Back to Journals » International Journal of Nanomedicine » Volume 14
Development and mechanistic study of a microemulsion containing vitamin E TPGS for the enhancement of oral absorption of celecoxib
Authors Subongkot T
Received 13 January 2019
Accepted for publication 8 March 2019
Published 30 April 2019 Volume 2019:14 Pages 3087—3102
DOI https://doi.org/10.2147/IJN.S201449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Thirapit Subongkot
Department of Pharmaceutical Technology, Faculty of Pharmaceutical Sciences, Burapha University, Chonburi, Thailand
Purpose: The purpose of this study was to develop a microemulsion containing D-α-tocopheryl polyethylene glycol 1000 succinate (vitamin E TPGS) as a biodegradable surfactant to increase the oral absorption of celecoxib.
Methods: This study investigated the intestinal absorption enhancement mechanism of this microemulsion by measuring transepithelial electrical resistance (TEER) values. This study also evaluated microemulsion particle–intestine interactions in terms of release and attachment processes using confocal laser scanning microscopy (CLSM).
Results: The prepared microemulsion particles had a size of <300 nm with a neutral surface charge. The celecoxib-loaded microemulsion release kinetic was classified as the zero-order model. This vitamin E TPGS-based microemulsion significantly increased the in vitro intestinal absorption of celecoxib compared to celecoxib solution. The CLSM study suggested that microemulsion particles with entrapped drugs might attach to the intestinal epithelium before releasing the entrapped drug into tissues. The TEER value of the intestinal tissues treated with the celecoxib-loaded microemulsion was significantly decreased compared to the value before treatment, indicating an increase in drug transport via the paracellular pathway. The evaluation of intestinal tissue cytotoxicity using lactate dehydrogenase cytotoxicity assay suggested that the prepared celecoxib-loaded microemulsion was safe for oral route administration.
Conclusions: The prepared celecoxib loaded microemulsion could increase the intestinal absorption of celecoxib compared to celecoxib solution. The intestinal absorption enhancement mechanism of this microemulsion resulted from the increase of the drug transport via the paracellular pathway.
Keywords: microemulsion, celecoxib, oral absorption enhancement mechanism, vitamin E TPGS, transepithelial electrical resistance
Introduction
Celecoxib is an NSAID that exhibits anti-inflammatory and analgesic effects. Celecoxib is approved for the treatment of osteoarthritis, primary dysmenorrhea, rheumatoid arthritis and acute pain. Since celecoxib is a specific COX-2 inhibitor, it has no gastrointestinal (GI) side effects, such as GI bleeding, in contrast to conventional NSAIDs.
To achieve maximal therapeutic efficacy of celecoxib from oral administration, the drug must be completely absorbed through the GI tract to the blood circulation. Celecoxib is highly hydrophobic (log P=3.5)1 and has an extremely low solubility in water (2.55–4.57 µg/mL).2,3 Due to its physicochemical properties, celecoxib exhibits poor oral bioavailability after oral administration.4,5
To improve the oral bioavailability of celecoxib, Subramanian et al6. developed self-microemulsifying drug delivery systems (SMEDDS) consisted of PEG-8 caprylic/capric glycerides as the oil phase, polysorbate 20 as the surfactant and propylene glycol monocaprylate ester as the cosurfactant. However, there was a report showing that surfactants such as sodium dodecyl sulfate, Tween 20, Tween 60 and sodium deoxycholate have adverse effects on the small intestines of rats by increasing the exfoliation of the brush border membrane and inhibiting disaccharidase activities.7
D-α-tocopheryl polyethylene glycol succinate (vitamin E TPGS) is a water-soluble derivative of vitamin E synthesized by the esterification of vitamin E and polyethylene glycol (PEG).8 The vitamin E TPGS structure is composed of vitamin E as the lipophilic component and PEG as the hydrophilic component. Therefore, vitamin E TPGS is a nonionic surfactant with a hydrophilic–lipophilic balance (HLB) value of 13.2 and a critical micelle concentration (CMC) of 0.02% w/w.9 Vitamin E TPGS has been approved with Generally Recognized As Safe (GRAS) status when used as an oral dietary supplement of vitamin E.10 In drug delivery systems, vitamin E TPGS has been used as an oral absorption enhancer of lipophilic drugs, such as paclitaxel,11,12 cyclosporine,13 l-sulpiride14and amprenavir.15
A microemulsion is a transparent colloidal system composing of an oil, surfactant, cosurfactant and water. Microemulsions offer many advantages for oral drug delivery system development, such as thermodynamic stability, simplicity of manufacturing, low viscosity and high solubilization ability for both lipophilic and hydrophilic drugs. To provide patient safety for the oral administration of celecoxib, this study formulated a microemulsion as a delivery system for the oral absorption enhancement of celecoxib using vitamin E TPGS as a surfactant, which can be degraded to vitamin E by esterase enzymes in vivo.
Theoretically, the major mechanisms of drug transport through the GI epithelium are the transcellular and paracellular pathways.16 Previous studies have shown that microemulsions can increase the oral absorption of drugs by increasing lymphatic transportation.17,18 However, the oral absorption enhancement mechanism of microemulsions, and in particular the role of the paracellular pathway, has not been fully clarified. These studies evaluated the intestinal absorption enhancement mechanism of microemulsions using transepithelial electrical resistance (TEER) measurements of treated intestinal tissues. Furthermore, basic knowledge and understanding of microemulsion particle–intestinal tissue interactions are lacking. This study also used a colocalization technique using multifluorescent compounds as microemulsion particle probes to investigate the interactions between microemulsion particles and intestinal tissues.
This study aimed to develop new microemulsion formulation using vitamin E TPGS as a surfactant with edible cosurfactants to improve the oral absorption of celecoxib, to investigate the intestinal absorption enhancement mechanism of microemulsions and to study microemulsion particle–intestinal tissue interactions.
Materials and methods
Materials
Celecoxib was purchased from Power Tech Chemical Industry (Bangkok, Thailand). Medium-chain triglyceride (MCT) oil (Lexol GT-865) was purchased from Inolex (Philadelphia, PA, USA). Vitamin E TPGS was purchased from Antares Health Products Inc. (Jonesborough, TN, USA). Nile red was purchased from Tokyo Chemical Industry (Tokyo, Japan). N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine triethylammonium salt (NBD-PE) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other reagents were of analytical reagent grade and were commercially available.
Solubility study
The solubility of celecoxib in different oils and cosurfactants was measured by mixing an excess amount of celecoxib into 2 mL of each solvent and stirred with magnetic stirrer at room temperature for 24 hrs. The celecoxib suspension was then transferred to a microcentrifuge tube and centrifuged at 10,000 rpm for 10 mins. The supernatant was withdrawn and filtered through a 0.45 µm pore size membrane. The filtrate was appropriately diluted with methanol and the celecoxib concentration was analyzed by HPLC. The oil providing the highest solubility of celecoxib will be used as oil phase to create the pseudoternary phase diagrams of microemulsions as described in the section “Construction of pseudoternary phase diagrams of microemulsions”.
Preparation of microemulsions
Construction of pseudoternary phase diagrams of microemulsions
Pseudoternary phase diagrams of microemulsions were devised in order to obtain the largest microemulsion region using water titration method. Microemulsion systems composed of the oil providing the highest solubility of celecoxib from the section “Solubility study” was used as oil phase, vitamin E TPGS was used as surfactant, whereas PEG 400, ethanol, propylene glycol or glycerine was used as cosurfactant with ultrapure water as the water phase.
The surfactant and each cosurfactant were weighted and mixed together (surfactant:cosurfactant weight ratio =1:1) until the clear solution was obtained to create the surfactant mixture or Sm. The oil and Sm were mixed, and the ratio of oil and Sm were varied from 9:1 to 1:9 (w/w). Each oil/Sm mixture was added dropwise with water and stirred gently with a magnetic stir bar to allow equilibration. After an aliquot of water, the change from clear to turbid was visually inspected and recorded. The microemulsion system was considered as the transparency mixtures.
Solubility of celecoxib in microemulsion
Celecoxib was added excessively into the microemulsion formulation obtaining from pseudoternary phase diagrams which provided the greatest microemulsion region as described in the section “Construction of pseudoternary phase diagrams of microemulsions” and stirred with a magnetic stirrer at room temperature for 24 hrs. The celecoxib suspension was then transferred to a microcentrifuge tube and centrifuged at 10,000 rpm for 10 mins to eliminate the excess drug. Afterward, the supernatant was filtered using a syringe filter through a 0.45 µm pore size membrane. The celecoxib concentration in the filtrate was analyzed by HPLC after proper dilution with methanol.
Characterization of microemulsions
Mean particle size, surface charge, particle size distribution and electrical conductivity
The mean particle size, surface charge (zeta potential), particle size distribution (polydispersity index, PDI) and electrical conductivity of both blank microemulsion and celecoxib-loaded microemulsion were evaluated using a photon correlation spectroscopy (PCS) particle size analyzer (Zetasizer Nano-ZS, Malvern Instruments, Worcestershire, UK) equipped with a He-Ne laser using a scattering angle of 173°. Each measurement was conducted under room temperature and in triplicate.
Rheological properties
The rheological properties of different celecoxib-loaded microemulsions were investigated with a rheometer (Kinexus Lab, Malvern Instruments) using cone and plate geometries. The experiments were conducted with a shear rate in the range of 1 to 100 s−1. Each measurement was performed at 25°C and in triplicate.
In vitro drug release
The in vitro drug release of celecoxib in PEG 400 as control and different celecoxib-loaded microemulsions was performed using light protective screw cap glass container. Four milliliters of each formulation was filled into a regenerated cellulose dialysis bag (CelluSep T2®, Membrane Filtration Products Inc., Seguin, TX, USA) having molecular weight cutoff between 6,000 and 8,000 daltons. Then, the dialysis bag with tested formulation was sealed tightly with the clamps. Afterward, the dialysis bag was immersed in a beaker which had 80 mL of release medium containing 50% v/v ethanol in PBS with pH 7.4. The beaker containing dialysis system was covered completely with aluminum foil and placed on a hot plate magnetic stirrer in order to sustain the release medium temperature at 37°C and stirred with a magnetic stir bar at a rate of 250 rpm. After the designated time at 0.5, 1, 2, 3, 4, 6, 8 and 24 hrs, 1 mL of release medium was withdrawn and replaced instantly with the same quantity of preheated release medium to continue a constant volume. The collected samples were analyzed for celecoxib concentration by HPLC. The percentage of cumulative amount of celecoxib was calculated and plotted as a function of time to determine the release rate. The release kinetics were evaluated using mathematical models by fitting to zero-order, first-order, and Higuchi diffusion models as follows:

where Q is the cumulative amount of drug released at time (t), t is time (hrs), Q0 is the initial amount of drug, K0 is the zero-order release constant, K1 is the first-order release constant and Kh is the Higuchi release constant.
In vitro intestinal absorption study
According to the previous reports which revealed that porcine intestine can be utilized for prediction of human intestinal absorption,19,20 this in vitro intestinal absorption study of celecoxib in PEG 400 as the control and various celecoxib-loaded microemulsions was conducted with Franz diffusion cells using porcine ileum as permeability barrier membrane. The sectioned porcine ileum obtained from a local slaughterhouse was gently cleaned with PBS to eliminate any lumen contents before intestinal absorption study. The tissue was placed between the donor and receiver compartment of diffusion cells which had penetration area around 2.3 cm2. The water jacketed receiver compartment, filled approximately with 6.5 mL of 50% v/v ethanol in PBS, was connected with a circulating water bath to maintain a temperature of 37°C. Two milliliters of each tested formulation was added in the donor compartment and then covered with parafilm®. At the predetermined times of 1, 2, 4, 6, 8, and 24 hrs, 0.5 mL of the receiver medium was withdrawn and immediately replaced with the same volume of new medium in order to preserve a constant volume. The concentration of celecoxib in the collected samples was analyzed by HPLC. All experiments were done in triplicate. The cumulative amount of celecoxib was plotted versus time, and the apparent permeability (Papp) was calculated using the following equation:
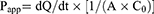
where dQ/dt is the linear appearance rate of celecoxib in the receiving compartment, A is the exposed tissue area and C0 is the donor concentration.
Evaluation of intestinal epithelium cytotoxicity
To ensure the safety of celecoxib-loaded microemulsions for oral administration, a lactate dehydrogenase (LDH) cytotoxicity assay was used to evaluate the intestinal tissue damage as previously reported by Subongkot et al.21 For this experiment, the tested formulations were blank PEG 400, celecoxib in PEG 400, blank microemulsion and the celecoxib-loaded microemulsion which provided the highest Papp, as mentioned in the section “In vitro intestinal absorption study” using PBS and 3% w/w Triton X-100 as negative and positive controls, respectively. This study was conducted in triplicate using Franz diffusion cells, as described in the section “In vitro intestinal absorption study”, with the ileum section as membrane without the presence of receiving medium. Two milliliters of each formulation was filled in the donor compartments and treated for 2 hrs. Next, each tested formulation inside the donor compartment was withdrawn and replaced with 2 mL of PBS. The washed PBS was pipetted into the microcentrifuge tube and centrifuged at 1,000× g for 10 mins at 4°C. The obtained supernatant was transferred into a 96-well plate and analyzed for the released LDH activity with an LDH cytotoxicity detection kit (Roche Diagnostics, Mannheim, Germany). The formed formazan in the supernatant was analyzed by measuring absorbance in triplicate at a wavelength of 492 nm with a microplate photometer (AccuReader M965, Taipei, Taiwan). The obtained percent cytotoxicity was calculated from the obtained absorbance according to the following equation:

Confocal laser scanning microscopy (CLSM) study
Visualization of lipophilic fluorescent compound penetration
To confirm the results of the in vitro intestinal absorption study, CLSM was applied to evaluate microemulsion particle penetration into the porcine intestinal tissue using a fluorescent dye instead of celecoxib. Nile red, a hydrophobic red fluorescent compound which has physicochemical characteristics (log partition coefficient =5.0 and molecular weight =318 Da)22 equivalent to those of celecoxib, was chosen as the fluorescent dye. In this experiment, two formulations, PEG 400 as a control and the microemulsion which yielded the highest Papp according to the experiments described in in vitro intestinal absorption study, were investigated using the same concentration of Nile red at 0.5 mg/mL.
In vitro intestinal absorption study. A penetration test was conducted using Franz diffusion cells, as described in the section “In vitro intestinal absorption study”, without the addition of receiving medium. Five hundred microliters of Nile red in PEG 400 or the Nile red-loaded microemulsion was added in the donor compartment. After treatment for 3 hrs, the obtained ileum was washed twice with PBS to remove the excess drug before visualization with CLSM.
CLSM evaluation. The treated ileum, described in the section “In vitro intestinal absorption study”, was placed on a coverslip with dimensions of 22×50 mm (MENZEL-GLÄSER®, Braunschweig, Germany) and observed using the 10× objective lens system of an inverted microscope (Zeiss LSM 800, Carl Zeiss, Jena, Germany) equipped with diode lasers (excitation wavelength =405, 488 and 561 nm).
To compare the penetration depths and fluorescence intensities of Nile red in the tissue, the treated ileum was scanned with a laser with an excitation wavelength of 561 nm in order to obtain serial x–z plane images. The fluorescence intensity was measured at the middle horizontal line from each x–z plane image using ZEN (blue edition) imaging software (Carl Zeiss). The obtained mean fluorescence intensity of each image was plotted versus the penetration depth.
Particle–intestinal tissue interaction
CLSM was used to investigate the absorption mechanism and characteristics of microemulsion particles into intestinal tissue using a colocalization technique. This technique utilized the difference in fluorescence color between the entrapped drug and the microemulsion particles. Nile red, a red fluorescent compound, represents a lipophilic drug entrapped in microemulsion particles. NBD-PE is a type of fluorescent phospholipid composed of phosphatidyl ethanolamine conjugated with NBD at the polar head group that exhibits green fluorescence. NBD-PE is classified as a fluorescent surfactant. NBD-PE was used as a marker of microemulsion particles.
To prepare the Nile red-loaded NBD-PE-labeled microemulsion particles, Nile red and NBD-PE were accurately weighed at 1 and 5.4 mg, respectively, in a 2 mL volumetric flask. The microemulsion formulation that provided the highest Papp according to the experiments described in the section “In vitro intestinal absorption study” was used and adjusted to 2 mL.
In vitro intestinal absorption study. A penetration test was conducted using Franz diffusion cells, as described in the section “In vitro intestinal absorption study”, except that the release medium was omitted. Five hundred microliters of the Nile red-loaded NBD-PE-labeled microemulsion particles was filled in the donor compartment and treated for 2 and 4 hrs. The treated intestinal tissue was washed twice with PBS to remove the excess drug before examination by CLSM.
Intestine histology preparation. The treated ileum, as described in the section “In vitro intestinal absorption study,” was sectioned to observe microemulsion particles and drug penetration in cross-sections using a cryomicrotome (Leica 1850, Leica Instruments GmbH, Nussloch, Germany). Each tissue sample was placed onto a metal sample holder inside the cryomicrotome, which was precooled at −30°C. Then, the tissue was mounted with a cryo-embedding medium (Neg 50, MICROM International, Waldorf, Germany). The frozen tissue was sectioned into 10 µm slices before being placed on positively charged slides (MENZEL-GLÄSER®, SuperFrostTM Plus, Braunschweig, Germany). To visualize the living cell layers of the intestine, the nucleic acids of these tissues were stained with an aqueous solution of 10 µg/mL 4ʹ,6-diamidino-2-phenylindole dihydrochloride (DAPI) for 15 s, washed with water, mounted with mounting medium and covered with a coverslip. The prepared slides were visualized by CLSM as described in the section “CLSM evaluation”.
Transepithelial electrical resistance (TEER) measurements
To evaluate the oral absorption enhancement mechanism of microemulsion particles by the paracellular pathway, small intestine epithelial cell barrier function was assessed by measuring the TEER values of treated tissues. The investigated formulations of this study included the microemulsion formulation that provided the highest Papp, as described in the section “In vitro intestinal absorption study”, while PBS and celecoxib in PEG 400 were used as controls. The measurements were performed using 6-well hanging insert plates that were placed on receiver trays (well diameter=24 mm, membrane area=4.5 cm2). A piece of ileum was placed on the semipermeable membrane of the hanging insert plate, which covered the entire membrane area and prevented the contact of medium between the apical and basolateral sides of the membrane. One milliliter of PBS was pipetted on the apical and basolateral sides. TEER values were measured using a Millicell® ERS-2 volt-ohm meter (Merck KGaA, Darmstadt, Germany). Only PBS on the apical side of the tested formulation was removed and replaced with 600 µL of microemulsion formulation. After treatment for 3 hrs, the microemulsion formulation was removed and replaced with 1 mL of PBS. The TEER values after treatment were measured and compared with those before treatment. The TEER values (R) were calculated as follows: 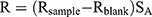
where Rsample is the electrical resistance value of treated tissue (Ω),
Rblank is the electrical resistance value of hanging insert plates (Ω) and
SA is the surface area of the hanging insert plate membrane (cm2).
HPLC assay
Celecoxib was analyzed by HPLC (Agilent 1260 infinity II LC system, Agilent Technologies, Santa Clara, CA, USA) using a 5 µm particle size of C18 reversed-phase column as stationary phase (VertiSep UPS C18, Vertical, Nonthaburi, Thailand) with dimensions of 4.6×250 mm. A mixture solution of acetonitrile:water (75:25% v:v) was used as mobile phase at a flow rate of 1 mL/min. Each sample was injected into the column at a volume of 20 µL with UV detection at a wavelength of 250 nm. A quantitative analysis of celecoxib was obtained from a standard curve, which was linear (r2≥0.9999) in the range of 0.1–500 µg/mL. Prior to sample analysis, the HPLC method was validated for accuracy, precision, limit of detection (LOD) and limit of quantification (LOQ). The LOD and LOQ were 0.05 µg/mL and 0.1 µg/mL, respectively. For accuracy and precision of this analysis condition, the percentage recovery and the percentage coefficient of variation (% CV) was 99.67±0.23 and 1.45±0.15, respectively.
Statistical analysis
All data were statistically analyzed using a paired t-test and ANOVA, followed by post-hoc test (LSD). Differences of p<0.05 were considered statistically significant.
Results and discussion
Solubility study
The solubilities of celecoxib in various types of oils and cosurfactants are shown in Table 1. Among the selected oils (MCT oil, fish oil and oleic acid), celecoxib had the greatest solubility in MCT oil. Therefore, MCT oil was selected as the oil phase for microemulsion preparation. Due to the highest solubility of celecoxib in PEG 400, this solvent was used as vehicle to prepare celecoxib solution as the control for in vitro release study and also intestinal absorption study. The solubility of celecoxib in vitamin E TPGS could not be determined because vitamin E TPGS is a waxy solid.
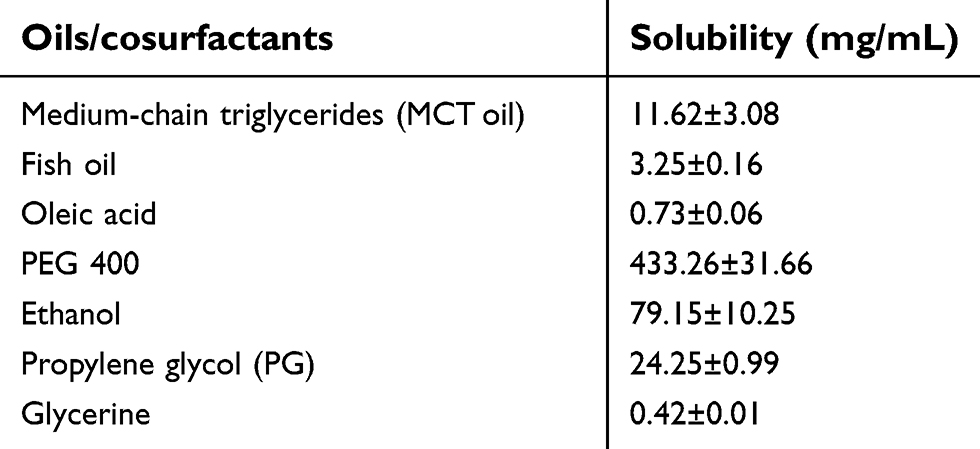 | Table 1 Solubility of celecoxib in various oils and cosurfactants |
Construction of pseudoternary phase diagrams of microemulsions
Screening of cosurfactants
Pseudoternary phase diagrams were devised to obtain the optimal concentration range of the microemulsion components. Microemulsion systems consisted of MCT oil as the oil phase, vitamin E TPGS as the surfactant and water as the aqueous phase and the variation of different cosurfactants was evaluated. The cosurfactants used for this study were PEG 400, ethanol, propylene glycol and glycerine. The microemulsion systems of MCT oil as the oil phase, vitamin E TPGS as the surfactant, water as the aqueous phase with PEG 400, ethanol and propylene glycol are shown in Figure 1.
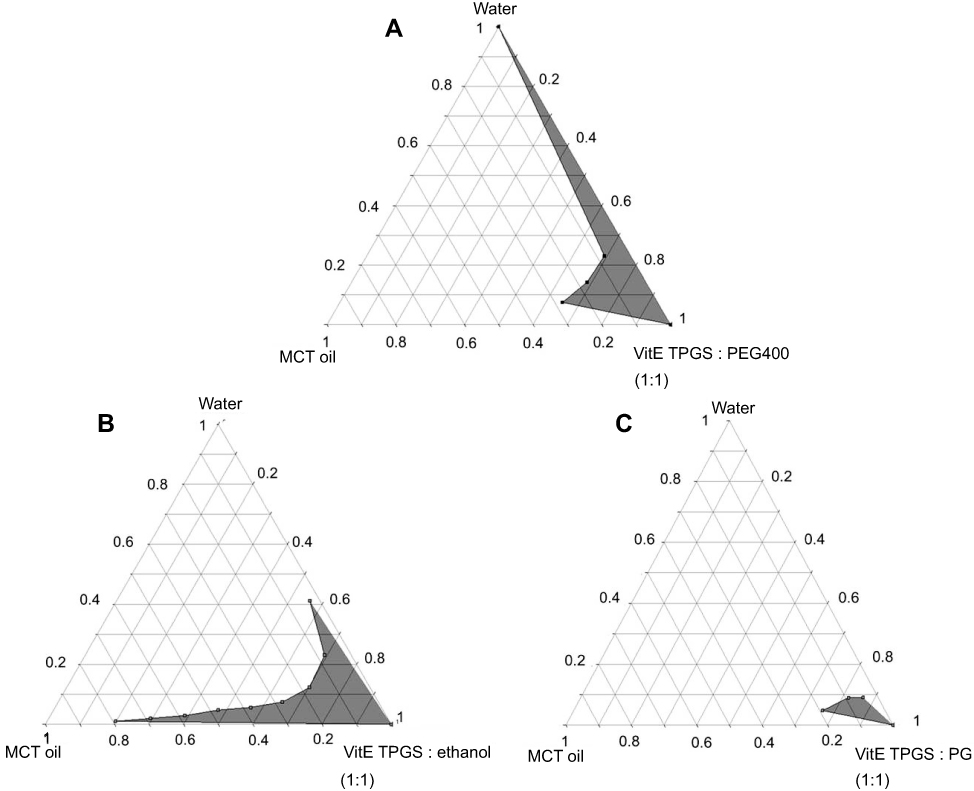 | Figure 1 |
The use of ethanol as a cosurfactant provided more microemulsion regions than PEG 400 and PG, whereas the use of glycerine as a cosurfactant did not cause microemulsion formation. Although Ke et al23 reported a pseudoternary phase diagram of a microemulsion system composed of MCT oil as an oil phase, vitamin E TPGS as a surfactant, PEG 400 as a cosurfactant and water as an aqueous phase, this study revealed that the addition of celecoxib in a microemulsion system composed of PEG 400 as a cosurfactant could reverse the microemulsion to a coarse emulsion. Therefore, a microemulsion system composed of MCT oil as the oil phase, vitamin E TPGS as the surfactant, ethanol as the cosurfactant and water as the aqueous phase was used for further study.
Solubility of celecoxib in the microemulsions
Celecoxib solubility was in the range of 131.4–201.1 mg/mL, as shown in Table 2. An increased volume of the surfactant mixture enhanced the solubility of celecoxib. The microemulsion system is a liquid dosage form that can be manufactured by encapsulation in soft gelatin capsules for convenient oral administration. Therefore, celecoxib was loaded at 90% of each formula’s solubility to reduce the size of the capsule for ease of swallowing, as shown in Table 2.
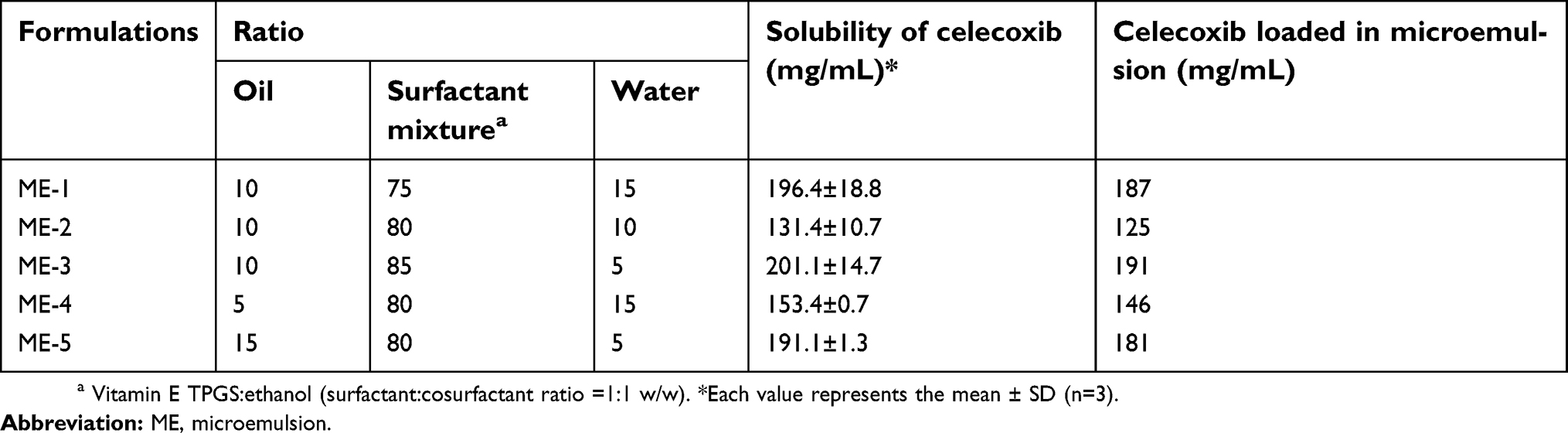 | Table 2 Formulations, component ratios, celecoxib solubilities and concentrations of celecoxib loaded in microemulsions |
Characterization of microemulsions
Mean particle size, surface charge, particle size distribution and electrical conductivity
The average particle size, zeta potential, PDI and conductivity of the microemulsion without celecoxib (blank ME) and the celecoxib-loaded microemulsion (celecoxib-loaded ME) are shown in Table 3. The mean particle size of the blank ME ranged from 162.03 to 229.07 nm, while the size of the celecoxib-loaded ME ranged from 106.12 to 268.57 nm. There were no significant differences in the mean particle size between the blank ME and the celecoxib-loaded ME (p>0.05). The addition of celecoxib in this microemulsion system did not affect the particle size. The zeta potentials of the blank ME and the celecoxib-loaded ME were −0.108–0.004 mV and −0.0706–0.0743 mV, respectively. Since the microemulsion components and celecoxib are nonionic molecules, the zeta potentials of the blank ME and the celecoxib-loaded ME were neutral. The PDI of the blank ME was between 0.329 and 0.765, while the PDI of the celecoxib-loaded ME was between 0.261 and 0.444. Celecoxib-loaded ME-4 and celecoxib-loaded ME-5 had significantly greater PDIs than blank ME-4 and blank ME-5, respectively.
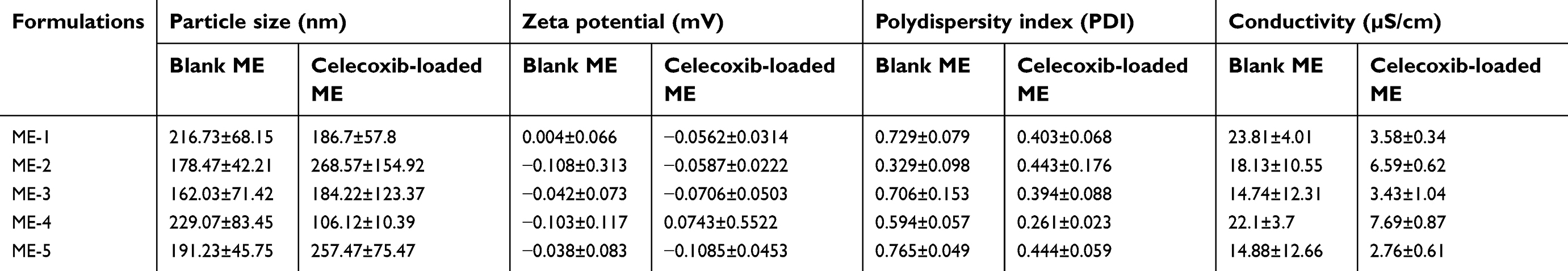 | Table 3 Particle size, zeta potential, polydispersity index (PDI) and electrical conductivity of microemulsions (MEs) without celecoxib (blank MEs) and celecoxib-loaded microemulsions (celecoxib-loaded MEs) |
Theoretically, the microemulsion systems can be divided into three main types of microemulsions as follows: oil-in-water microemulsion, bicontinuous microemulsion and water-in-oil microemulsion.24,25,26 The classification of microemulsion types depends on the ratio of oil to water phases. An oil-in-water microemulsion is formed when the proportion of water is greater than that of oil. On the contrary, a water-in-oil microemulsion is formed when the proportion of oil is greater than that of water. When the proportion of oil is equal to water, a bicontinuous microemulsion is formed. Droplets of oil-in-water microemulsions and water-in-oil microemulsions are spherical nanoparticles, as indicated by TEM,27,28 whereas bicontinuous microemulsions have been proposed as nonspherical nanoparticles.29 Electrical conductivity is a beneficial parameter which can be used for the evaluation of different microemulsion types.
According to the classification of microemulsion types by Krauel et al,30 the authors concluded that microemulsion which had conductivity <1 µS/cm was water-in-oil microemulsion. The conductivity of bicontinuous microemulsion was in the range of 1–10 µS/cm, while the conductivity of oil-in-water microemulsion was >10 µS/cm.
The conductivity of the blank microemulsion was in the range of 14.74–23.81 µS/cm, while that of the celecoxib-loaded microemulsion was in the range of 2.76–7.69 µS/cm. In accordance with the above criterion, this study suggested that blank ME-1, blank ME-2, blank ME-3, blank ME-4 and blank ME-5 were oil-in-water microemulsions, whereas celecoxib-loaded ME-1, celecoxib-loaded ME-2, celecoxib-loaded ME-3, celecoxib-loaded ME-4 and celecoxib-loaded ME-5 were bicontinuous microemulsions.
All celecoxib-loaded microemulsions had average conductivities that were significantly less than that of the blank microemulsion (p>0.05). The addition of celecoxib, which is a lipophilic drug into microemulsion, resulted in a decrease in the conductivity by enhancing the proportion of oil phase. These findings agreed with the results reported by Djekic et al31 and Subongkot et al21 who found that the incorporation of a hydrophobic drug led to a decrease in the conductivity of oil-in-water microemulsions compared to blank microemulsions.
Rheological properties
The rheological profiles of celecoxib-loaded ME-1, celecoxib-loaded ME-2, celecoxib-loaded ME-3, celecoxib-loaded ME-4 and celecoxib-loaded ME-5 are shown in Figure 2. The rheograms showed that all celecoxib-loaded microemulsions demonstrated a linear function between shear stress versus shear rate. Therefore, the rheological properties of celecoxib-loaded microemulsions were Newtonian flow behavior.
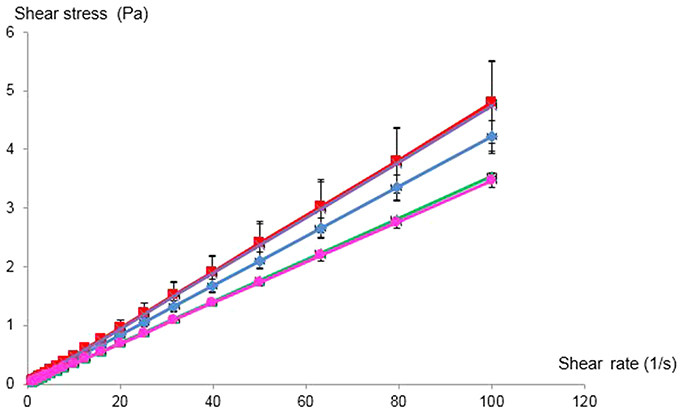 | Figure 2 The rheological properties of celecoxib-loaded ME-1( |
 ), celecoxib-loaded ME-2(
), celecoxib-loaded ME-2( ), celecoxib-loaded ME-3(
), celecoxib-loaded ME-3( ), celecoxib-loaded ME-4(
), celecoxib-loaded ME-4( ) and celecoxib-loaded ME-5 (
) and celecoxib-loaded ME-5 ( ). All data represent the mean±standard deviation (n=3).
). All data represent the mean±standard deviation (n=3).In vitro drug release
The release characteristics of celecoxib from the control and various microemulsion formulations are illustrated in Figure 3. According to the coefficient of determination (R2) as shown in Table 4, the zero-order model provided the greatest fit for the celecoxib release graphs of every microemulsion formulation and the control. The release rate for PEG 400 was significantly higher than those of all microemulsion formulations. There were no significantly differed in the release rates among ME-1, ME-2, ME-3, ME-4 and ME-5. This study agreed with the results previously reported by Panapisal et al32 and Cavalcanti et al,33 in which microemulsions had lower release rates than solutions. Since celecoxib has high solubility in surfactant mixtures, the slow release rate of the microemulsions resulted from the partitioning ability of drugs between the surfactant mixture and the oil/aqueous phase.
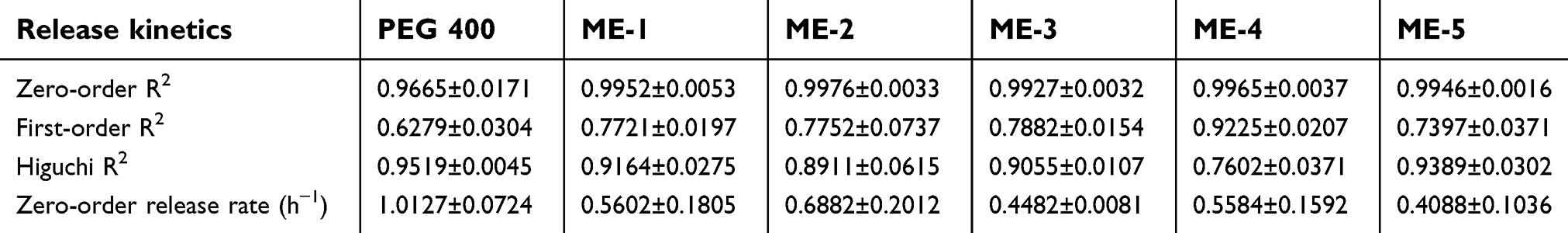 | Table 4 In vitro release kinetics of celecoxib in PEG 400 and celecoxib-loaded microemulsions (ME) |
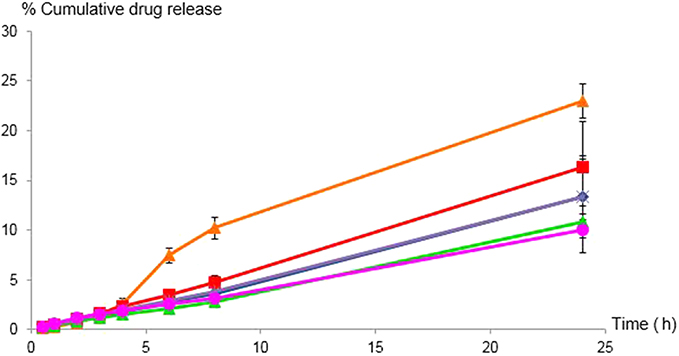 | Figure 3 In vitro cumulative release profiles (%) of celecoxib from PEG 400 ( |
 ) and the following microemulsion formulations: ME-1(
) and the following microemulsion formulations: ME-1( ), ME-2 (
), ME-2 ( ), ME-3 (
), ME-3 ( ), ME-4 (
), ME-4 ( ) and ME-5 (
) and ME-5 ( ). All data represent the mean±standard deviation (n=3).
). All data represent the mean±standard deviation (n=3).In vitro intestinal absorption study
The cumulative intestinal absorption profiles and the Papp values of celecoxib in PEG 400 as the control with various celecoxib-loaded microemulsions are shown in Figure 4 and Table 5, respectively. Papp for celecoxib-loaded ME-4 was significantly greater than celecoxib in PEG 400 (p=0.015). Therefore, celecoxib-loaded ME-4 was selected for further study. Theoretically, colloidal drug delivery systems can improve oral absorption of lipophilic drug by enhancing solubility, in which the drug molecules can be penetrated via enterocytes of the intestinal tissue to systemic blood circulation as free drug or as drug entrapped in particles. Regarding the in vitro drug release results reported in the section “In vitro drug release”, in which the microemulsion had a slower release rate than that of the solution, the increase in intestinal drug absorption might involve microemulsion particles as drug carriers with the surfactant/cosurfactant acting as an absorption enhancer.
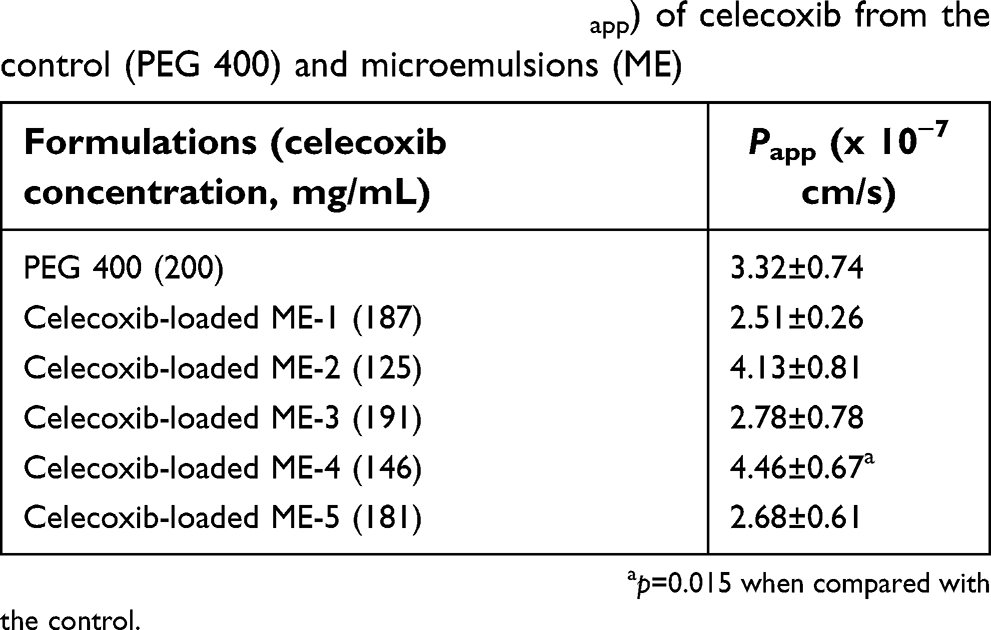 | Table 5 The apparent permeability (Papp) of celecoxib from the control (PEG 400) and microemulsions (ME) |
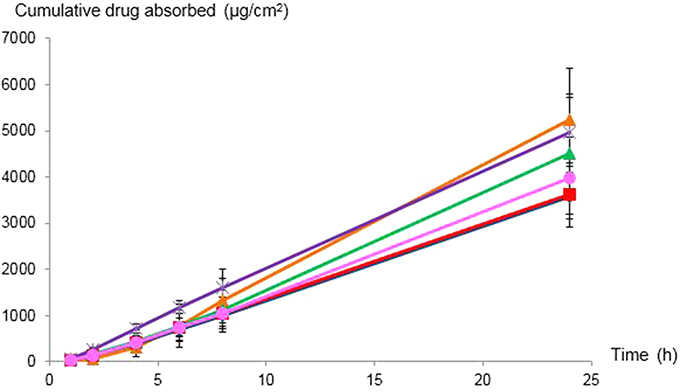 | Figure 4 In vitro cumulative intestinal absorption profiles of celecoxib from PEG 400 ( |
 ) and the following microemulsion formulations: ME-1(
) and the following microemulsion formulations: ME-1( ), ME-2 (
), ME-2 ( ), ME-3 (
), ME-3 ( ), ME-4 (
), ME-4 ( ) and ME-5 (
) and ME-5 ( ). All data represent the mean±standard deviation (n=3).
). All data represent the mean±standard deviation (n=3).Evaluation of intestinal epithelium cytotoxicity
To investigate the safety of this novel microemulsion formulation as an oral dosage form, LDH cytotoxicity assay was utilized to evaluate the intestinal cell membrane damage by comparing with PEG 400, a widely used solvent in oral pharmaceutical products. The percent cytotoxicity of blank ME-4, celecoxib-loaded ME-4, blank PEG 400 and celecoxib in PEG 400 are shown in Table 6. There were no significant differences among the percent cytotoxicity of blank ME-4, celecoxib-loaded ME-4, blank PEG 400 and celecoxib in PEG 400. The percent cytotoxicity of blank PEG 400 did not significantly differ from celecoxib in PEG 400 (p=0.854), and also the percent cytotoxicity of the blank ME-4 did not significantly differ from the celecoxib-loaded ME-4 (p=0.458), which revealed that the addition of celecoxib did not result in intestinal epithelium cytotoxicity. This study suggested that the celecoxib-loaded ME-4 was safe for oral route administration because the percent cytotoxicity of celecoxib-loaded ME-4 was not significantly different from that of PEG 400 (p=0.482), an edible solvent, that is commonly employed as a cosolvent for oral liquid dosage forms. This study, therefore, suggested that celecoxib-loaded ME-4 was safe for oral intake.
 | Table 6 The percent cytotoxicity of blank ME-4, celecoxib-loaded ME-4, blank PEG 400 and celecoxib in PEG 400 |
CLSM study
Visualization of lipophilic fluorescent compound penetration
Based on the in vitro intestinal absorption study mentioned in the section “In vitro intestinal absorption study”, ME-4 had the highest Papp values. To confirm the results of the absorption study, ME-4 was selected for encapsulation with Nile red by comparing the penetration depths and also fluorescent intensities with Nile red in PEG 400 as control group.
The sequential x–z plane images at various penetration depths of Nile red in PEG 400 and Nile red-loaded ME-4 are shown in Figure 5A and B, respectively. The red fluorescence of Nile red in PEG 400 was observed from 15 µm to 70 µm, while ME-4 was clearly observed from 15 µm to 95 µm. The fluorescent intensity graphs of Nile red at different penetration depths for the PEG 400 as control and ME-4 are revealed in Figure 5C. ME-4 also exhibited a greater fluorescent intensity of Nile red than PEG 400. These CLSM analyses using Nile red agreed with the results obtained from the intestinal absorption study in the section “In vitro intestinal absorption study”.
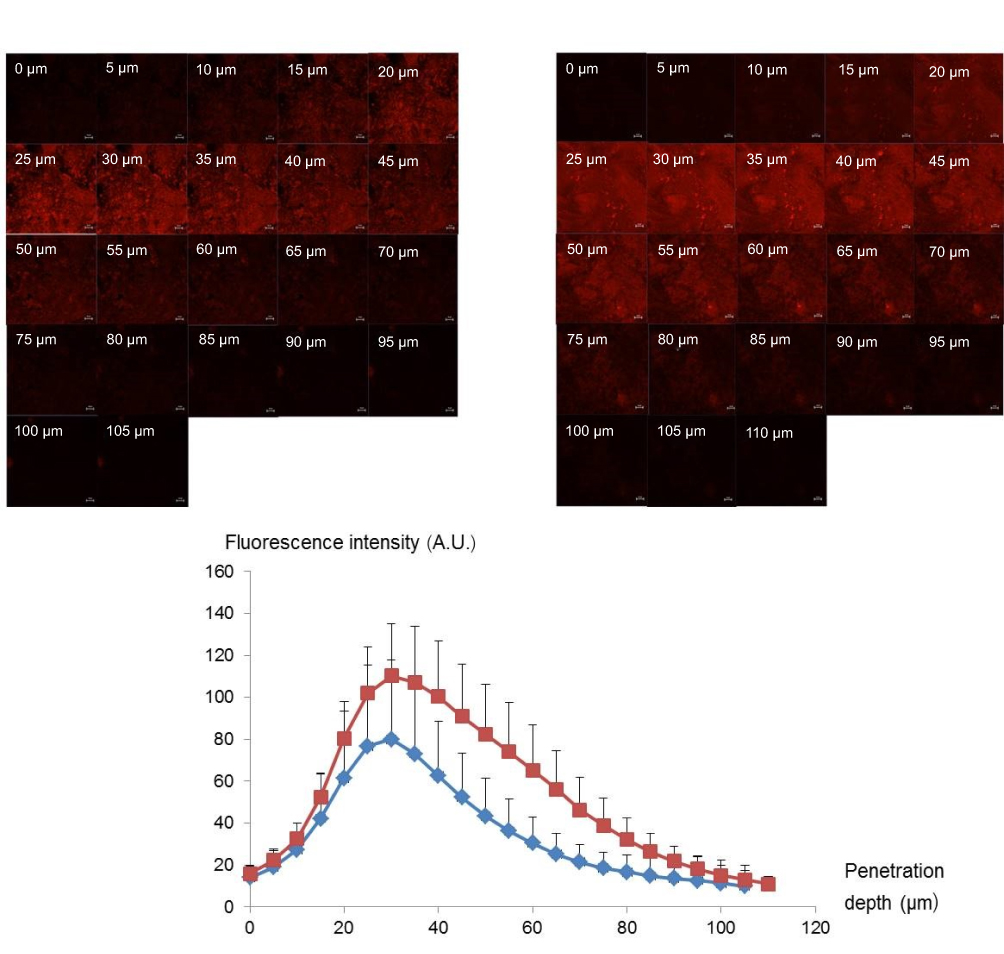 | Figure 5 CLSM images show x-z plane sequential penetration of a porcine intestine treated with (A) Nile red in PEG 400 and (B) Nile red-loaded ME-4 after 3 h. Scale bar represents 50 µm. (C) Comparison of the fluorescence intensity profiles of Nile red at various depths in the intestine shown in Figure 5a ( |
 ) and Figure 5b (
) and Figure 5b ( ). All data represent the mean±standard deviation.Abbreviation: A.U., arbitrary units.
). All data represent the mean±standard deviation.Abbreviation: A.U., arbitrary units.Particle–intestinal tissue interactions
CLSM was used to observe the intestinal tissue distribution and penetration of microemulsion particles using a fluorescence colocalization technique. Nile red, which was used as the entrapped drug, exhibited red fluorescence, whereas NBD-PE, which was used to label the microemulsion particles, exhibited green fluorescence. A schematic diagram of Nile red-loaded NBD-PE-labeled microemulsion particle is shown in Figure 6A, and an intact porcine intestine cross-section stained with DAPI is presented in Figure 6B.
 | Figure 6 (A) Schematic representation of Nile red-loaded NBD-PE-labeled microemulsion particles and (B) CLSM image of an intact porcine intestine cross section stained with DAPI. MCT oil = medium-chain triglycerides oil. The scale bar represents 50 µm. |
Drug absorption mechanisms into intestinal tissues from particles can be divided into 2 types of mechanisms, which include free drug and drug carrier mechanisms. For the free drug mechanism, drug molecules were absorbed independently into intestinal tissue as free drugs after being released from the particles. In the drug carrier mechanism, drug-loaded particles attached to the surface of intestinal tissue before the entrapped drugs were gradually released and absorbed into the intestinal tissue. To evaluate the absorption mechanism of the drug into intestinal tissue as either a free drug or drug carrier mechanism, a colocalization technique involving Nile red-loaded NBD-PE-labeled microemulsion particles was used. If the entrapped drugs were released before the particles attached to the tissues, the fluorescence from the entrapped drugs and the fluorescence from the particles would not be observed in the same area throughout the entire tissue surface. If the particles attached to the tissues before releasing the entrapped drugs, the 2 fluorescent signals would be observed in the same area.
Based on the possibility of the deposition of fluorescent compounds, this study assumed that if the red and green fluorescence were observed in the same area on the intestinal tissue surface, it indicates a drug carrier mechanism. If the red and green fluorescence were observed at different sites on intestinal tissue surface, it indicates a free drug mechanism. Since red and green fluorescence of the entrapped drug and microemulsion particles were observed at the same site on intestinal tissues at both 2 hrs and 4 hrs, as shown in Figure 7A-1–A-3 and B-1–B-3, respectively, this result suggests that the intestinal drug absorption mechanism of the microemulsion particles was a drug carrier mechanism, in which particle-bearing drugs attached to the intestinal tissue surface before the entrapped drugs were gradually released into intestinal tissue.
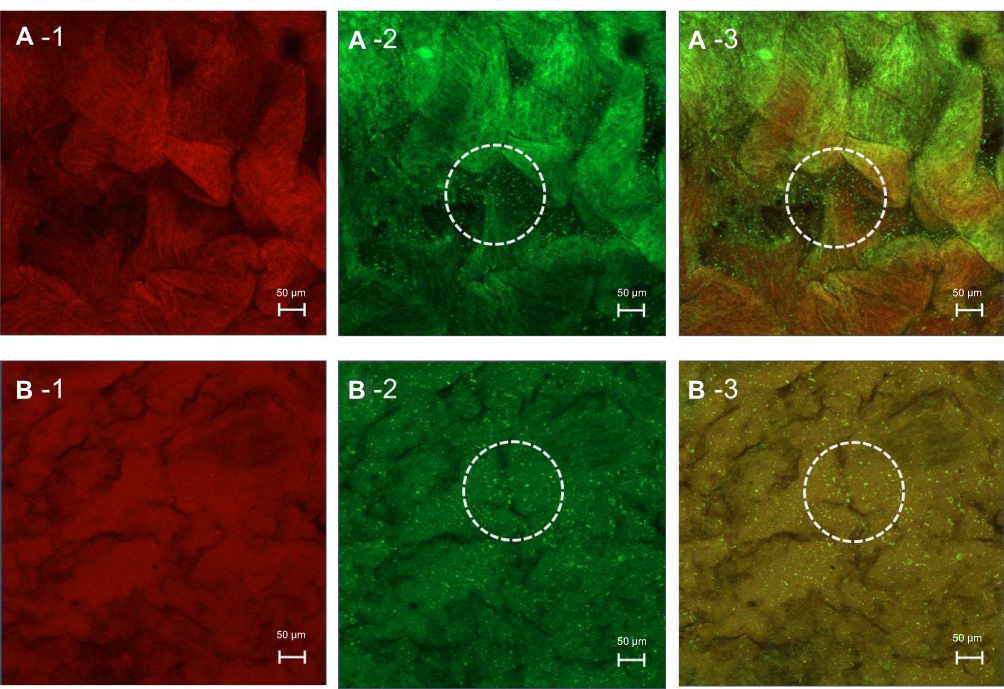 | Figure 7 CLSM (x–y plane) images show a porcine intestine treated with Nile red-loaded NBD-PE-labeled microemulsion after (A) 2 h and (B) 4 h. Each image is divided into three frames as follows: (1) red fluorescence of Nile red, (2) green fluorescence of NBD-PE-labeled microemulsion particles and (3) overlay of 1 and 2. The scale bar represents 50 µm. The dash circles indicate a group of microemulsion particles from which the entrapped drugs were released into the tissues. |
Many green fluorescence signals were observed, as shown in the white circle of Figure 7A-2, A-3 and B-2, B-3, but red fluorescent signals were not observed in the same area, as shown in Figure 7A-1 and B-1. These signals might indicate a group of microemulsion particles from which the entrapped drugs were released into the tissues.
The cross-section images of a porcine intestine treated with Nile red-loaded NBD-PE-labeled microemulsion and stained with DAPI at 4 h from 2 areas are shown in Figure 8A and B. The red fluorescence of Nile red can be seen throughout the tissue (Figure 8A-1 and B-1), whereas the green fluorescence of NBD-PE had a strong intensity at the villi compared with the muscular externa, as shown in Figure 8A-2 and B-2. This indicated that Nile red, as an entrapped drug, could penetrate throughout the tissue, while most of the microemulsion particles accumulated in villi. The results from the cross-section images also agreed with the results that are shown in Figure 7; many of the blank microemulsion particles were found on the surface of the tissues without the entrapped drugs.
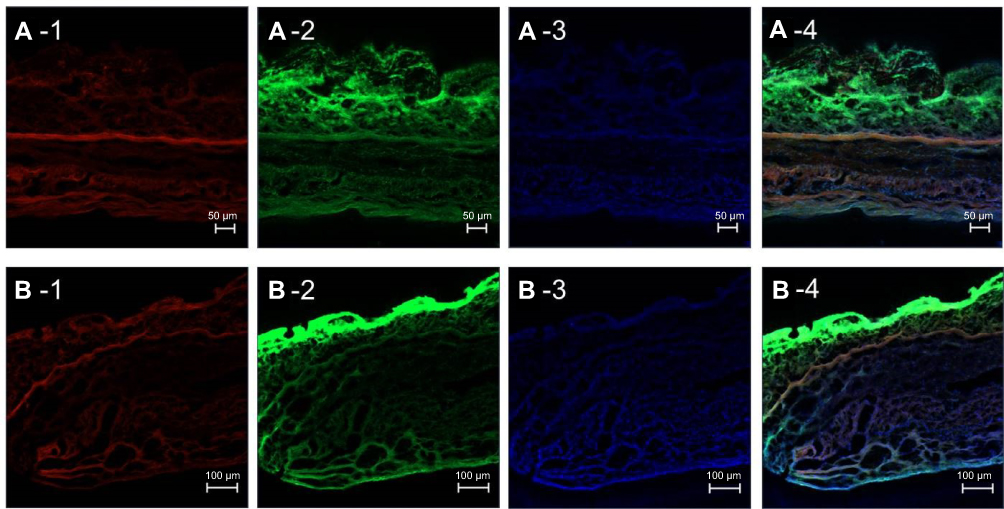 | Figure 8 CLSM images show a cross-section image of a porcine intestine stained with DAPI and treated with Nile red-loaded NBD-PE-labeled microemulsion particles after 4 h from different areas (A) and (B). Each image is divided into four frames as follows: (1) red fluorescence of Nile red, (2) green fluorescence of NBD-PE, (3) blue fluorescence of DAPI and (4) overlay of 1, 2 and 3. The scale bar represents 50 µm and 100 µm for A and B, respectively. |
In summary, this study suggested that drug-loaded microemulsion particles attached to intestinal tissue before releasing the entrapped drug into the tissues. The released drug molecules could penetrate throughout the intestinal tissue, whereas most of the empty microemulsion particles remained on the villi.
Transepithelial electrical resistance (TEER) evaluation
The TEER values of porcine intestinal tissue before and after treatment with PBS, celecoxib in PEG 400 and celecoxib loaded ME-4 are shown in Table 7. The TEER values of the tissues after treatment with PBS or celecoxib in PEG 400 were not significantly different from those before treatment, whereas the TEER value of the tissue treated with celecoxib-loaded ME-4 was significantly decreased compared to the value before treatment. This indicated that celecoxib-loaded ME-4 reduced tight junction integrity, whereas celecoxib in PEG 400 did not.
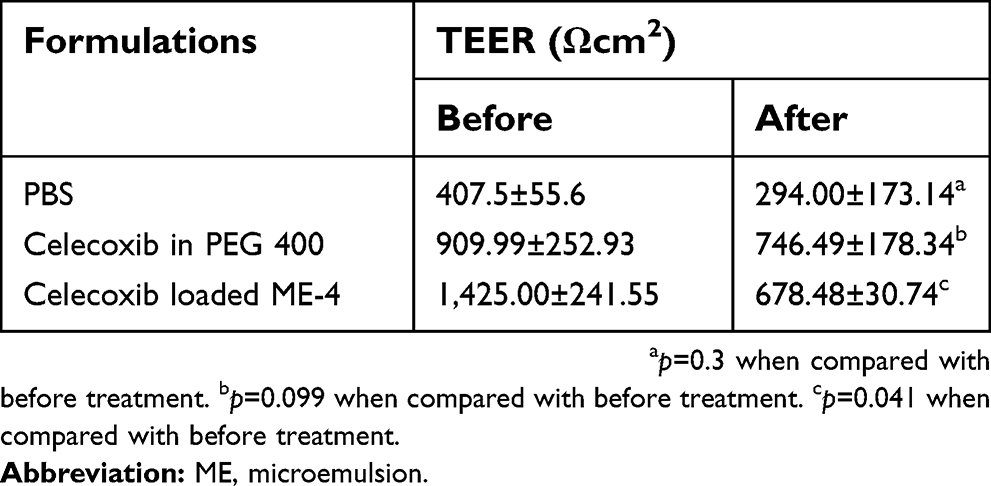 | Table 7 Transepithelial electrical resistance (TEER) of porcine intestines before and after treatment with PBS, celecoxib in PEG 400 and celecoxib-loaded ME-4 |
For intestinal epithelium drug absorption, molecules were absorbed through enterocyte cells on intestinal epithelium into blood circulation by passive diffusion through cell membranes (transcellular pathway), by diffusion between adjacent cells (paracellular pathway) and by carrier-mediated transport (carrier-mediated transcellular pathway).34 Absorption via the paracellular route is important for the diffusion of ions and the transport of glucose, amino acids, peptides and small, hydrophilic and ionized drugs. Tight junctions composed of transmembrane proteins, which are occludin, claudins, tricellulin and junctional adhesion molecules, act as semipermeable barriers to the paracellular transport of ions, water and drugs.35 Transport via the paracellular or intercellular pathways is limited by the integrity of cellular tight junctions. To increase permeability through the paracellular pathway, many chemicals have been reported to act as paracellular permeability enhancers, such as bile salts, dimethyl β-cyclodextrin, surfactants, medium-chain fatty acids, ethanol and chitosan derivatives.34,36,37,38
TEER measurement is a generally accepted quantitative technique used to evaluate the integrity of tight junction dynamics in tissues and cell culture models of epithelial monolayers. The decrease in the TEER value of the tissue treated with celecoxib-loaded ME-4 resulted from the decrease of the integrity of cellular tight junctions. It is suggested that the reduction of tight junction integrity is affected by the chemical components of the microemulsion formulation, which are vitamin E TPGS and ethanol.
Since celecoxib is a lipophilic molecule, celecoxib can diffuse into tissues by the transcellular pathway. The decrease in the TEER value of the tissue treated with celecoxib-loaded ME-4 indicated that besides transcellular pathway transport, microemulsion particles also increased the intestinal absorption of celecoxib by enhancing the diffusion of the drug through the paracellular pathway. The TEER value of the tissue treated with celecoxib in PEG 400 did not differ from that before celecoxib treatment, indicating that the diffusion of celecoxib from this solution into the tissues occurred only by the transcellular pathway.
P-glycoprotein (P-gp), an efflux pump found on hepatocytes, epithelial cells of blood-brain barrier, renal proximal tubular cells and also intestinal epithelial cells, can decrease oral drug absorption by pumping drug molecules back into the intestinal lumen.39 There are evidences showing that vitamin E TPGS could inhibit P-gp in Caco-2 cell mode and increase oral absorption of drug in healthy volunteers by inhibiting P-gp ATPase enzyme inhibition.40,41
In conclusion, microemulsion containing vitamin E TPGS increased intestinal absorption of celecoxib by enhancing drug transport via transcellular pathway, paracellular pathway and inhibiting P-gp.
Conclusions
A celecoxib-loaded microemulsion containing vitamin E TPGS as the surfactant was successfully developed. The physicochemical properties of the microemulsion were also characterized. This microemulsion increased the intestinal absorption of celecoxib by acting as a drug carrier system, in which the microemulsion particles attached to the intestinal tissue surface before the entrapped drugs were released and penetrated the tissues. The advantage of microemulsion particles compared to a solution dosage form is that the microemulsion particles could increase intestinal drug absorption by the enhancement of drug diffusion through the paracellular pathway. The LDH cytotoxicity test indicated that the safety of celecoxib-loaded microemulsion containing vitamin E TPGS delivered to the intestinal membrane was equivalent to an edible solvent (PEG 400).
Acknowledgments
The author is grateful to Burapha University through the National Research Council of Thailand (Grant number 127/2561) and the Faculty of Pharmaceutical Sciences, Burapha University, for financial support.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Garti N, Avrahami M, Aserin A. Improved solubilization of Celecoxib in U-type nonionic microemulsions and their structural transitions with progressive aqueous dilution. J Colloid Interface Sci. 2006;299:352–365. doi:10.1016/j.jcis.2006.01.060
2. Gupta P, Kakumanu VK, Bansal AK. Stability and solubility of celecoxib-PVP amorphous dispersions: a molecular perspective. Pharm Res. 2004;21:1762–1769.
3. Gupta VR, Mutalik S, Patel MM, Jani GK. Spherical crystals of celecoxib to improve solubility, dissolution rate and micromeritic properties. Acta Pharm. 2007;57:173–184.
4. Paulson SK, Vaughn MB, Jessen SM, et al. Pharmacokinetics of celecoxib after oral administration in dogs and humans: effect of food and site of absorption. J Pharmacol Exp Ther. 2001;297:638–645.
5. Morgen M, Bloom C, Beyerinck R, et al. Polymeric nanoparticles for increased oral bioavailability and rapid absorption using celecoxib as a model of a low-solubility, high-permeability drug. Pharm Res. 2012;29:427–440. doi:10.1007/s11095-011-0558-7
6. Subramanian N, Ray S, Ghosal SK, Bhadra R, Moulik SP. Formulation design of self-microemulsifying drug delivery systems for improved oral bioavailability of celecoxib. Biol Pharm Bull. 2004;27:1993–1999.
7. Kimura T, Imamura H, Hasegawa K, Yoshida A. Mechanisms of toxicities of some detergents added to a diet and of the ameliorating effect of dietary fiber in the rat. J Nutr Sci Vitaminol (Tokyo). 1982;28:483–489.
8. Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49:175–186. doi:10.1016/j.ejps.2013.02.006
9. Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33:4889–4906. doi:10.1016/j.biomaterials.2012.03.046
10. Yan A, Bussche A, Kane A, Hurt R. Tocopheryl polyethylene glycol succinate as a safe, antioxidant surfactant for processing carbon nanotubes and fullerenes. Carbon N Y. 2007;45:2463–2470. doi:10.1016/j.carbon.2007.08.035
11. Varma MV, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci. 2005;25:445–453. doi:10.1016/j.ejps.2005.04.003
12. Ho PY, Yeh TK, Yao HT, et al. Enhanced oral bioavailability of paclitaxel by D-alpha-tocopheryl polyethylene glycol 400 succinate in mice. Int J Pharm. 2008;359:174–181. doi:10.1016/j.ijpharm.2008.04.013
13. Chang T, Benet LZ, Hebert MF. The effect of water-soluble vitamin E on cyclosporine pharmacokinetics in healthy volunteers. Clin Pharmacol Ther. 1996;59:297–303. doi:10.1016/S0009-9236(96)80007-5
14. Kim DS, Kim DW, Kim K, et al. Development of a novel l-sulpiride-loaded quaternary microcapsule: effect of TPGS as an absorption enhancer on physicochemical characterization and oral bioavailability. Colloids Surf B Biointerfaces. 2016;147:250–257. doi:10.1016/j.colsurfb.2016.08.010
15. Yu L, Bridgers A, Polli J, Vickers A, Long S, Roy A. Vitamin E-TPGS increases absorption flux of HIV protease inhibitor by enhancing its solubility and permeability. Pharm Res. 1999;16:1812–1817.
16. Zhu L, Lu L, Wang S, et al. Oral absorption basics: pathways and physicochemical and biological factors affecting absorption. In: Qiu Y, Chen Y, Zhang GGZ, Mantri RV, Yu L, editors. Developing Solid Oral Dosage Forms.
17. Xing Q, Song J, You X, et al. Microemulsions containing long-chain oil ethyl oleate improve the oral bioavailability of piroxicam by increasing drug solubility and lymphatic transportation simultaneously. Int J Pharm. 2016;511:709–718. doi:10.1016/j.ijpharm.2016.07.061
18. Tang TT, Hu XB, Liao DH, Liu XY, Xiang DX. Mechanisms of microemulsion enhancing the oral bioavailability of puerarin: comparison between oil-in-water and water-in-oil microemulsions using the single-pass intestinal perfusion method and a chylomicron flow blocking approach. Int J Nanomedicine. 2013;8:4415–4426. doi:10.2147/IJN.S51469
19. Nolte K, Backfisch G, Neidlein R. In vitro studies of poorly absorbed drugs using porcine intestine in the ring model RIMO. Arzneimittelforschung. 2000;50:664–668. doi:10.1055/s-0031-1300269
20. Westerhout J, van de Steeg E, Grossouw D, et al. A new approach to predict human intestinal absorption using porcine intestinal tissue and biorelevant matrices. Eur J Pharm Sci. 2014;63:167–177. doi:10.1016/j.ejps.2014.07.003
21. Subongkot T, Ngawhirunpat T. Development of a novel microemulsion for oral absorption enhancement of all-trans retinoic acid. Int J Nanomedicine. 2017;12:5585–5599. doi:10.2147/IJN.S142503
22. Bader CA, Shandala T, Carter EA, et al. A molecular probe for the detection of polar lipids in live cells. PLoS One. 2016;11:e0161557. doi:10.1371/journal.pone.0161557
23. Ke WT, Lin SY, Ho HO, Sheu MT. Physical characterizations of microemulsion systems using tocopheryl polyethylene glycol 1000 succinate (TPGS) as a surfactant for the oral delivery of protein drugs. J Control Release. 2005;102:489–507. doi:10.1016/j.jconrel.2004.10.030
24. Acharya DP, Hartley PG. Progress in microemulsion characterization. Curr Opin Colloid Interface Sci. 2012;17:274–280.
25. Callender SP, Mathews JA, Kobernyk K, Wettig SD. Microemulsion utility in pharmaceuticals: implications for multi-drug delivery. Int J Pharm. 2017;526:425–442. doi:10.1016/j.ijpharm.2017.05.005
26. Spernath A, Aserin A. Microemulsions as carriers for drugs and nutraceuticals. Adv Colloid Interface Sci. 2006;128–130:47–64. doi:10.1016/j.cis.2006.11.016
27. Fouad SA, Basalious EB, El-Nabarawi MA, Tayel SA. Microemulsion and poloxamer microemulsion-based gel for sustained transdermal delivery of diclofenac epolamine using in-skin drug depot: in vitro/in vivo evaluation. Int J Pharm. 2013;453:569–578. doi:10.1016/j.ijpharm.2013.06.009
28. Zhao JH, Ji L, Wang H, et al. Microemulsion-based novel transdermal delivery system of tetramethylpyrazine: preparation and evaluation in vitro and in vivo. Int J Nanomedicine. 2011;6:1611–1619. doi:10.2147/IJN.S23597
29. Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery systems. Adv Drug Deliv Rev. 2012;64:175–193. doi:10.1016/j.addr.2012.09.018
30. Krauel K, Davies NM, Hook S, Rades T. Using different structure types of microemulsions for the preparation of poly(alkylcyanoacrylate) nanoparticles by interfacial polymerization. J Control Release. 2005;106:76–87. doi:10.1016/j.jconrel.2005.04.013
31. Djekic L, Primorac M, Filipic S, Agbaba D. Investigation of surfactant/cosurfactant synergism impact on ibuprofen solubilization capacity and drug release characteristics of nonionic microemulsions. Int J Pharm. 2012;433:25–33. doi:10.1016/j.ijpharm.2012.04.070
32. Panapisal V, Charoensri S, Tantituvanont A. Formulation of microemulsion systems for dermal delivery of silymarin. AAPS PharmSciTech. 2012;13:389–399. doi:10.1208/s12249-012-9762-y
33. Cavalcanti AL, Reis MY, Silva GC, et al. Microemulsion for topical application of pentoxifylline: in vitro release and in vivo evaluation. Int J Pharm. 2016;506:351–360. doi:10.1016/j.ijpharm.2016.04.065
34. Ward PD, Tippin TK, Thakker DR. Enhancing paracellular permeability by modulating epithelial tight junctions. Pstt. 2000;3:346–358.
35. Chiba H, Osanai M, Murata M, Kojima T, Sawada N. Transmembrane proteins of tight junctions. Biochim Biophys Acta. 2008;1778:588–600. doi:10.1016/j.bbamem.2007.08.017
36. Ates M, Kaynak MS, Sahin S. Effect of permeability enhancers on paracellular permeability of acyclovir. J Pharm Pharmacol. 2016;68:781–790. doi:10.1111/jphp.12551
37. Yu Q, Wang Z, Li P, Yang Q. The effect of various absorption enhancers on tight junction in the human intestinal Caco-2 cell line. Drug Dev Ind Pharm. 2013;39:587–592. doi:10.3109/03639045.2012.692376
38. Kowapradit J, Opanasopit P, Ngawhirunpat T, Rojanarata T, Ruktanonchai U, Sajomsang W. Methylated N-(4-N,N-dimethylaminocinnamyl) chitosan enhances paracellular permeability across Caco-2 cells. Drug Deliv. 2010;17:301–312. doi:10.3109/10717541003706273
39. Canaparo R, Finnström N, Serpe L, et al. Expression of CYP3A isoforms and P-glycoprotein in human stomach, jejunum and ileum. Clin Exp Pharmacol Physiol. 2007;34:1138–1144. doi:10.1111/j.1440-1681.2007.04691.x
40. Bogman K, Zysset Y, Degen L, et al. P-glycoprotein and surfactants: effecton intestinal talinolol absorption. Clin Pharmacol Ther. 2005;77:24–32. doi:10.1016/j.clpt.2004.09.001
41. Collnot EM, Baldes C, Wempe MF, et al. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: influence on ATPase activity and membrane fluidity. Mol Pharm. 2007;4:465–474. doi:10.1021/mp060121r
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.