Back to Journals » International Journal of Nanomedicine » Volume 11
Development and evaluation of vitamin E D-α-tocopheryl polyethylene glycol 1000 succinate-mixed polymeric phospholipid micelles of berberine as an anticancer nanopharmaceutical
Authors Shen R, Kim J, Yao M, Elbayoumi T
Received 29 December 2015
Accepted for publication 4 March 2016
Published 26 April 2016 Volume 2016:11 Pages 1687—1700
DOI https://doi.org/10.2147/IJN.S103332
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Roger Shen,1 Jane J Kim,2 Mingyi Yao,2,3 Tamer A Elbayoumi2,3
1Department of Family Medicine, Northeastern Health Systems-Tahlequah City Hospital, Tahlequah, OK, USA; 2Department of Pharmaceutical Sciences, College of Pharmacy-Glendale, Midwestern University, 3Nanomedicine Center of Excellence in Translational Nanomedicine, Midwestern University, Glendale, AZ, USA
Abstract: Berberine (Brb) is an active alkaloid occurring in various common plant species, with well-recognized potential for cancer therapy. Brb not only augments the efficacy of antineoplastic chemotherapy and radiotherapy but also exhibits direct antimitotic and proapoptotic actions, along with distinct antiangiogenic and antimetastatic activities in a variety of tumors. Despite its low systemic toxicity, several pharmaceutical challenges limit the application of Brb in cancer therapy (ie, extremely low solubility and permeability, very poor pharmacokinetics (PKs), and oral bioavailability). Among lipid-based nanocarriers investigated recently for Brb, stealth amphiphilic micelles of polymeric phospholipid conjugates were studied here as a promising strategy to improve Brb delivery to tumors. Specifically, physicochemically stable micelles made of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (PEG-PE) mixed with D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) (PEG-succinate ester of vitamin E), in a 3:1 M ratio, increased Brb solubilization by 300%. Our PEG-PE/TPGS-mixed micelles firmly retained the incorporated Brb, displaying extended-release profile in simulated media, with up to 30-fold projected improvement in simulated PKs of Brb. Owing to the markedly better uptake of Brb-containing mixed micelles in vitro, our Brb-mixed micelles nanoformulation significantly amplified apoptosis and overall cytotoxic effectiveness against monolayer and spheroid cultures of human prostate carcinomas (16- to 18-fold lower half-maximal inhibitory concentration values in PC3 and LNPaC, respectively), compared to free Brb. Mixed PEG-PE/TPGS micelles represent a promising delivery platform for the sparingly soluble anticancer agent, Brb, encouraging further pharmaceutical development of this drug for cancer therapy.
Keywords: mixed micelles, polymer–phospholipid conjugates, vitamin E TPGS, berberine hydrochloride, apoptosis, prostatic adenocarcinoma
Introduction
Berberine (Brb, Figure 1) is a common isoquinoline quaternary alkaloid (also known as Natural Yellow 18) isolated from a variety of medicinal plants, including the Berberidaceae, Ranunculaceae, and Rutaceae families, many of which are used in traditional medicines.1–3 This biologically important alkaloid skeleton of Brb has drawn extensive attention owing to its diverse pharmacological effects, including anti-inflammatory, antimicrobial, antipyretic, and antihyperlipidemic activities.1–5 So far, Brb has been widely investigated as a potential therapeutic agent in a broad spectrum of clinical applications, such as hyperlipidemia, diabetes, metabolic syndrome, obesity, and mycotic infections.6–9 Furthermore, in recent years, many accumulated preclinical reports have extensively established potent antitumor activities of Brb, namely, inhibition of proliferation, induction of apoptosis, arrest of angiogenesis, and suppression of metastasis.1,6,9,10 The significant impact of Brb on tumor progression and metastasis has been primarily linked to inhibition of NF-κB, MMP-1, -2, and -9, activation of AMP-activated protein kinase signaling, and reduction of ERK and COX-2 activities.2,11 Inhibition of cancer cell division and arrest of cell cycle at the G0/G1 or G2/M phases have been attributed to direct interaction of Brb with several molecular targets such as DNA, along with telomerase, topoisomerase I/II, p53, and COX-2 proteins.2,9 Evident proapoptotic activity of Brb, in various cancer cell lines, has been mostly mediated via direct mitochondrial depolarization, inducing cytosolic cytochrome C release and reactive oxygen species generation, plus modulation of Bcl-2 and Bcl-xL expression, activation of caspases, as well as induction of PARP-1 cleavage.2,3,10 In a dose-dependent manner, Brb has shown induction of autophagy and apoptosis, as nonmutually exclusive events, signaling cell death activation.3,9 A canonical autophagic cell death is likely driven by Brb through inhibition of mTOR signaling pathway, mediated by both MAPK activation and AKt inhibition.2,3,9 Interestingly, the hypoglycemic and antihyperlipidimic effects of Brb appear to be interconnected with adipocyte involvement in breast cancer tumorigenesis and tumor microenvironment.3,5 By inhibiting the adipogenesis-positive regulator, PPARγ, and upregulating PPARα, Brb has been shown to suppress adipogenesis, potentially limiting cancer cell invasion, and decreasing metastatic breast cancer risk.2,9
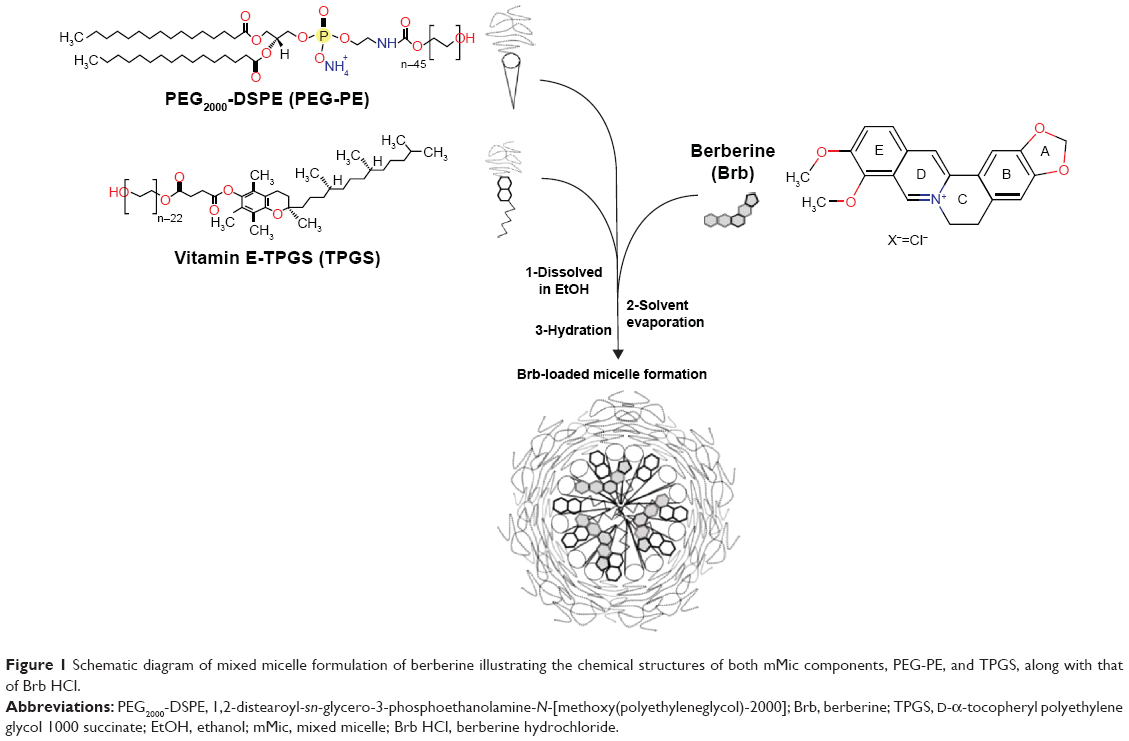 | Figure 1 Schematic diagram of mixed micelle formulation of berberine illustrating the chemical structures of both mMic components, PEG-PE, and TPGS, along with that of Brb HCl. |
The widespread occurrence of Brb in various common plant species, combined with its low toxicity, further promotes its clinical prospects to become an effective antitumor drug in the foreseeable future.3,9 However, further medical applications of Brb have encountered a few obstacles in pharmaceutical development.1,12 Preclinical studies have shown that Brb has a very limited oral bioavailability (BA) (<5% in plasma), largely due to its poor aqueous solubility,13 combined with low gastrointestinal absorption, and rapid metabolism.1,12 Being an excellent substrate for P-glycoprotein (Pgp) efflux pumps, intestinal Pgp transporters would account for almost 50%–90% of Brb excretion back into luminal side.1,6,12,14,15 Furthermore, it has been well reported that the extremely poor absolute oral BA of Brb noted in rats (0.36%–0.68%) can be additionally attributed to high hepatic extraction and rather dominant distribution of Brb in the liver.1,6,8,16–18 The systemic administration difficulty with such extensive first-pass metabolism is even more complicated because of the fact that Brb is a very slightly soluble compound, and thus would be classified as a class IV drug.1,12,16–18 Although intravenous (IV) administration of Brb (5 mg/kg) would certainly avoid the intestinal elimination barrier, the main clinical issue of its very short plasma half-life (0.5 hours ≤ t1/2<1 hours) remains.4,16,19 Parenteral Brb has also been associated with muscle protein atrophy in mice by stimulating breakdown and inhibiting synthesis.4,20,21 Other adverse reactions of Brb include gastrointestinal side effects and serious drug interactions, which can be attributed to its high plasma protein binding, hepatic metabolism inhibition, and systemic distribution throughout the body.2–4,9,12,18,21
Recently, several nanoparticulate delivery systems for Brb have been reported, attempting to address the major pharmaceutical concerns associated with its systemic administration, with lipid-based nanocarriers being the most investigated.1,6–8,12,16 Currently, lipopolymeric micelles are popular pharmaceutical delivery vehicles for poorly water-soluble active compounds, such as Brb, which can be solubilized within the hydrophobic core of a micelle. Micelles prepared from conjugates of polyethylene glycol (PEG) and diacyllipids, such as phosphatidylethanolamine (PE) – ie, PEG–PE conjugates – have demonstrated several pharmaceutical advantages such as small size (10–100 nm), good solubilization efficacy, low toxicity, controlled drug release, and extremely high stability due to very low critical micelles concentration (CMC) (typically in the micromolar range).22–24 In vivo, owing to their extended plasma circulation half-life (typically >36 hours in mice), the small size of PEG-PE micelles allows also for their effective accumulation in tumorous tissues with leaky vasculature, via the enhanced permeability and retention effect.23–25
Here, mixed micelle (mMic) formulation of Brb is investigated in order to enhance the solubilization efficacy of PEG-PE micellar carriers, by including D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) as an additional component – a PEGylated derivative of natural Vitamin E (VE), (α-tocopherol) commonly utilized as a pharmaceutical solubilizer, absorption enhancer, and a vehicle for lipidic drug formulations (Figure 1).25–29 Such a mixed micellar system would not only improve Brb solubilization capacity, owing to increased core volume of mMics, created by the planar chromane moiety of VE, but would also provide better protection for the active isoquinolone structure of Brb (Figure 1), effectively enhancing both Brb encapsulation and stability.30 Based on recent reports, TPGS has shown notable proapoptotic activity as well as a strong ability to overcome drug resistance by inhibiting Pgp efflux pumps.31,32 In fact, TPGS-incorporating mMic systems have been successfully used to sensitize multidrug-resistant tumors to chemotherapy drugs, including doxorubicin, paclitaxel, gemcitabine, and vinblastine.22,26,33,34
The main objectives of our study are to develop mMics made of TPGS and PEG-PE as efficient Brb nanocarriers, allowing for superior drug encapsulation and delivery. Consequently, the potential enhancement in anticancer efficacy of such combined Brb-micelle formulation is also investigated.
Materials and methods
Materials and cell lines
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (mPEG2000-DSPE, or PEG-PE) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA) and used without further purification. Berberine hydrochloride (Brb HCl) was purchased from Sigma-Aldrich (St Louis, MO, USA). Seziol® TPGS-Pharma (NF-grade Vitamin E polyethyleneglycol succinate, TPGS) was received as a gift from Cognis (Cincinnati, OH, USA). All other reagents and components of buffer solutions were analytical grade preparations. Trypsin/ethylenediamine tetraacetic acid, penicillin/streptomycin, and fetal bovine serum (FBS) were obtained from Thermo Fisher Scientific (Waltham, MA, USA), while CellTiter Fluor® cell viability assay kit and Apo-One™ homogenous caspase 3/7 assay kit were purchased from Promega Corporation (Fitchburg, WI, USA). Both eight-chamber polystyrene tissue culture-treated glass slides and Black 96-well tissue culture plates were purchased from BD Biosciences (San Jose, CA, USA). PC3, LNCaP, and Caco2 cells were purchased from American Type Culture Collection (Manassas, VA, USA). Milli-Q (MQ) water was utilized for all preparations. Since none of the studies involved people, patient medical records or tissues, the work included in this study did not require any approvals from Midwestern’s University Institutional Review Board.
Methods
Preparation of Brb-loaded PEG-PE/TPGS-mMics
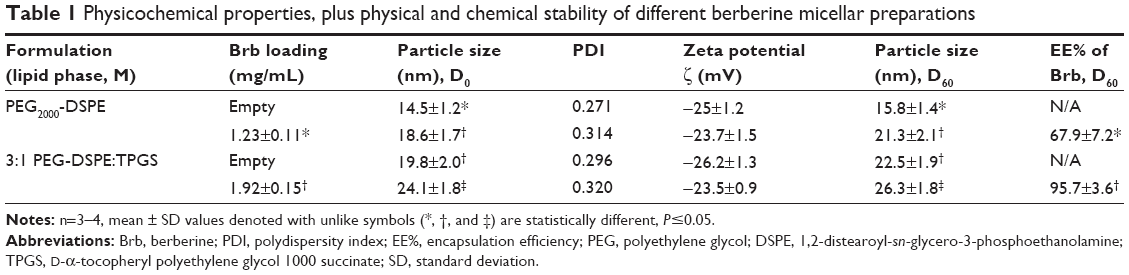
A fixed molar ratio (1:3) mixture of vitamin E TPGS and PEG2000-DSPE were stirred with Brb HCl previously dissolved in warmed ethanol (200 proof) at 2 mg/mL concentration, followed by solvent evaporation; then, HEPES buffered saline (HBS) solution, pH 7.4, was added to the formed lipid film, leading to formation of a clear Mic solution after 10 minutes of vortexing at 800 rpm. The resulting Mic dispersions were filtered twice with a 0.22-μm pore size polycarbonate membrane filter to initially remove any nonincorporated Brb and to sterilize Mic formulations, and then the filtrates were stored in vials under argon until further use.27,35 Alternatively, filtered Mic samples can be freeze-dried by storing aliquots of the filtrates in 5 mL glass vials, following freezing in liquid nitrogen and vacuum-drying (VirTis 2KP Lyophilyzer, SP Scientific, Stone Ridge, NY, USA, P<128×10−3 mbar, condenser temperature ≤−50°C). Following lyophilization, samples were sealed under argon and stored at 4°C until use. Table 1 lists some of the properties of the produced Mic formulations.22,34
 | Table 1 Physicochemical properties, plus physical and chemical stability of different berberine micellar preparations |
Physicochemical characterization of micelles
Particle size analysis
The Mic formulations were diluted with deionized distilled water before analysis, and the numbered average particle hydrodynamic diameter and the polydispersity index (PDI) were determined using dynamic light scattering technique, using a Malvern Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK).27,35,36
Zeta potential (ζ) measurements
All formulation samples were diluted with deionized distilled water, pH 6.8, and placed in the electrophoretic cell of the Malvern Zetasizer Nano-ZS (Nano-ZS, Malvern Instruments), and the average surface charge was determined.27,35,36
Encapsulation efficiency determination
The amount of Mic-encapsulated drug, Brb, was determined using a modified reversed-phase high-performance liquid chromatography (HPLC) method (immediately and then after 60 days of cold storage at 4°C−8°C, D60). Briefly, clear aqueous Mic dispersion was dissolved in methanol, and then after dilution in the mobile phase, a sample volume of 20 μL was injected through ACE PFP C-18 column (4.6×250 mm, 4 μm packing vol). The mixture of acetonitrile and 0.05 mol/L NaH2PO4 (final pH 2.5, adjusted by phosphoric acid), in the ratio of 30:70 v/v, was utilized as the mobile phase, after filtration and degassing. The flow rate was 1.0 mL/min, and UV detection was performed at λmax =346 nm, while the column temperature was maintained at 27°C±1°C. The retention time of Brb was ~7 minutes, and the encapsulation efficiency (EE%) of Brb in the Mic samples was calculated based on standard curves constructed in the range of 4–200 μg/mL (r2=0.998, n=3), which were validated for linearity, precision, and accuracy.7,8,16
Physical stability of Brb-micelles
The Brb-loaded PEG-PE micelles, TPGS-mixed or pure, were monitored over 2 months of storage in refrigerated conditions (4°C–8°C) for time-dependent changes in the physical characteristics (drug precipitation, change in micelle size, and surface charge post-60 days, D60) of the formulations. The chemical stability of Brb in micelles was evaluated by HPLC as described earlier.37
CMC determination
The CMC of both PEG-PE micelle types, TPGS-mixed or pure, was estimated via the pyrene method, as reported earlier.22 Briefly, exactly 10−4–10−7 M of micellar solution of PEG2000-DSPE, TPGS, and 1:3 molar mix of TPGS/PEG2000-DSPE in HBS were added to tubes containing 1 mg of pyrene crystals. The mixtures were incubated at room temperature (RT), for 24 hours, with continuous shaking at 200 rpm. Free pyrene was removed by filtration through 0.22 μm pore size polycarbonate membrane filters, and the fluorescence of filtrated samples (λ excitation/emission: 390 nm/505 nm) was measured using Synergy 2 Biotek fluorescence plate reader (BioTek Instruments Co., Winooski, VT, USA). CMC values corresponding to the concentration of the polymer at which a sharp increase in fluorescence is observed.22,28
In vitro release of Brb from micelles at “sink” conditions and modeled physiological media
To study the time-dependent release of Brb – from both mixed PEG–PE/TPGS and “pure” PEG-PE micelles – the dialysis bag method was employed. Drug-loaded micelles (0.3 mL of ~2 mM of Brb) were placed in Spectra/Por® dialysis cellulose ester membranes (MWCO =25 or 100 kDa, Spectrum Laboratories Inc., Rancho Dominguez, CA, USA), after soaking overnight in respective release media. Closed dialysis bags were submerged in 200 mL of test external release media, for up to 48 hours of incubation with continuous stirring at 200 rpm speed, as follows: phosphate-buffered saline solution (PBS, pH 7.4), while maintaining pseudo-sink conditions in a limited volume; fasting state simulated gastric fluid (SGF), pH 1.6; fasting state simulated intestinal fluid (SIF) with 5 mM of bile salts, pH 6.5; and finally FBS albumin (pH 7.4), to test the stability in blood serum. At specific time intervals, 0.5 mL samples of each release medium were withdrawn and replaced with an equal volume of fresh medium, and then samples were filtered through 0.22 μm syringe membrane filters. The Brb content in samples was finally determined by HPLC, as described earlier.22,27,35,37
Simulation of clinical in vivo profile of Brb-mMic formulation
To study the applicability of the Brb-mMic formulation, both its oral and intravenous administration were simulated (A4S Simulator version 2.0, Accelera S.r.l., Nerviano, Italy) using an open, single-compartment model.38,39 In case of oral administration scenario, a single human dose of 300 mg was considered for free Brb solution, given as bolus ingestion (first-order absorption and elimination, Ka =0.81 h−1, Ke =0.24 h−1, respectively) with a volume of distribution (Vd) of 66.5 L and plasma half-life (t1/2α) of 0.87 hours.40 For comparison purposes, the simulation of oral doses of 300 and 100 mg for Brb-mMic were also fit using the same equation of compartmental analysis, considering the same exact oral PK model parameters, except only for an improved oral BA, BA (F=0.05 in case of free Brb, while F=0.15 for Brb-mMic). Similarly, an IV infusion administration scenario was simulated using an open, two-compartment model with extensive distribution, where a single canine dose of 100 mg was considered for free Brb solution, given as bolus IV infusion (zero-order absorption and first-order elimination, Ke =4.62 h−1, and a volume of distribution [Vd] of 699.54 L).4,8,17 For comparison purposes, the simulation of IV bolus doses of 100 and 25 mg for Brb-mMic were also fit using the same equation of compartmental analysis, considering the same exact IV bolus model PK parameters (K12=0.055 h−1, K21=0.9 h−1, except t1/2=6 hours for Brb-mMic in comparison to t1/2=0.15 hours for free Brb).4,8,17,19,39
In vitro hemolysis assay
Hemolysis studies were carried out for empty mMic, along with different Brb-containing samples, according to previous report.30 The release of hemoglobin from the erythrocytes was used for toxicity measurements of these carriers. Briefly, defibrinated rabbit blood (Thermo Fisher Scientific) was diluted ten times with PBS and centrifuged at 2,000 rpm for 15 minutes. The supernatant was decanted, and the precipitate was rinsed three times with PBS, followed by centrifugation at 2,000 rpm for 15 minutes. The concentration of the resulting blood cells was adjusted to 2% (v/v). At a physiologically relevant dilution scenario of IV bolus administration (10 μg/mL of Brb-equivalent concentration), 50 μL of test samples were mixed with 500 μL of blood cells, and the resulting suspensions were incubated at 37°C for 3 hours. The samples were then centrifuged at 2,000 rpm for 15 minutes. The absorbance of the supernatant was measured at 540 nm to determine the amount of hemoglobin release. Zero hemolysis and 100% hemolysis consisted of red blood cells suspended in physiological saline (−ve ctrl) and distilled water (+ve ctrl), respectively.
The percentage of hemolysis was determined using the following equation:
|
|
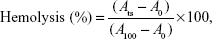
where Ats is the absorbance of the test sample, A100 is the absorbance of completely lysed red blood cells in distilled water, and A0 is the absorbance of zero hemolysis.30
In vitro cell culture and formation of spheroids
PC3, LNCaP, and Caco2 cells were grown in ATCC-formulated F-12K, RPMI-1640, and Eagle’s Minimum Essential Medium culture media, respectively, containing 10%–20% FBS and penicillin (100 U/mL)/streptomycin (100 μg/mL) in a humidified environment of 37°C, 5% CO2. Spheroids of 400–500 μm diameter were formed from 20,000 cells in 96-well microplates, as described earlier, where Dulbecco’s Modified Eagle’s Medium/F12 with 1.5% agarose was used – instead of poly (2-hydroxyethyl) methacrylate – to prevent cell adhesion, followed by microplate centrifugation for 20 minutes at 900 rpm. Spheroid formation was monitored using a Nikon Eclipse 800 epifluorescent upright microscope, equipped with Kodak RT slider camera (Nikon Corporation, Tokyo, Japan) at 10× magnification.41,42
Accumulation and Pgp inhibition in Caco2 cells
The intracellular accumulation and Pgp efflux inhibition were studied using Caco2 cells monolayers, grown in 24-well tissue culture plates, as previously described.22 When the cells reached 70% confluence, they were washed twice with Hank’s balanced buffer (HBSS), and then exposed to 20 μM of Brb, either encapsulated in our PEG-PE/TPGS-mMic or freely dissolved in HBS (alone or simply admixed with empty mMic), with or without 100 μM of verapamil hydrochloride. After 2 hours of incubation at 37°C, 5% CO2, cells were washed twice with HBSS, pH 7.4, and further incubated with or without 100 μM of verapamil hydrochloride for 1 hour, at 37°C, 5% CO2. Following this, cells were washed twice with sterile PBS, then lysed with 100 μL of dimethyl sulfoxide (DMSO), and finally diluted to 1 mL with PBS. The fluorescence of Brb-samples was measured (λ excitation/emission: 465 nm/520 nm) using Synergy 2 Biotek fluorescence plate reader (Biotek instruments Co.).22,27
Microscopic and spectroscopic evaluation of cellular uptake
The enhanced tumor-cell association of Brb-loaded mMic was demonstrated using fluorescence microscopy.22,27 After an initial passage in tissue culture flasks, ~2×105 of PC3 cells/well were grown in in an eight-chamber tissue culture glass slide system (BD Biosciences). After reaching 70% confluence, the plates were washed with HBSS, pH 7.4, and then treated with complete F12K medium (300 μL/well) and incubated for 2 hours at 37°C, 5% CO2. Different fluorescent Brb formulations – free/unencapsulated drug and Brb-loaded mMic – were added and incubated for 4 hours at 37°C, in 5% CO2. After incubation, cells were washed with HBSS, pH 7.4, and then immediately fixed and mounted on glass slides using the fluoromount G-DAPI mounting medium. Finally, slides were viewed with a Nikon Eclipse 800 epifluorescent upright microscope equipped with Kodak RT slider camera (Nikon Corporation), under bright light or epifluorescence with fluorescein/FITC filter (λ excitation/emission: 460 nm/510 nm) and DAPI filter (excitation/emission: 330 nm/435 nm) at 40× magnification. Composite FL-images were created by overlapping the images from the individual FL-channels.35,36
Quantitatively, semiconfluent LNCaP cells, grown in 12-well tissue culture plates, were exposed to 20 μM of Brb, either encapsulated in our PEG-PE/TPGS-mMic or freely dissolved in HBS (alone or simply admixed with empty mMic), for 4 hours at 37°C, in 5% CO2. After incubation, cells were washed twice with sterile HBSS, with centrifugation for 5 minutes at 900 rpm and lysed with 100 μL of DMSO and then diluted to 1 mL with HBSS. The fluorescence of Brb-samples was measured (λ excitation/emission: 465 nm/520 nm) using Synergy 2 Biotek fluorescence plate reader (BioTek Instruments Co.).22,27,35,36
Cytotoxicity assays
PC3 and LNCaP cells were seeded at 10×103 cells/well in 96-well microplates in five replicates for 24 hours. Then, respective complete culture media were exchanged for serum-free media containing several twofold serial dilutions of unencapsulated Brb as positive drug control treatments (either alone or simply admixed with empty mMic), and drug-equivalent concentrations of Brb-loaded mMic formulation plus blank mMic vehicle control, all compared with plain serum-free media, serving as negative control. Following 48 hours of coincubation at 37°C in 5% CO2, the total cell viability was determined using CellTiter Fluor® cell viability assay Kit (Promega Corporation), after washing twice with HBSS and reading sample plate fluorescence (λ excitation/emission: 390 nm/505 nm), using Synergy 2 Biotek fluorescence plate reader (BioTek Instruments Co.), according to manufacturer’s instructions.22,27,29,36
Apoptosis assays
Approximately 1.5×106 cells – used from each of PC3 or LNCaP human prostate carcinomas – were grown overnight to attach into 25 cm2 flasks. Cells were incubated for 24 hours with Brb-loaded mMic and blank mMic vehicle control, along with unencapsulated drug controls (either free Brb solution or simple mix of: Brb solution + empty mMic), at concentration equivalent to 20 μM of drug. In addition, CCCP was added to induce apoptosis as positive control for the same period and incubation conditions. Cells were then washed, trypsinized, collected, and normalized by their protein content, determined as per standard BCA protein assay kit protocol (Thermo Fisher Scientific). Afterward, 100 μL of the Apo-ONE™ Homogenous caspase 3/7 assay (Promega Corporation) substrate solution was added to each well containing 100 μL of sample media, and after 30 seconds of mixing, the plate contents were incubated for 3 hours. Finally, the sample plate fluorescence values were measured (λ excitation/emission: 480 nm/530 nm) using Synergy 2 Biotek fluorescence plate reader (BioTek Instruments Co.), according to manufacturer’s instructions. The caspase-3/7 activity was reported as percent (%) activation relative to untreated control.29,36
Data analysis
A minimum of triplicates were run for each experiment, unless indicated differently. Data were reported as mean ± standard error (SE), unless noted otherwise. Comparisons between two groups were made using Student’s t-test, and for more than two groups, Kruskal–Wallis test with Tukey’s post hoc analysis was used to compare results. P<0.05 was considered statistically significant. All statistical analyses were performed using Sigma Plot® software, version 11.0 (Systat Software Inc., San Jose, CA, USA).22,27,28,36
Results and discussion
With its great potential in cancer therapy, exhibited via marked blockade of cell progression, induction of autophagy and apoptosis, plus antiangiogenic and metastasis suppression activities, Brb not only augments the efficacy of antineoplastic chemotherapy and radiotherapy, but it also unveils substantial prospects for cancer treatment, in a variety of solid and hematological tumors. Hence, TPGS-containing mMics were investigated as Brb delivery vehicles to improve the systemic administration and BA of this drug and also to enhance its specific antiproliferative and proapoptotic efficacies against tumor cells.
Brb-micelle incorporation and physicochemical stability
Earlier, we first reported mMic-based preparation of camptocethin in PEG-PE/D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) micelles.27 Here, for optimal encapsulation of our Brb HCl drug, a new combination of the micelle-forming components was adopted to develop stable nanosized mMic from the mixture of PEG-PE and vitamin E-PEG ester, TPGS (Figure 1). It is well established that within micelles in aqueous systems, nonpolar molecules will be solubilized within the hydrophobic core, while polar molecules will be adsorbed on the micelle surface. Substances with intermediate polarity – such as our active alkaloid, Brb HCl – are typically distributed mostly surrounding the core and along the surfactant molecules in certain intermediate positions within the micelle’s corona.27,28,32 The addition of the PEGylated vitamin E, TPGS, was thus intended to enhance the Brb drug loading due to the hydrophobic fragment in its molecules. This should increase both the core and corona volumes and the overall micellar capacity (Figure 1). Hence, its ability to encapsulate and solubilize Brb molecules within such mMic structure would proportionally improve, especially in comparison to smaller and more packed micelles made solely of PEG conjugates of saturated DSPE phospholipids.22,26,27,32,36
At 15 mM concentration of mMic components (PEG2000-DSPE: VE TPGS molar ratio 3:1) and with ~5 wt% of Brb HCl added to the mixture, the mMic formulations were produced, containing 1.92±0.15 mg of Brb per 1 mL of micelle formulation (Table 1). Therefore, this mMic system attained ~300% increase in Brb solubilization, in comparison to free drug in aqueous solution. Indeed, the Brb-loading capacity of such an mMic platform (~4.3% w/w) also represents >150% increase in drug incorporation capacity of single-component micelles (almost 2.7% w/w) – made solely of PEG-PE, similar to earlier mMic reports.22,27,34 Furthermore, the dilution of the micelle sample influenced only the amplitude of the HPLC peak, without affecting its position or shape, confirming the high stability of the mMic. At the quantities used, Brb loading did not substantially influence the uniform mMic small size (Figure 2), indicating that most of the drug molecules were located in or close to nonpolar core space of the micelles, showing minimal impact on the whole particle size (z-average =24.1±1.8 nm) and charge (average ζ-potential =−23.5±0.9 mV) (Table 1) (Figure 2). Both drug-free and Brb-loaded mMic formulations did not show any marked changes in their mean particle size over 2 months at either RT or at 4°C (Table 1), which suggests the high stability of Brb-loaded PEG-PE/TPGS micelles.22,27
 | Figure 2 Representative analysis of berberine-loaded mixed micelles. |
Simulated in vitro release, PKs, Pgp inhibition in colonic cells, and hemocompatibility
The markedly higher and faster in vitro release profiles of Brb from PEG-PE-only micelles (PEGMic) provide further evidence relative to Brb-loaded mMic (Figure 3). It appears that our mixed PEG-PE/TPGS micelle system (mMic) retains the drug even better than “pure” PEG-PE micelles, indicating that the presence of TPGS moieties facilitates not only improved Brb incorporation into micelles but also the stability of its entrapment, within more spacious micellar carriers. This can be explained by possible additional hydrophobic interactions of Brb alkaloid molecules with the aromatic chromane structure in nonpolar region of TPGS.22,27,28,32 The Brb release from our PEG-PE/TPGS micelles, presented in Figure 3, generally shows an initial burst of ~20% of drug within the first 4 hours, followed by a slow-release phase over the remaining 24- or 48-hour-periods, irrespective of the pH or composition of the test release media. This overall in vitro release behavior of Brb from mMic reflects the drug incorporation stability and can be explained through the localization of Brb within the micelles. The initial burst that happened within the first few hours of incubation can be attributed to the relatively small quantity of the drug associated within the mMic corona, closer to the micelle–water interface, which can proceed via both the hydration of the interfacial drug molecules and their passive diffusion.22,27,36 The drug incorporated at the inner core compartment remains firmly inside the mMic, showing the slowest release profile, even at aqueous buffered conditions with low pH values.22,36 The incorporated Brb remains mostly associated with the micelles, even after 48 hours of incubation at 37°C (Figure 3A). On the other hand, the amount of Brb released from both micelle types (PEGMic and mMic) was somewhat high (61% and 48%, respectively), after only 24 hours in the presence of blood serum (FBS, pH 7.4) as a release medium (Figure 3B). This relatively faster in vitro release behavior illustrated the potential impact of plasma proteins on Brb-loaded micelles (almost equally on PEGMic and mMic), along with predicted superior stability of the mMic nanocarrier, following systemic IV administration (after 12 hours, ~38% of Brb was released from PEGMic vs 23% from mMic). As to the oral route, the SGF- and SIF-mediated release profiles (Figure 3, panels C and D) appeared quite similar to each other (Brb-loaded PEGMic released drug much faster than Brb-loaded mMic, over the 24-hour test period), with fairly greater Brb release from micellar vehicles in the presence of bile salts and duodenal pH value 6.5, as expected.22
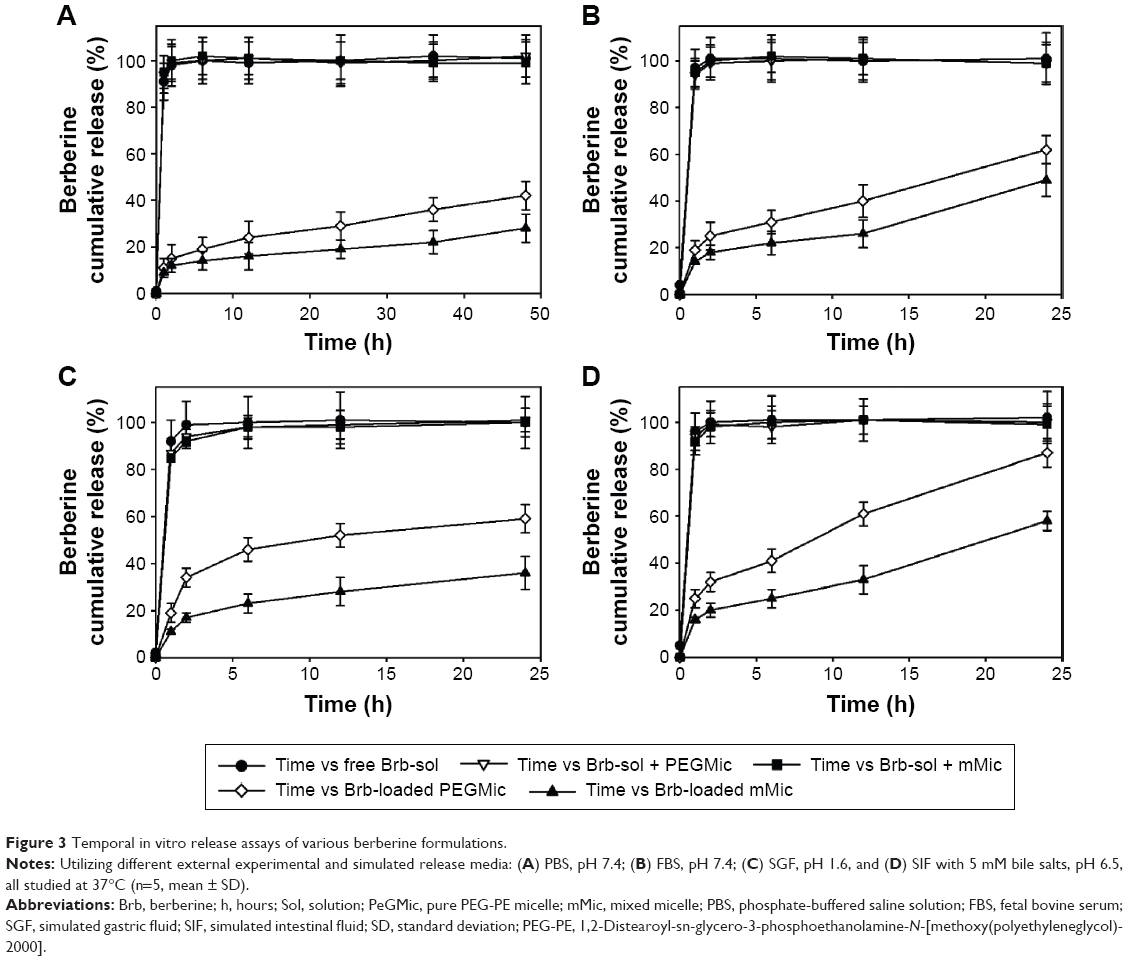 | Figure 3 Temporal in vitro release assays of various berberine formulations. |
The CMC value of any micelle-forming formulation influences both its in vitro and in vivo stabilities, and the low CMC values of PEG-PE underlie the high stability of PEG-PE micelles in solutions upon dilution.28,43 The determined CMC value of our prototype mMic carriers (composed of PEG2000-DSPE and TPGS in 3:1 molar ratio) was also found to be low, ~2.2×10−5 M. (The CMC for the Vitamin E-free plain PEG2000-DSPE is determined as 1.1×10−5 M.)22,27 Such low CMC values of our mMic system promise sufficient stability of and ability to maintain structural integrity upon strong dilution in body.26,32
Apart from the native, very poor aqueous solubility of Brb, another major concern is its extremely limited oral BA, because of strong intestinal Pgp counterefflux. Furthermore, Brb is rapidly cleared from blood circulation (plasma t1/2α =6–15 minutes, following IV administration), as a result of extensive liver metabolism combined with efficient renal clearance.4,12,14,17 Clinical PK simulation can help us realize the positive impact of mMic encapsulation of Brb on its plasma concentration–time curves (Figure 4), which can be better delineated based on the potential route of drug administration.38,39 Figure 4A represents the linear simulated profile of Brb-micelles following a single oral-dose administration (at either 100 or 300 mg/kg) in human subjects, which is compared to the only reported clinical PK profile of an oral dose of 300 mg/kg of free Brb solution (Brb-sol).40 The same PK model input parameters (single-compartment model with first-order absorption and elimination) were applied for our TPGS-mixed micellar carriers of Brb, except for calculated oral BA. Based on the well-established intestinal absorption enhancement effect of TPGS molecules present in our mMic system, ~3- to-5-fold improvement in their oral BA of Brb can be predicted because of the significant intestinal Pgp-impairment action of TPGS.22,31 Hence, a maximum plasma concentration (Cmax) of 1.2257 mg/kg can be projected for Brb-mMic (estimated oral BA =15%), compared to only Cmax =0.3977 mg/L obtained for the same 300 mg/kg dose of free Brb (reported oral BA =5%). Therefore, effective oral doses of Brb-mMic can be quite lower than those of free drug, as illustrated in Figure 4A, where an example of 100 mg/kg oral dose of Brb-Mic would be predicted to yield the same Cmax as that reported for three times larger dose of free Brb solution.
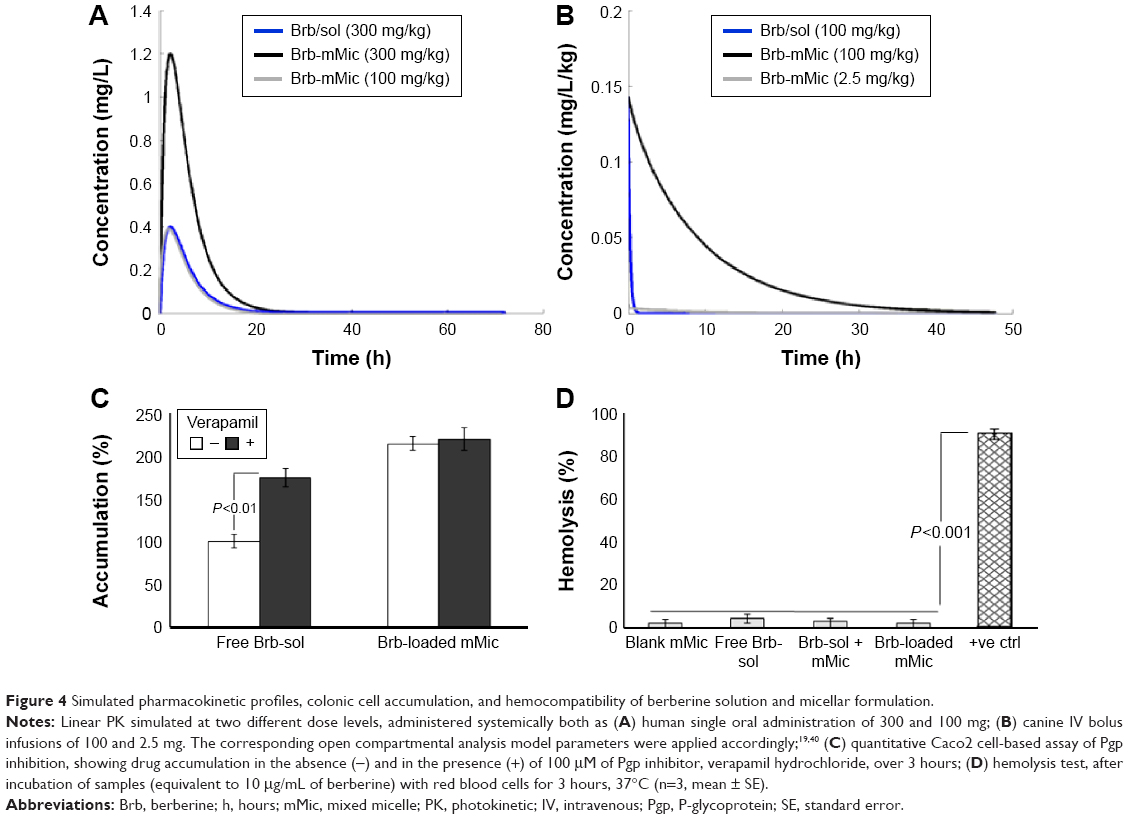 | Figure 4 Simulated pharmacokinetic profiles, colonic cell accumulation, and hemocompatibility of berberine solution and micellar formulation. |
In another similar simulation scenario utilizing clinical PK data of single IV bolus administration of free Brb solution, reported only in healthy canine model,4,19 absolute BA (100%) of the drug was then applied (Figure 4B). As demonstrated, the AUC0–∞ attained after a single 100 mg/kg bolus IV dose of Brb-sol was 1.979 mg/h/L, which displayed a very short plasma half-life, t1/2=0.15 hours. In contrast, simulated PK profile of Brb loaded in PEGylated mMic carriers, with an estimated midrange plasma t1/2=6 hours (reported in vivo t1/2 of 4–10 hours of similar micelle carriers),23,24,43 achieved a projected AUC0–∞=69.308 mg/h/L, ~37-fold higher compared to free drug injection. For comparison purposes, a simulated 40-fold lower IV dose of Brb-mMic (2.5 mg/kg) was projected to attain AUC0–∞ value of 1.7754 mg/h/L, very close to that of the originally high dose of free drug. This simply revealed one immense advantage of mMic for Brb, to potentially reduce the effective injected dose of the drug dramatically (>35-fold), while significantly increasing its plasma half-life and thus enhancing the clinical potential of Brb-mMic, owing to possibly less-frequent dosing regimen.1,3,23
In vitro verification of the strong potential of the mMic system for oral delivery of Brb (an excellent substrate of intestinal Pgp, which severely limits its oral BA) is shown in Figure 4C. Indicative of Pgp activity in cultured human Caco2 colorectal cells, a significantly higher accumulation of Brb was observed in cells treated with verapamil hydrochloride versus nontreated cells, evidently owing to the inhibition of Pgp efflux by verapamil. Nevertheless, when Caco2 cells were incubated with Brb encapsulated into PEG-PE/vitamin E TPGS-mMic, the treatment with verapamil did not influence the drug accumulation in model colonic cell culture. Similar to earlier reports of TPGS-containing micelles, this can be attributed to the strong inhibition of Pgp by TPGS, as well as the ability of Brb-mMic to bypass the drug efflux by Pgp.22,26,33,34 This additional property of TPGS-mMic essentially eliminates the need for coadministration of Pgp inhibitors, which would be very important for the development of such prototype micellar platform of Brb, especially for oral administration.
Furthermore, the biocompatibility of our proof-of-concept mMic formulation of Brb in the presence of red blood cells (RBCs) was examined by simple hemolysis assay. As shown in Figure 4D, free Brb in solution caused a marginal increase in hemolysis compared to empty mMic vehicle, as well as Brb encapsulated in mMic, when compared to the negative control. Clearly, the developed mMic platform, alone or encapsulating Brb, demonstrated excellent hemocompatibiltiy, which is a requisite for potential IV administration of the drug.
Uptake and cytotoxicity of Brb-mMic in prostate cancer cell and spheroid models, mediated via enhanced proapoptotic activity
The concentration-dependent cytotoxicity against two human cancer cell spheroid models, PC3 and LNPaC, was investigated (Figures 5 and 6, respectively) to evaluate the effects of mMic nanocarrier encapsulation of Brb on its anticancer pharmacological activity versus Brb controls. In all studied cell spheroids, Brb in PEG-PE/vitamin E TPGS-mMic demonstrated significantly superior concentration-dependent cytotoxicity profiles, compared to those of the free drug and nonencapsulated drug controls (Figures 5A and 6A), starting at Brb concentrations ≤0.5 μM. The cytotoxicity “cross-section” (Figures 5B and 6B), at Brb concentrations of 5 μM, reveals that Brb-mMic resulted in 2.5–3 times higher cancer cell kill than unencapsulated drug, while empty mMic vehicles showed marginal activity. For instance, after 48 hours, the Brb-mMic formulation killed >60% of PC3 cells, while the best Brb-containing treatments (free Brb solution) killed only ~20% of cancer cells. As the biological properties of Brb remain the same, such enhanced cytotoxic effect obtained with TPGS-mixed nanocarriers of Brb is evidently attributed to better drug solubilization inside micelles. At the same time, this marked increase in spheroid cell killing can be further explained by the known ability of PEG-PE-based micellar nanocarriers to penetrate into the cancer spheroid mass due to their small size, as reported in previous studies.41,42 The half-maximal inhibitory concentration values for various formulation toward PC3 cell spheroids were calculated as 87±11.3 μM for free Brb (extrapolated) and only 4.86±0.28 μM for Brb-loaded PEG-PE/TPGS-mMic (Figure 5A). In LNPaC spheroids, these values were 107±16.5 μM for free Brb (extrapolated) and only 6.4±0.63 μM for Brb-mMic (Figure 6A). There was virtually no difference in cytotoxicity between the two treatments containing free Brb solution and extemporaneous/simple mixture of Brb + mMic.
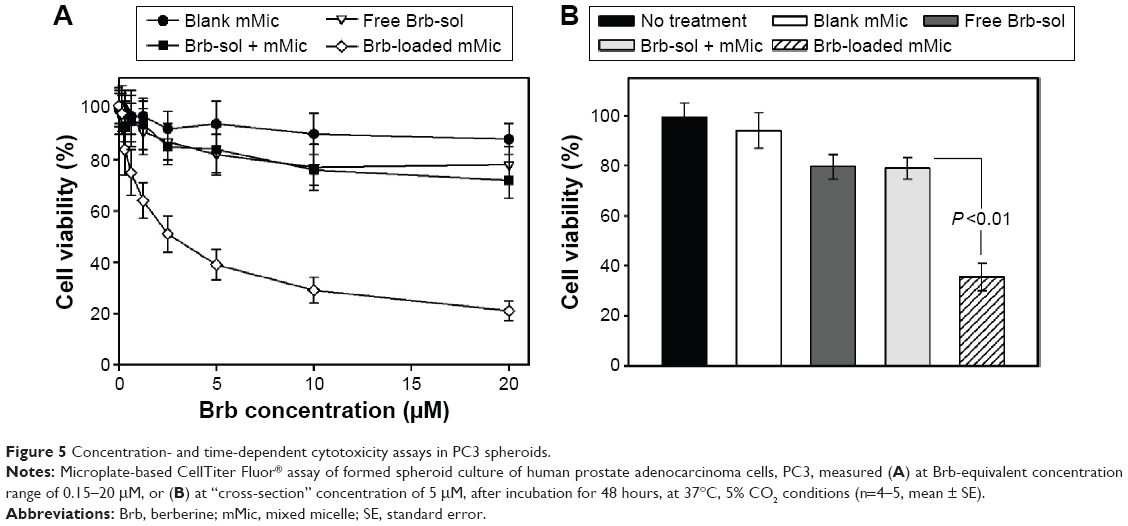 | Figure 5 Concentration- and time-dependent cytotoxicity assays in PC3 spheroids. |
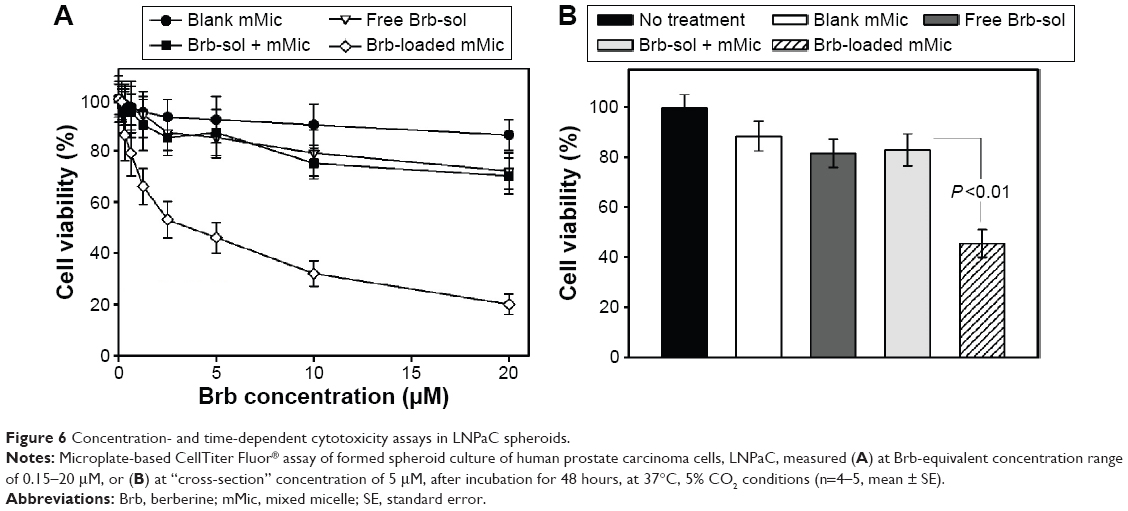 | Figure 6 Concentration- and time-dependent cytotoxicity assays in LNPaC spheroids. |
Moreover, fluorescent tracking of Brb formulation binding and uptake by human PC3 cells (Figure 7A–E) clearly demonstrated higher cell-associated fluorescence in case of Brb-loaded mMic treatment (Figure 7D), compared to unincorporated Brb controls (Figure 7B and C). As noted in earlier reports, such enhanced intracellular uptake of drug-containing micelles by the cells (quantitatively evident in case of Brb-mMic after 2–4 hours, Figure 7E) – mostly mediated by endocytosis – generally results from multiple drug molecules entering cells with each internalized micelle, in comparison to slow diffusion of single drug molecules.23,27,34,42 Collectively, such in vitro enhancements observed for nanocarrier-facilitated Brb solubilization, spheroid penetration, and cell uptake – in our mMic formulation – seem to have strongly supported a greater increase in the anticancer efficacy of the drug, up to 18-fold higher than equivalent quantities of the free Brb.
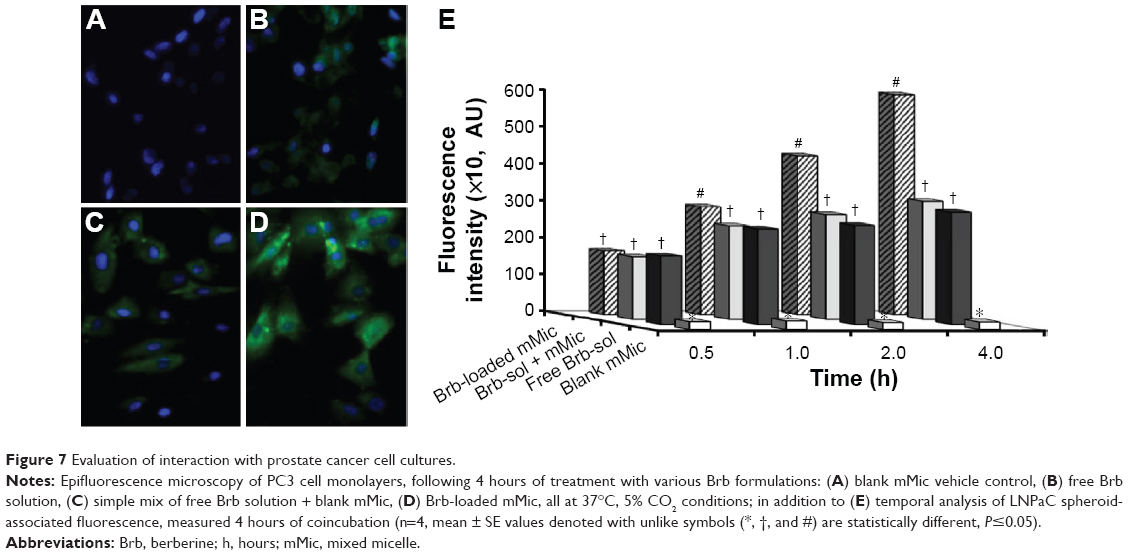 | Figure 7 Evaluation of interaction with prostate cancer cell cultures. |
Along with our cell-uptake analysis, the intracellular apoptosis induction markers were examined to better elucidate the multiple combined roles involved in mediating such distinctive anticancer efficacy of Brb, noted with its mMic encapsulation (Figure 8). Marked increase in the levels of major executioner caspases, caspase-3 and -7, in PC3 and LNPaC cells (illustrated in Figure 8A and B, respectively),29 directly implicate strong apoptosis induction with nanoencapsulated Brb-mMic as a major underlying antineoplastic mechanism.25,26,32 The efficient cellular delivery of Brb, via TPGS-mMics, seems to have triggered apoptotic death machinery in both cancer models, potentially with the involvement of mitochondrial depolarization, as emphasized previously.10,11,29,35 In fact, analogous to the apoptosis assay positive control, CCCP, the Brb-mMic treatment was also associated with maximum elevation of influential caspase proteins, caspase-3 and -7, which are commonly recognized as the chief effector caspases involved in driving the different pathways of apoptosis in tumors.
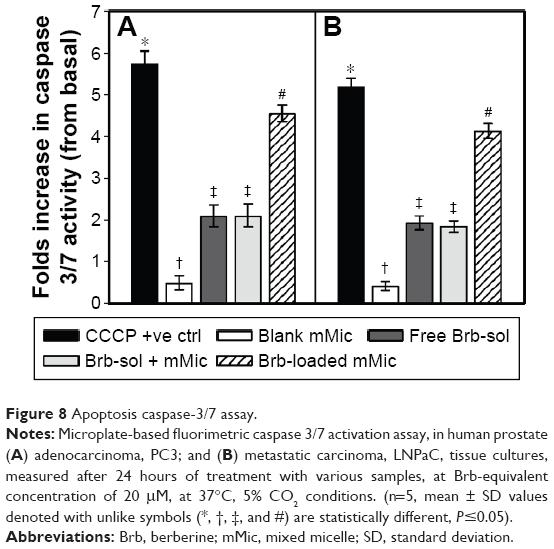 | Figure 8 Apoptosis caspase-3/7 assay. |
Conclusion
Despite significant solubility, PK, and pharmacodynamic limitations, Brb can not only augment the efficacy of antineoplastic chemotherapy and radiotherapy but also exhibits substantial antiproliferative and proapoptotic activities for cancer treatment in a variety of human tumors. To exploit the great clinical potential of Brb in cancer therapy, stealth micelles comprising a mixture of vitamin E TPGS and PEG2000-DSPE lipid conjugates were used for efficient solubilization of this poorly soluble active alkaloid to potentially improve systemic PK profiles of Brb.
Moreover, the current work clearly demonstrates that such an mMic platform resulted in remarkable enhancement of the proapoptotic action and overall anticancer efficacy of Brb, against various in vitro models of prostate cancers. Altogether, our data strongly suggest TPGS-mixed phospholipid micelles as an effective pharmaceutical system for Brb and warrants further development of these Brb nanocarriers for antitumor drug studies.
Acknowledgments
This work was supported by intramural faculty development fund of Midwestern University, College of Pharmacy-Glendale. The authors also thank Ms P Iacoban for her technical support.
Disclosure
The authors report no conflicts of interest in this work.
References
Tan W, Li Y, Chen M, Wang Y. Berberine hydrochloride: anticancer activity and nanoparticulate delivery system. Int J Nanomedicine. 2011;6:1773–1777. | ||
Jabbarzadeh Kaboli P, Rahmat A, Ismail P, Ling KH. Targets and mechanisms of berberine, a natural drug with potential to treat cancer with special focus on breast cancer. Eur J Pharmacol. 2014;740:584–595. | ||
Tillhon M, Guamán Ortiz LM, Lombardi P, Scovassi AI. Berberine: new perspectives for old remedies. Biochem Pharmacol. 2012;84:1260–1267. | ||
Ye M, Fu S, Pi R, He F. Neuropharmacological and pharmacokinetic properties of berberine: a review of recent research. J Pharm Pharmacol. 2009;61:831–837. | ||
Kong W, Wei J, Abidi P, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins.Nat Med. 2004;10:1344–1351. | ||
Pund S, Borade G, Rasve G. Improvement of anti-inflammatory and anti-angiogenic activity of berberine by novel rapid dissolving nanoemulsifying technique. Phytomedicine. 2014;21:307–314. | ||
Zhu JX, Tang D, Feng L, et al. Development of self-microemulsifying drug delivery system for oral bioavailability enhancement of berberine hydrochloride. Drug Dev Ind Pharm. 2013;39:499–506. | ||
Wang T, Wang N, Song H, et al. Preparation of an anhydrous reverse micelle delivery system to enhance oral bioavailability and anti-diabetic efficacy of berberine. Eur J Pharm Sci. 2011;44:127–135. | ||
Vuddanda PR, Chakraborty S, Singh S. Berberine: a potential phytochemical with multispectrum therapeutic activities. Expert Opin Investig Drugs. 2010;19:1297–1307. | ||
Patil JB, Kim J, Jayaprakasha GK. Berberine induces apoptosis in breast cancer cells (mcf-7) through mitochondrial-dependent pathway. Eur J Pharmacol. 2010;645:70–78. | ||
Ho YT, Lu CC, Yang JS, et al. Berberine induced apoptosis via promoting the expression of caspase-8, -9 and -3, apoptosis-inducing factor and endonuclease g in scc-4 human tongue squamous carcinoma cancer cells. Anticancer Res. 2009;29:4063–4070. | ||
Battu SK, Repka MA, Maddineni S, Chittiboyina AG, Avery MA, Majumdar S. Physicochemical characterization of berberine chloride: a perspective in the development of a solution dosage form for oral delivery. AAPS PharmSciTech. 2010;11:1466–1475. | ||
Lu Y-C, Lin Q, Luo G-S, Dai Y-Y. Solubility of berberine chloride in various solvents. J Chem Eng Data. 2006;51:642–644. | ||
Pan GY, Wang GJ, Liu XD, Fawcett JP, Xie YY. The involvement of P-glycoprotein in berberine absorption. Pharmacol Toxicol. 2002;91:193–197. | ||
Shitan N, Tanaka M, Terai K, Ueda K, Yazaki K. Human MDR1 and MRP1 recognize berberine as their transport substrate. Biosci Biotechnol Biochem. 2007;71:242–245. | ||
Xue M, Yang MX, Zhang W, et al. Characterization, pharmacokinetics, and hypoglycemic effect of berberine loaded solid lipid nanoparticles. Int J Nanomedicine. 2013;8:4677–4687. | ||
Tsai PL, Tsai TH. Hepatobiliary excretion of berberine. Drug Metab Dispos. 2004;32:405–412. | ||
Liu YT, Hao HP, Xie HG, et al. Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats. Drug Metab Dispos. 2010;38:1779–1784. | ||
Li BX, Yang BF, Hao XM, Zhou H, Sun MZ. Study on the pharmacokinetics of berberine in single dosage and coadministration with oryzanol in rabbits and healthy volunteers. Chin Pharm J. 2000;35:33–35. | ||
Wang H, Liu D, Cao P, Lecker S, Hu Z. Atrogin-1 affects muscle protein synthesis and degradation when energy metabolism is impaired by the antidiabetes drug berberine. Diabetes. 2010;59:1879–1889. | ||
Kheir MM, Wang Y, Hua L, et al. Acute toxicity of berberine and its correlation with the blood concentration in mice. Food Chem Toxicol. 2010;48:1105–1110. | ||
Dabholkar RD, Sawant RM, Mongayt DA, Devarajan PV, Torchilin VP. Polyethylene glycol-phosphatidylethanolamine conjugate (PEG-PE)-based mixed micelles: some properties, loading with paclitaxel, and modulation of p-glycoprotein-mediated efflux. Int J Pharm. 2006;315:148–157. | ||
Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 2007;24:1–16. | ||
Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv Drug Deliv Rev. 2004;56:1273–1289. | ||
Wang Y, Fan W, Dai X, et al. Enhanced tumor delivery of gemcitabine via PEG-DSPE/TPGS mixed micelles. Mol Pharm. 2014;11:1140–1150. | ||
Duhem N, Danhier F, Préat V. Vitamin E-based nanomedicines for anti-cancer drug delivery. J Control Release. 2014;182:33–44. | ||
Mu L, Elbayoumi TA, Torchilin VP. Mixed micelles made of poly(ethylene glycol)-phosphatidylethanolamine conjugate and D-α-tocopheryl polyethylene glycol 1000 succinate as pharmaceutical nanocarriers for camptothecin. Int J Pharm. 2005;306:142–149. | ||
Sawant RR, Sawant RM, Torchilin VP. Mixed PEG-PE/vitamin E tumor-targeted immunomicelles as carriers for poorly soluble anti-cancer drugs: improved drug solubilization and enhanced in vitro cytotoxicity. Eur J Pharm Biopharm. 2008;70:51–57. | ||
Brownlow B, Nagaraj VJ, Nayel A, Joshi M, Elbayoumi T. Development and in vitro evaluation of vitamin E-enriched nanoemulsion vehicles loaded with genistein for chemoprevention against UVB-induced skin damage. J Pharm Sci. 2015;104:3510–3523. | ||
Vuddanda PR, Rajamanickam VM, Yaspal M, Singh S. Investigations on agglomeration and haemocompatibility of vitamin E TPGS surface modified berberine chloride nanoparticles. Biomed Res Int. 2014;2014:951942. | ||
Dintaman JM, Silverman JA. Inhibition of P-glycoprotein by D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS). Pharm Res. 1999;16:1550–1556. | ||
Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33:4889–4906. | ||
Jin Y, Zhang Z, Zhao T, Liu X, Jian L. Mixed micelles of doxorubicin overcome multidrug resistance by inhibiting the expression of P-glycoprotein. J Biomed Nanotechnol. 2015;11:1330–1338. | ||
Abouzeid AH, Patel NR, Torchilin VP. Polyethylene glycol-phosphatidylethanolamine (PEG-PE)/vitamin E micelles for co-delivery of paclitaxel and curcumin to overcome multi-drug resistance in ovarian cancer. Int J Pharm. 2014;464:178–184. | ||
Pham J, Grundmann O, Elbayoumi T. Mitochondriotropic nanoemulsified genistein-loaded vehicles for cancer therapy. Methods Mol Biol. 2015;1265:85–101. | ||
Pham J, Brownlow B, Elbayoumi T. Mitochondria-specific pro-apoptotic activity of genistein lipidic nanocarriers. Mol Pharm. 2013;10:3789–3800. | ||
Pham J, Nayel A, Hoang C, Elbayoumi T. Enhanced effectiveness of tocotrienol-based nano-emulsified system for topical delivery against skin carcinomas. Drug Deliv. Epub July 14, 2015. | ||
Germani M, Del Bene F, Rocchetti M, Van Der Graaf PH. A4s: a user-friendly graphical tool for pharmacokinetic and pharmacodynamic (PK/PD) simulation. Comput Methods Programs Biomed. 2013;110:203–214. | ||
Araújo FA, Kelmann RG, Araujo BV, Finatto RB, Teixeira HF, Koester LS. Development and characterization of parenteral nanoemulsions containing thalidomide. Eur J Pharm Sci. 2011;42:238–245. | ||
Bao LH. The study of pharmacokinetics of berberine after oral administration in human. Chin Pharmacol Bull. 1997;13:95–96. | ||
Patel NR, Aryasomayajula B, Abouzeid AH, Torchilin VP. Cancer cell spheroids for screening of chemotherapeutics and drug-delivery systems. Ther Deliv. 2015;6:509–520. | ||
Perche F, Torchilin VP. Cancer cell spheroids as a model to evaluate chemotherapy protocols. Cancer Biol Ther. 2012;13:1205–1213. | ||
Sawant RR, Torchilin VP. Polymeric micelles: polyethylene glycol-phosphatidylethanolamine (PEG-PE)-based micelles as an example. Methods Mol Biol. 2010;624:131–149. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.