hydrophobic core; however, with a high amount of P(Asp), both the neutralized PEI blocks as well as the P(Asp) blocks could be involved in the formation of the hydrophobic core. Nevertheless, the increased CAC values, despite the decrease in particle sizes due to the interaction between the PLA block and the neutralized P(Asp) and PEI block, may be due to the increase in hydrophilicity as the charge of PEI block increases at a high C/A ratios. However, when compared with other types of amphiphiles (critical micelle concentration =5–1,000 μg/mL) or low-MW surfactants (eg, sodium dodecyl sulfate [SDS] =2.0 mg/mL), PICs have very low CAC values.31,32 This indicates that the micelle-like structures of PIC have high stability in in vivo conditions, since sudden dilution upon injection can destabilize drug-loading micelles at concentrations below their critical micelle concentration.
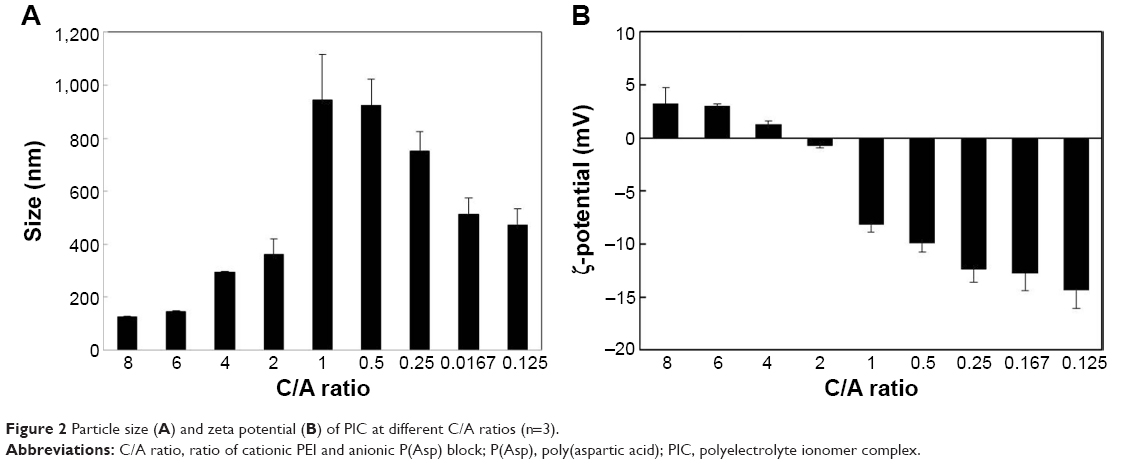 | Figure 2 Particle size (A) and zeta potential (B) of PIC at different C/A ratios (n=3). |
 | Table 1 The CAC value of the PEG–PLA–PEI/P(Asp) complex according to the cationic/anionic ratios (n=3) |
The surface charge of particles is an important determinant in particle stability and the electrostatic interaction with cells. Thus, the zeta potentials of PIC at different C/A ratios were measured (Figure 2B). The zeta potentials of PIC show that as the C/A ratio increases, the values of PIC were initially negative, with approximately −15 mV at a C/A ratio of 0.125, and became positively charged particles with a zeta potential of +4.5 mV at a C/A ratio of 8. Interestingly, the value of PIC at a C/A ratio of 2 shows a neutral charge compared with a negative charge at a C/A ratio of 1 (−8 mV). This may be due to the PLA blocks present in PEG–PLA–PEI.33–35 The zeta potential of micelles prepared with PEG (5 kDa)-PLA (6 kDa) was −12.5±3.4 mV. A positive zeta potential of the complexes was more favorable to ensure the uptake of nanoparticles into cells, since a positive surface charge could allow an electrostatic interaction between the negatively charged cellular membranes and the positively charged complexes.
For the potent drug delivery platform, the PIC system with a C/A ratio of 8 and a small particle size, positive zeta potential, and comparatively low CAC value was selected for further studies.
pH sensitivity of PIC
The pH sensitivity of drug delivery systems (DDS) is a significant property for targeting the extracellular pH of cancers and for triggering the drug release from DDS at lower than physiological pH, such as that in endosomes or lysosomes.36,37 In the present study, PIC with a C/A ratio of 8 was studied with respect to particle size and zeta potential in relation to pH-sensitive properties (Figure 3). At pH 8–9, the particle sizes were ~160–130 nm, which were slightly higher than those of PIC at pH 7.4 (126 nm). The slight increase in particle size may result from the decrease in hydrophobicity through the deprotonation of the P(Asp) blocks and protonation of the PEI blocks. The zeta potential at pH 9 is negative values, reflecting the predominant deprotonation of P(Asp) and PEI. At pH 8, the zeta potentials became positive, and the particle sizes of PIC increased due to an increase in the protonation of PEI blocks, which could then associate with the negatively charged P(Asp) blocks. The decreased pH (pH 7–4) due to acidic conditions, compared with physiological pH, induced an increase in particle sizes and positive zeta potentials of PIC. This suggests that the increased particle sizes and positive zeta potentials of PIC may be due to the loosening of particle formation by the lower electrostatic interactions between protonated PEI blocks (positive) and P(Asp) blocks (neutral).
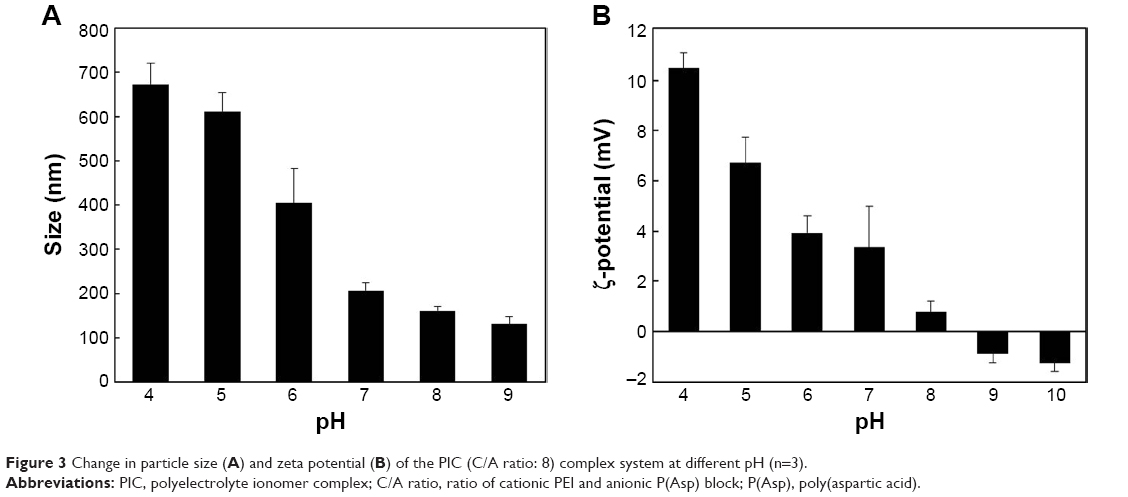 | Figure 3 Change in particle size (A) and zeta potential (B) of the PIC (C/A ratio: 8) complex system at different pH (n=3). |
Development of a pH-dependent nanomedicine
Based on the results shown earlier, a pH-dependent anticancer nanomedicine was developed using dox as a model drug. Dox was incorporated into the PIC (C/A ratio: 8) using the flat bottom flask method, and the unloaded drug was removed by filtration at 0.45 μm. The loading capacity of PIC for dox was 8.3% and the particle size was ~130 nm, with a narrow size distribution (Figure 4) in the DLS measurement. The size and particle distribution of dox-loaded PIC were smaller and narrower than those of empty PIC, as a result of the formation of a compact core in PIC by the addition of the hydrophobic drug, dox.21 The morphology under FE-SEM investigation revealed regular spherical discrete particles with smooth surfaces, and the particle sizes were relatively similar to the results obtained by the DLS technique. The sizes of dox-loaded PIC did not change for more than a week (data not shown).
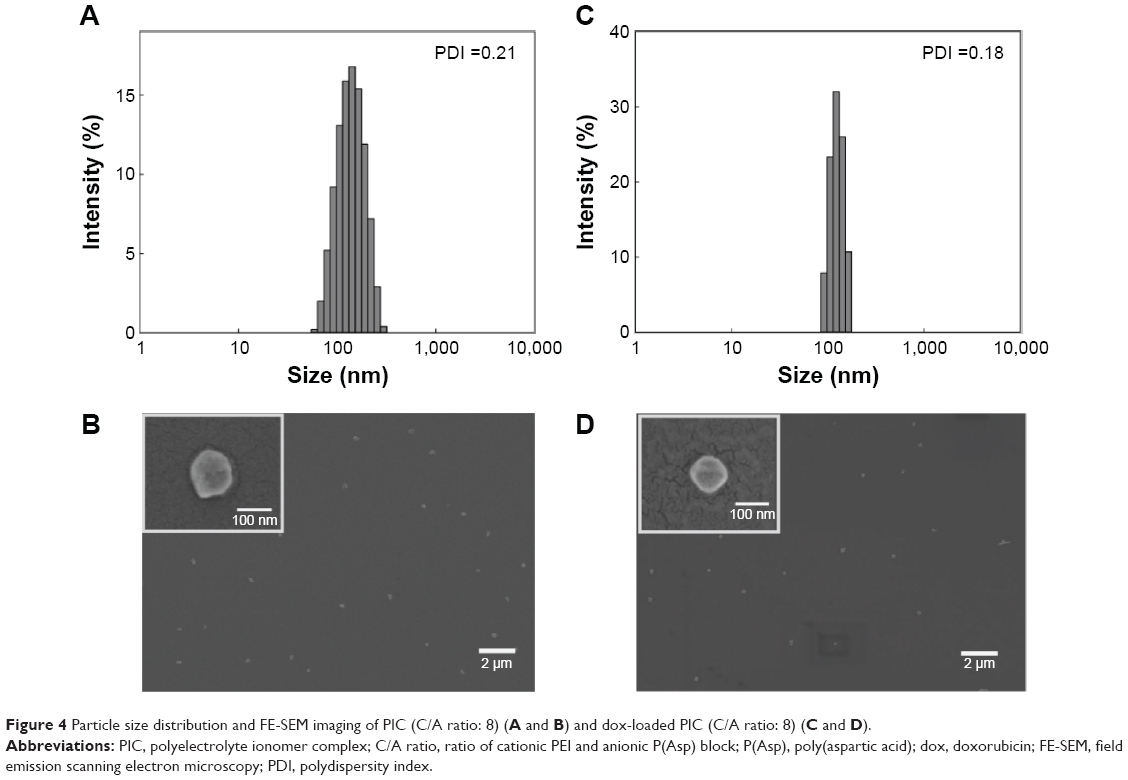 | Figure 4 Particle size distribution and FE-SEM imaging of PIC (C/A ratio: 8) (A and B) and dox-loaded PIC (C/A ratio: 8) (C and D). |
The release behavior of drug-loaded PIC with 8.3% drug loading was studied at different pH (Figure 5A). The dox release from PIC at different pH showed a burst effect in the very initial states, which may result from the release of drug located at the surface of PIC.38,39 For the first 4 hours, even though only 20% of the loaded drug was released from PIC at physiological pH and pH 9.0, 40% was released at acidic pH. For 24 hours, the amount and rate of dox release from PIC at pH 7.4 and pH 9.0 were much lower than those at pH 4.0 and pH 6.0. This result demonstrates that the hydrophobic drug was tightly incorporated into the hydrophobic core and was released by simple diffusion at physiological pH. At acidic pH, however, PIC initiates the disintegration of the core composed of PLA blocks and neutralized PEI/P(Asp) complexes by protonation of the charged molecules, resulting in rapid drug release from the loosened core of PIC. These results are consistent with the cell viability study shown in Figure 5B. The pH-sensitive cytotoxicity of dox-loaded PIC against MCF-7 cells was compared at pH 6.0 and pH 7.4. Under acidic conditions, the anticancer activity of PIC against the MCF-7 cells was enhanced compared with that at pH 7.4. The IC50 values of the complex system were 129.7 ng/mL and 239.7 ng/mL at pH 6.0 and pH 7.4, respectively. However, the cellular uptake of dox at the different pHs appeared similar (data not shown). These suggested that the acidic pH condition could trigger the drug release from PIC and the increased dox near MCF-7 cells could enhance the anticancer toxicity.19 These drug release profiles and cell viability support the notion that PIC may be useful for targeting cancer microenvironment-associated pH, and be considered for the triggering of drug release at endosomal/lysosomal pH.
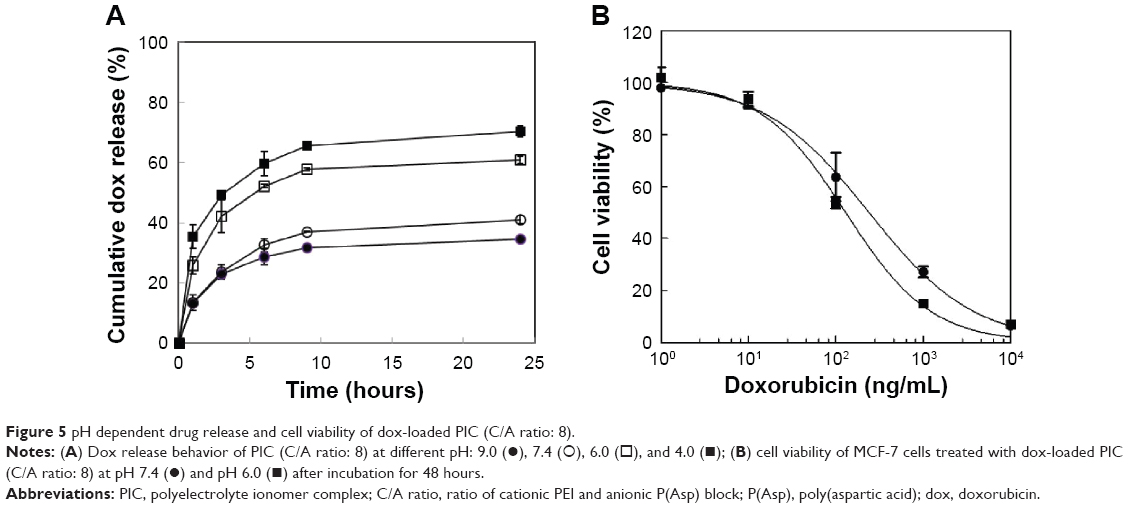 | Figure 5 pH dependent drug release and cell viability of dox-loaded PIC (C/A ratio: 8). |
The tumor targeting ability of PIC was evaluated in tumor-bearing nude mice by high-resolution fluorescent imaging using Cy5.5-labeled PIC (Figure 6), revealing that by 24 hours, PIC gradually accumulated at the tumor sites.
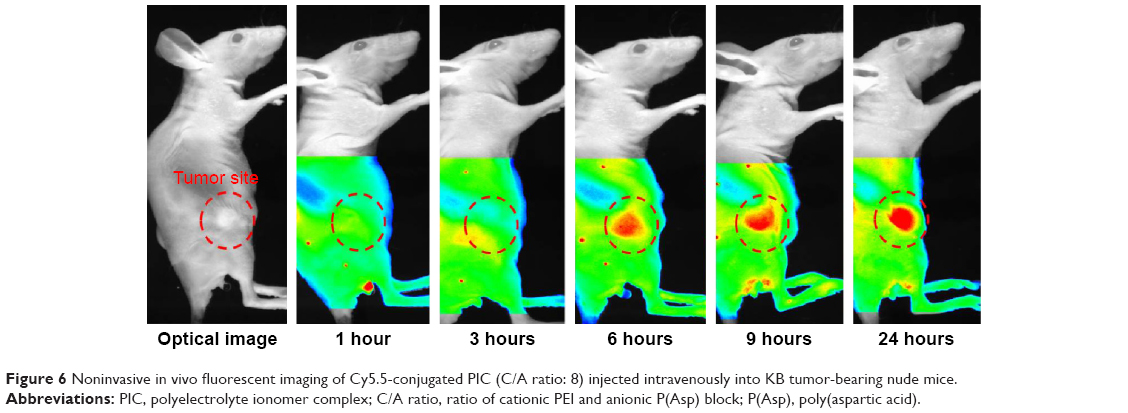 | Figure 6 Noninvasive in vivo fluorescent imaging of Cy5.5-conjugated PIC (C/A ratio: 8) injected intravenously into KB tumor-bearing nude mice. |
From these overall results, we were able to hypothesize a concept of the nanosized pH-sensitive PIC (Figure 7). The cationic PEG–PLA–PEI and anioic P(Asp) can be complexed by electrostatic interactions, resulting in the formation of a stable hydrophobic core with PLA and the neutralized blocks, and a hydrophillic corona with PEG and the charged polyelectrolyte blocks at physiological pH. The hydrophobic core can provide a pool for hydrophobic and charged drugs. When the environmental pH changes to acidic conditions, the deprotonated polyelectrolytes can initiate protonation, leading to the destabilization of the hydrophobic core formed by the electrostatic interactions (concept box in Figure 7). In vivo, the PIC in normal blood vessels can be circulated without recognition by the immune system due to the nanosize and surface charge properties,40 and the decrease in opsonization by PEG. At the tumor site, the leaky vasculature allows an increase in PIC accumulation via the enhanced permeability and retention effect.41 The extracellular pH of the tumor (pH <7.0) can trigger drug release by destabilization of PIC through the protonation of the PEI and P(Asp) blocks. Furthermore, PIC can enter the tumor cells by endocytosis and accelerate drug release at endosomal pH (pH <6.0) due to drastic destabilization. In addition, PIC with mobile cations may evade exocytosis and rupture the endosome by the proton sponge effect, which can increase cytotoxicity.
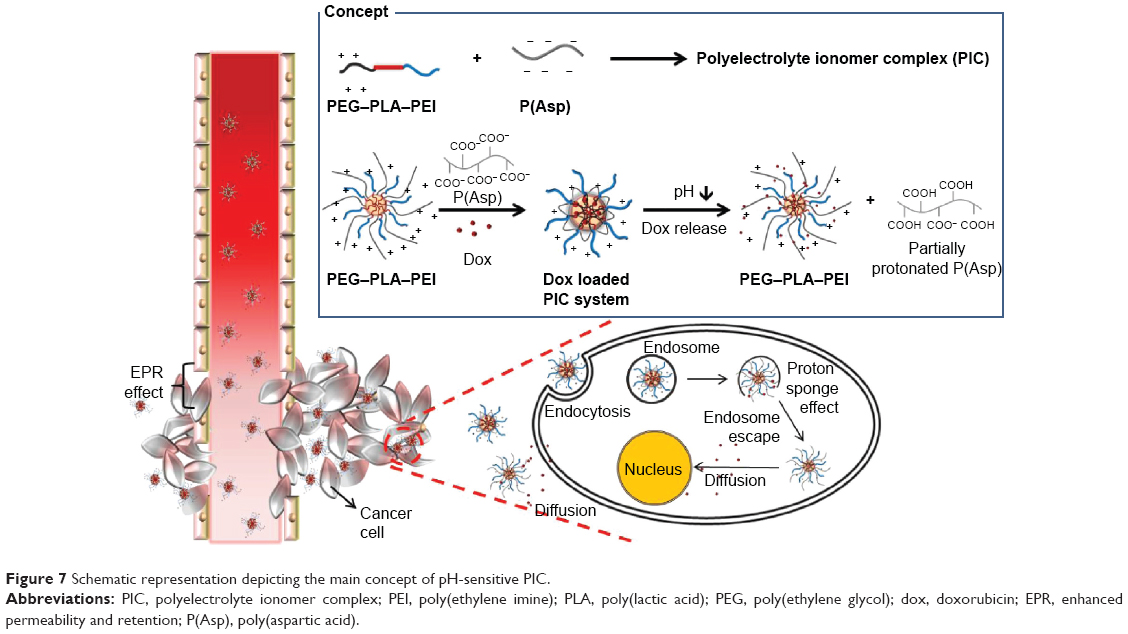 | Figure 7 Schematic representation depicting the main concept of pH-sensitive PIC. |
Conclusion
Here, the newly designed PIC formed from positively charged PEG–PLA–PEI and negatively charged P(Asp) was successfully prepared. PIC spontaneously self-assembled in aqueous solution at various C/A ratios through electrostatic interactions. When the C/A ratio was increased, the CAC and zeta potential values were increased; however, the size decreased up to 150 nm. Such a self-aggregated system at a C/A ratio of 8 showed pH-dependent particle properties with respect to size and zeta potential. The PIC nanomedicine prepared using dox as a model anticancer drug showed pH-dependent drug release and cytotoxicity. Overall, the 150 nm sized PIC nanomedicine was stable, with a low level of drug release at physiological pH, such as in the systemic circulation, and a rapid disintegration and drug release at acidic pH, such as extracellular tumor pH or endosomal pH. The animal imaging following intravenous administration of PIC revealed accumulation by passive targeting at the tumor site. In vivo tumor inhibition studies using these systems are in progress to demonstrate the enhanced antitumor activity by comparing with other nanosystems. In conclusion, the PIC nanomedicine may have considerable potential as a novel class of DDS for anticancer therapy.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the South Korean government (Ministry of Science, ICT, and Future Planning) (NRF-2014R1A2A1A11050094 and 2015R1A 5A1008958).
Disclosure
The authors report no conflicts of interest in this work.
References
He H, Xiao H, Kuang H, et al. Synthesis of mesoporous silica nanoparticle–oxaliplatin conjugates for improved anticancer drug delivery. Colloids Surf B Biointerfaces. 2014;117:75–81. | ||
Chacko RT, Ventura J, Zhuang J, Thayumanavan S. Polymer nanogels: a versatile nanoscopic drug delivery platform. Adv Drug Deliv Rev. 2012;64(9):836–851. | ||
Huang S-L. Liposomes in ultrasonic drug and gene delivery. Adv Drug Deliv Rev. 2008;60(10):1167–1176. | ||
Gu Z, Biswas A, Zhao M, Tang Y. Tailoring nanocarriers for intracellular protein delivery. Chem Soc Rev. 2011;40(7):3638–3655. | ||
Wu J, Kamaly N, Shi J, et al. Development of Multinuclear Polymeric Nanoparticles as Robust Protein Nanocarriers. Angew Chem Int Ed Enql. 2014;53(34):8975–8979. | ||
Kamimura M, Kim JO, Kabanov AV, Bronich TK, Nagasaki Y. Block ionomer complexes of PEG-block-poly(4-vinylbenzylphosphonate) and cationic surfactants as highly stable, pH responsive drug delivery system. J Control Release. 2012;160(3):486–494. | ||
Birch NP, Schiffman JD. Characterization of Self-Assembled Polyelectrolyte Complex Nanoparticles Formed from Chitosan and Pectin. Langmuir. 2014;30(12):3441–3447. | ||
Bronich TK, Nehls A, Eisenberg A, Kabanov VA, Kabanov AV. Novel drug delivery systems based on the complexes of block ionomers and surfactants of opposite charge. Colloids Surf B Biointerfaces. 1999;16 (1–4):243–251. | ||
Nakashima K, Bahadur P. Aggregation of water-soluble block copolymers in aqueous solutions: recent trends. Adv Colloid Interface Sci. 2006;123–126(0):75–96. | ||
Sun T-M, Du J-Z, Yan L-F, Mao H-Q, Wang J. Self-assembled biodegradable micellar nanoparticles of amphiphilic and cationic block copolymer for siRNA delivery. Biomaterials. 2008;29(32): 4348–4355. | ||
Itaka K, Yamauchi K, Harada A, Nakamura K, Kawaguchi H, Kataoka K. Polyion complex micelles from plasmid DNA and poly(ethylene glycol)–poly(l-lysine) block copolymer as serum-tolerable polyplex system: physicochemical properties of micelles relevant to gene transfection efficiency. Biomaterials. 2003;24(24):4495–4506. | ||
Kishimura A, Koide A, Osada K, Yamasaki Y, Kataoka K. Encapsulation of myoglobin in PEGylated polyion complex vesicles made from a pair of oppositely charged block ionomers: a physiologically available oxygen carrier. Angew Chem Int Ed Enql. 2007;46(32): 6085–6088. | ||
Kim JO, Sahay G, Kabanov AV, Bronich TK. Polymeric micelles with ionic cores containing biodegradable cross-links for delivery of chemotherapeutic agents. Biomacromolecules. 2010;11(4):919–926. | ||
Merdan T, Callahan J, Petersen H, et al. Pegylated polyethylenimine-Fab’ antibody fragment conjugates for targeted gene delivery to human ovarian carcinoma cells. Bioconjug Chem. 2003;14(5):989–996. | ||
Huang F-W, Wang H-Y, Li C, et al. PEGylated PEI-based biodegradable polymers as non-viral gene vectors. Acta Biomater. 2010;6(11):4285–4295. | ||
Oh KT, Bronich TK, Bromberg L, Hatton TA, Kabanov AV. Block ionomer complexes as prospective nanocontainers for drug delivery. J Control Release. 2006;115(1):9–17. | ||
Shuai X, Merdan T, Unger F, Wittmar M, Kissel T. Novel biodegradable ternary copolymers hy-PEI-g-PCL-b-PEG: synthesis, characterization, and potential as efficient nonviral gene delivery vectors. Macromolecules. 2003;36(15):5751–5759. | ||
Liu R, He B, Li D, et al. Effects of pH-sensitive chain length on release of doxorubicin from mPEG-b-PH-b-PLLA nanoparticles. Int J Nanomedicine. 2012;7:4433–4446. | ||
Lee ES, Kim JH, Sim T, et al. A feasibility study of a pH sensitive nanomedicine using doxorubicin loaded poly(aspartic acid-graft-imidazole)-block-poly(ethylene glycol) micelles. J Mat Chem B. 2014;2(9):1152–1159. | ||
Kim J, Oh Y, Lee K, Yun J, Park B, Oh K. Development of a pH-sensitive polymer using poly(aspartic acid-graft-imidazole)-block-poly(ethylene glycol) for acidic pH targeting systems. Macromol Res. 2011;19(5):453–460. | ||
Yun J, Park S-Y, Lee E, et al. Physicochemical characterizations of amphiphilic block copolymers with different MWs and micelles for development of anticancer drug nanocarriers. Macromol Res. 2012;20(9):944–953. | ||
Heald CR, Stolnik S, Kujawinski KS, et al. Poly(lactic acid)-poly(ethylene oxide) (PLA-PEG) nanoparticles: NMR studies of the central solidlike PLA core and the liquid PEG corona. Langmuir. 2002;18(9):3669–3675. | ||
Han So, Mahato RI, Kim SW. Water-soluble lipopolymer for gene delivery. Bioconjug Chem. 2001;12(3):337–345. | ||
Oh NM, Oh KT, Youn YS, Lee ES. Poly(l-aspartic acid) derivative soluble in a volatile organic solvent for biomedical application. Colloids Surf B Biointerfaces. 2012;97(0):190–195. | ||
Oh KT, Lee ES. Cancer-associated pH-responsive tetracopolymeric micelles composed of poly(ethylene glycol)-b-poly(L-histidine)-b-poly(L-lactic acid)-b-poly(ethylene glycol). Polymers for Advanced Technologies. 2008;19(12):1907–1913. | ||
Koo H, Jin G-W, Kang H, et al. Biodegradable branched poly(ethylenimine sulfide) for gene delivery. Biomaterials. 2010;31(5):988–997. | ||
Kircheis R, Wightman L, Wagner E. Design and gene delivery activity of modified polyethylenimines. Adv Drug Deliv Rev. 2001;53(3):341–358. | ||
Kanayama N, Fukushima S, Nishiyama N, et al. A PEG-based biocompatible block catiomer with high buffering capacity for the construction of polyplex micelles showing efficient gene transfer toward primary cells. ChemMedChem. 2006;1(4):439–444. | ||
Lin C-AJ, Sperling RA, Li JK, et al. Design of an amphiphilic polymer for nanoparticle coating and functionalization. Small. 2008;4(3):334–341. | ||
Anraku Y, Kishimura A, Oba M, Yamasaki Y, Kataoka K. Spontaneous formation of nanosized unilamellar polyion complex vesicles with tunable size and properties. J Am Chem Soc. 2010;132(5):1631–1636. | ||
Alexandridis P, Holzwarth JF, Hatton TA. Micellization of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) triblock copolymers in aqueous solutions: Thermodynamics of Copolymer Association. Macromolecules. 1994;27(9):2414–2425. | ||
Wilhelm M, Zhao CL, Wang Y, et al. Poly(styrene-ethylene oxide) block copolymer micelle formation in water: a fluorescence probe study. Macromolecules. 1991;24(5):1033–1040. | ||
Vila A, Gill H, McCallion O, Alonso MaJ. Transport of PLA-PEG particles across the nasal mucosa: effect of particle size and PEG coating density. J Control Release. 2004;98(2):231–244. | ||
Doiron AL, Chu K, Ali A, Brannon-Peppas L. Preparation and initial characterization of biodegradable particles containing gadolinium-DTPA contrast agent for enhanced MRI. Proc Natl Acad Sci. 2008;105(45):17232–17237. | ||
Jacobson GB, Shinde R, Contag CH, Zare RN. Sustained release of drugs dispersed in polymer nanoparticles. Angew Chem Int Ed Enl. 2008;120(41):7998–8000. | ||
Liu N, Li B, Gong C, Liu Y, Wang Y, Wu G. A pH- and thermo-responsive poly(amino acid)-based drug delivery system. Colloids Surf B Biointerfaces. 2015;136:562–569. | ||
Lin L, Xu W, Liang H, et al. Construction of pH-sensitive lysozyme/pectin nanogel for tumor methotrexate delivery. Colloids Surf B Biointerfaces. 2015;126:459–466. | ||
Allen C, Maysinger D, Eisenberg A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf B Biointerfaces. 1999;16 (1–4):3–27. | ||
Teng Y, Morrison ME, Munk P, Webber SE, Procházka K. Release kinetics studies of aromatic molecules into water from block polymer micelles. Macromolecules. 1998;31(11):3578–3587. | ||
He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666.014. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.