Back to Journals » Drug Design, Development and Therapy » Volume 19
Development and Characterization of a Cationic Polyurethane Carrier for Enhanced Topical DNA Delivery
Authors Borcan F ![]() , Len A, Vlase T, Vlase G, Dudas ZI, Popescu R
, Len A, Vlase T, Vlase G, Dudas ZI, Popescu R ![]() , Albulescu RC, Dehelean CA, Szuhanek CA
, Albulescu RC, Dehelean CA, Szuhanek CA
Received 20 December 2024
Accepted for publication 13 June 2025
Published 8 July 2025 Volume 2025:19 Pages 5781—5799
DOI https://doi.org/10.2147/DDDT.S510803
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Florin Borcan,1 Adel Len,2,3 Titus Vlase,4 Gabriela Vlase,4 Zoltan I Dudas,2 Roxana Popescu,5 Ramona C Albulescu,6 Cristina A Dehelean,1 Camelia A Szuhanek7
1Department I, Faculty of Pharmacy, “Victor Babes” University of Medicine and Pharmacy, Timisoara, 300041, Romania; 2Budapest Neutron Centre, Institute for Energy Security and Environmental Safety, HUN-REN Centre for Energy Research, Budapest, 1121, Hungary; 3Department of Civil Engineering, Faculty of Engineering and Information Technology, University of Pécs, Pécs, 7624, Hungary; 4Department of Chemistry (Research Center “Thermal Analysis in Environmental Problems”), Faculty of Chemistry, Biology, Geography, West University Timisoara, Timisoara, 300115, Romania; 5Department II (Cellular and Molecular Biology), Faculty of Medicine, “Victor Babes” University of Medicine and Pharmacy, Timisoara, 300041, Romania; 6Department XI (Paediatrics II), Faculty of Medicine, “Victor Babes” University of Medicine and Pharmacy, Timisoara, 300594, Romania; 7Department II (Research Center “Ortho-Center”), Faculty of Dental Medicine, “Victor Babes” University of Medicine and Pharmacy, Timisoara, 300041, Romania
Correspondence: Zoltan I Dudas, Budapest Neutron Centre, Institute for Energy Security and Environmental Safety, HUN-REN Centre for Energy Research, Budapest, 1121, Hungary, Tel +36-1-392-2222/1414, Email [email protected]
Background: Nucleic acids are increasingly being recognized for their potential as therapeutic agents for the treatment of a variety of pathologies, such as genetic diseases, viral infections, and cancer. However, the safe delivery of these negatively charged macromolecules to their intended sites of action remains a major challenge.
Purpose: This study aimed to design and characterize cationic particles for use as nonviral vectors for nucleic acid delivery; another primary objective was to evaluate the biocompatibility between the particles and DNA.
Methods: The particles were synthesized via a polyaddition process between isophorone diisocyanate and a mixture of polyethylene glycol and polycaprolactone diol. The structural characteristics were assayed using a variety of techniques, including measurements of pH, refractive index, and Zetasizer, drug release and penetration through an artificial membrane, SEM, FTIR and Raman spectroscopy, thermal analyses, cell viability, and in vivo evaluation of skin parameters.
Results: The findings revealed that nearly neutral-pH particles were successfully synthesized, displaying a broad size distribution ranging from 400– 900 nm, a prolonged release profile, and an encapsulation efficacy of 72.5%. Thermal analyses demonstrated that the samples remained stable at temperatures up to 200 °C, and the results of the spectroscopy, cell assay, and evaluations on mouse skin suggest that the obtained particles are safe for use as DNA carriers.
Conclusion: Cationic polyurethane carriers present a potential alternative to the more established polyethylenimine. However, additional studies are necessary to fully assess the therapeutic effectiveness of these formulations.
Keywords: cell cultures, FT-IR, mice skin, Raman, thermal analysis, Zetasizer
Introduction
Usually, nucleic acids cannot penetrate the cell membrane unless their entry is facilitated by different active cellular uptake mechanisms (pinocytosis, endocytosis, phagocytosis) or by the creation of transient holes by physical meaning.1 Thus, two major delivery approaches have been intensively investigated: viral vectors based on retrovirus, herpes virus, pox virus, adenovirus, lentivirus, human foamy virus, and non-viral vectors which include physical methods (microinjection, electroporation, laser, controlled short electric impulses, elevated temperature, and ultrasounds) and natural or synthetic compounds that promote the transfer of exogenous nucleic acids into target cells.2,3 Various nonviral delivery systems have been developed in the last four decades owing to their multiple advantages: they are often less immunogenic and toxic than their viral counterparts, the possibility to repeat the administration, and the ease pathway to obtain. The chemical vectors consist of inorganic particles based on gold, silica, calcium phosphate, supermagnetic iron oxide, carbon nanotubes, quantum dots, and cationic polymers (polyplexes), lipids (lipoplexes), cationic polymer/lipid (lipo-polyplexes).4
The use of nano- and micro-particles in transmembrane transport systems has gained significant importance in contemporary medical practice. These systems offer a promising approach for treating human diseases by enabling targeted delivery of various compounds while ensuring constant therapeutic doses. Polymeric materials are frequently used in this field of research because of their ease of synthesis at various sizes and degradation rates as well as their inert and biodegradable nature. They improve the pharmacokinetic and pharmacodynamic properties of the encapsulated compound, reduce immunogenicity, and increase its stability. All of these factors are crucial for improving the bioavailability of transported biologically active agents.
The delivery of nucleic acids is often crucial for the treatment of rare skin diseases, and the development of new and effective carriers is essential for genetic manipulation. Introduced by Behr in 1995 as a non-viral vector, branched polyethyleneimine (PEI) represents the gold standard in this field.5 Poly-L-(lysine) and its derivatives are among the earliest polymers used in non-viral delivery of genetic materials. Previous research has focused on the use of polymer particles for DNA delivery; however, the development of cationic polyurethane particles offers a new and potentially superior option. The acquisition and characterization of cationic polyurethanes based on N,N-diethylethylenediamine commenced at the dawn of the new millennium.6
Polyurethane (PU) is a synthetic material obtained by the reaction between di- or poly-isocyanates and mixtures of polyesters and/or polyethers (Figure 1). The urethane linkages (-NH-CO-O-), which also appear in carbamates, are specific to PU but are present in the macromolecular chains alongside different terminal groups or to allophanate and biuret groups obtained from side reactions. The variety of functional groups in a PU structure and the different aliphatic/aromatic character of the raw materials opens the possibility of designing new biomaterials with applications in different industries, medical, and pharmaceutical fields, as the final products exhibit a diverse range of hardness, high resistance, and tensile properties, as well as blood compatibility and biocompatibility. Despite the toxicity of isocyanates used as raw materials, PU safety for humans has been certified for a large variety of biomedical applications by the US Food and Drug Administration.7,8
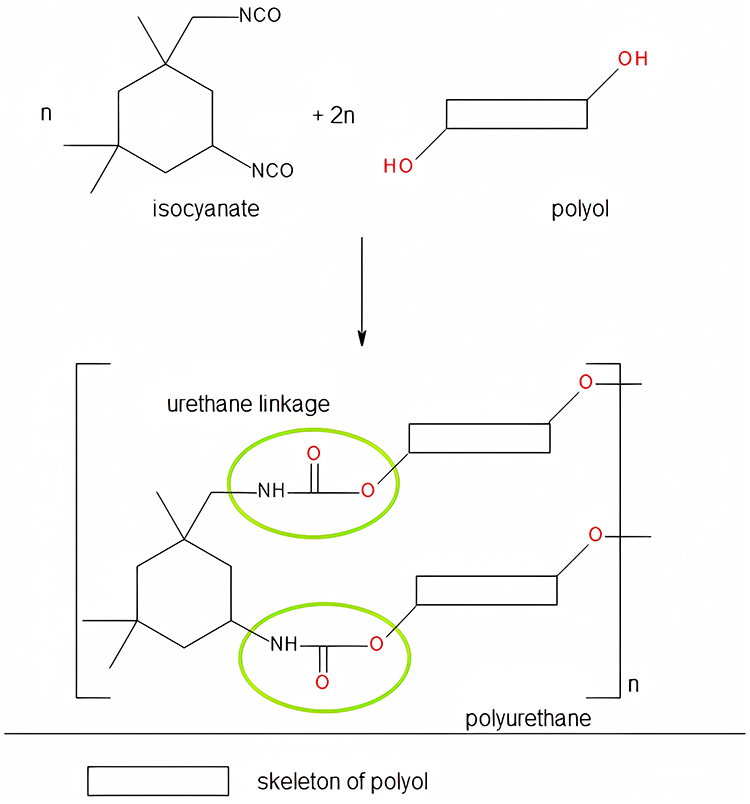 |
Figure 1 General scheme for the formation of PU chains. |
The earliest syntheses of PU delivery systems were based on aromatic compounds, such as methylene-diphenyl diisocyanate and toluene diisocyanate. These substances are routinely employed as raw materials for the fabrication of foams and elastomers. However, their propensity for causing cancer and the generation of particles with large sizes and low penetration rates presented significant shortcomings, hindering their viability for further use in clinical settings.9 A set of studies conducted by Bouchemal et al10–12 describes the creation of PU capsules that have considerably smaller sizes than previous models. To create the capsules, isophorone diisocyanate, polyethylene glycol, ethylene glycol, butanediol, hexanediol, and two surfactants - Span® 85, a lipophilic agent, and Tween® 20, a hydrophilic agent - were used in spontaneous emulsification and interfacial polyaddition processes.
Cationic PU were initially synthesized for fabric processing, the treatment of wood and metal surfaces, as a leather primer and fiberglass infiltrating agent, used tertiary amines to obtain salts that present enhanced compatibility, optical, mechanical, thermal, and static stability compared to non-ionic products.13 Nowadays, novel antibacterial and nonhemolytic biomaterials based on cationic PU and foams for wound healing have been developed for medical applications;14,15 these studies reveal the versatility of PU and the obtaining of safety products that can be used as medical devices in health care.
PU particles suitable as delivery systems for different extracts and drugs have been developed by our research team in the recent years;16–21 the raw materials and synthesis recipes have been continuously modified to enhance encapsulation efficacy, membrane permeation, and release time. The advantages of developing a PU-type vector over PEI carriers for DNA include reduced toxicity (immune response and adverse reactions) and versatility in design to optimize the degradability rate, DNA binding, and cellular uptake. PU can be tailored to achieve a controlled release of DNA, potentially improving gene delivery efficiency. To our understanding, the employment of PU as carriers in genetic therapies has been quite restricted.
The present research aims to design, characterize, and check the biocompatibility of PU particles based on N-methyldiethanolamine, with possible applications in the transport of DNA. After a “classical” synthesis, the bare and the DNA containing capsules were chemically (FT-IR, RAMAN), morphologically (SEM, SANS), thermally (thermogravimetry) and biologically characterized (cell viability, and in vivo evaluation of skin parameters) by using a set of complementary methods. The novelty of this study lies in the utilization of inexpensive, biodegradable, and biocompatible PU particles derived from aliphatic raw materials, without the use of heavy metal salts as catalysts, as a delivery system for nucleic acids.
Materials and Methods
Raw Materials and Animals
Chemicals used in the study were purchased from various sources, as follows: Tween® 20 and caffeine powder were obtained from Merck (Darmstadt, Germany), while isophorone diisocyanate (IPDI), polyethylene glycol (PEG 400), polycaprolactone diol (PCL Mn~530), 1,6-hexanediol (HD), N-Methyldiethanolamine (MDEA), and inorganic salts (sodium, potassium, and magnesium chlorides, Na2HPO4, K2HPO4, KH2PO4, and sodium bicarbonate) were purchased from Sigma-Aldrich (St. Louis, MI, USA). Acetone was obtained from Honeywell Riedel-de Haen (Seelze, Germany). All of the reagents utilized in the experiment were of analytical-grade purity and were employed without having undergone any prior purification procedures. The preparation of double-distilled water was undertaken in-house using a JP Selecta Dest-4 distiller.
In vitro cell culture experiments used human dermal fibroblasts (HDFa), which were obtained from Invitrogen (Waltham, MA, USA), along with reagents and culture supplies from Thermo Fischer Scientific (Waltham, MA, USA). These reagents included culture media, fetal bovine serum (FBS), antibiotics, phosphate-buffered saline (PBS), 0.25% trypsin-EDTA solution, MTT solution, and dimethyl sulfoxide. The culture supplies comprised of culture flasks, Pasteur pipettes, pipette tips, conical tubes, and 96-well microplates.
The study utilized female 6-8-week-old CB17SCID and Nu/Nu BALB/c mice, which were purchased from Charles River (Sulzfeld, Germany).
DNA Extraction
The procedure was already published in the literature.22 Briefly, a solution of peeled banana fruit (25 g) and water (50 mL) was prepared to separate the fruit’s cells, which was then strained to obtain a banana-fruit solution. Next, 10 mL of a 10% Sodium Dodecyl Sulfate (SDS) solution was added to break down the cells and release the DNA. The mixture was stirred for 10 minutes at room temperature, and then centrifuged for 5 minutes at 8000 rpm to separate the debris from the solution. The clear supernatant was then stirred for 5 minutes with sodium chloride salt to obtain a 4.00 mM salt concentration. The next step was to add 2 mL of 99.5 mM isopropanol to precipitate the DNA. The mixture was then applied to the QIAamp Mini column and centrifuged at 8000 rpm for 1 minute to bind the DNA to the silica column. The DNA was washed using 0.5 mL of Qiagen DNA wash buffers, and eluted in 100 μL of water. The DNA concentration was then determined using a spectrophotometer.
Synthesis of the Carrier
The process of obtaining a polyurethane delivery system involves an exothermic reaction between an active hydrogen-containing component (a mix of PEG 400, PCL, HD, MDEA) and an organic component based on IPDI. In order to determine an optimal ratio between the reagents and identify the most effective synthesis parameters, the synthesis process was conducted 12 times, varying the quantities and temperature settings. Regrettably, the majority of the resulting samples were unsuccessful due to the increased adhesiveness of the final material, which manifested as a duromer that was insoluble in common solvents. During the trials stage, it was found that MDEA allows for better control and improved characteristics in the final polyurethane product compared to other alternatives. Thus, the first component (3.65 g PEG 400, 3.00 g PCL, 2.60 mL HD and 1.20 mL MDEA), 40.00 mg caffeine as a catalyst of PU synthesis, and 90 mL of distilled water were added to a Duran® beaker and stirred on a Velp hot plate stirrer (Usmate, Italy) at 400 rpm and room temperature overnight; the selection of these reagents and their proportions was informed by the exceptional outcomes achieved by our laboratory in synthesizing a polyurethane carrier for isosorbide derivatives, which demonstrated superior performance compared to carriers employed for ginger extract and triclosan.18–20 The other component (6.00 mL IPDI in 50.00 mL acetone) was homogenized in a cone-shaped flask in the same conditions according to previous researches on PU materials based on isophorone- and hexamethylene-diisocyanate.16,17,21 Then, the content of the beaker was rapidly injected in the flask and the reaction mixture was stirred at 400 rpm and 40±0.2 °C for 4 hours to complete the synthesis of the macromolecular chains in the absence of any polymerization and chain propagation initiators; the temperature was monitored using a P700 Universal thermometer from Dostmann electronic GmbH (Wertheim, Germany). Our previous comparative studies served as the basis for the selection of the current temperature-time regime.18–21 The content of the flask was sonicated for 5 min using a Bandelin Sonorex Digitec bath sonicator (Berlin, Germany). The procedure was repeated twice in order to obtain a reference sample, without DNA (labeled as sample PU_0), and another sample containing 2.00 mL DNA extract in the aqueous component, that was labeled as sample PU_1.
The suspensions were thoroughly cleaned using a 1:1.35 (v/v) mixture of water and acetone, then were centrifuged at 2000 G using an Ika Mini G micro-centrifuge (Staufen, Germany) to increase the concentration of samples, and analyzed to determine the free DNA content. Finally, the samples were dried in borosilicate glass Petri dishes at 45 °C in-side a PolEko SL115 drying oven to a constant mass, taking approximately 48 hours.
Pre-Formulation Characteristics
The pH of the sample was measured using a Mettler Toledo FiveGo F2 portable pH meter, InLab® Expert Go Sensor (Schwerzenbach, Switzerland), and aqueous solutions with a concentration of 2.0 mg mL-1 at a temperature of 25 °C. The instrument was previously calibrated using standard solutions with pH values of 4.01, 7.00, 9.21, and 11.00, which were provided by the same manufacturer. The refractive index of the synthesized samples was determined using a few drops of the aqueous solutions on a Kern digital refractometer (Balingen, Germany) at 25 °C.
DNA Encapsulation Efficacy
The percentage of DNA successfully encapsulated within PU particles was determined by comparing the amount of free nucleic acid to the total amount added. The quantity of free DNA was determined by measuring the absorbance at 260 nm relative to the concentration. The quantity of free DNA collected in the washing solution from the final step of the synthesis process was calculated using a previously established method,23 which involved plotting the absorption values against its concentration to construct a calibration curve, as described by Equation 1:

where y = absorbance, x = dilution (ng/mL), R2 = coefficient of determination.
The Release Profile
The total cumulative release percentage has been determined by keeping the sample of particles containing DNA (PU_1) in a simulated body fluid (SBF) based on a recipe by T. Kokubo for 60 hours:24 2.00 mL solution of the PU_1 sample (2.0 mg mL-1) were added to 30 mL SBF, and every 12 hours, 3 aliquots of 0.50 mL each were replaced with fresh medium. This SBF, formulated based on the ionic composition and physiological conditions of human blood plasma, serves as a valuable in vitro model for assessing the behavior of biomaterials in an environment that mimics physiological conditions; it enables the examination of degradation kinetics and drug release profiles under simulated physiological conditions.25,26 The maximum absorption was measured at 260 nm according to the literature,27 and the average concentrations ± standard deviations were used to plot the release profile. The investigation into release was conducted by characterizing the kinetic profiles using zero-order and first-order models, as well as the Higuchi, Korsmeyer-Peppas, and Hickson-Crowell models.
Penetrability Measurements
The permeability of PU particles, both with and without DNA, was evaluated using a Spectra/Por® PVDF artificial membrane and a Franz-type vertical static diffusion cell system having a diffusion area of 1.77 cm2 and receptor volume of 12.0 mL. Phosphate buffer saline was poured into the receptor compartment, and the experiment was run at 25±1 °C; after every 12 hours, 1.0 mL of the receptor compartment’s liquid was replaced with fresh buffer, and the liquid was analyzed using a UVi Line 9400 (SI Analytics, Germany) at 362 nm.
FTIR-UATR Spectroscopy
The FTIR-UATR analysis was conducted utilizing a Shimadzu FT-IR IRTracer-100 spectrometer with the Attenuated Total Reflectance (ATR) methodology, without the need for sample pretreatment. The data was collected over 20 consecutive readings, with a resolution of 4 cm−1 within the range of 4000–280 cm−1.
Raman Spectra
The spectra were acquired using a LabRAM Soleil V1.0 – Confocal Raman Microscope (Horiba Scientific, Kyoto, Japan). Dried samples were transformed into circular discs by applying pressure with a hydraulic press, and a 785 nm (red) laser with a deviation angle and a diffraction grating of 500 nm was utilized. The Raman shift range of 30 to 2000 cm−1 was probed using a 5× objective lens, and each spectrum was recorded with an acquisition time of 30 seconds and two accumulations.
Thermal Stability
Using the Mettler Toledo Thermal Analyzer TGA/DSC3+ STARe System (Melbourne, Australia), the samples’ thermal analysis was investigated. The studies were performed in a dynamic air environment (100 mL/min, synthetic air) with a heating rate of 10°/min, and the temperature range covered was 25–500 °C. A DSC-204 Netzsch calorimeter (Selb, Germany) was used to measure the very small quantities of each sample (around 4 mg) in aluminum melting crucibles with punctured lids to seal them under the same temperature range, heating rate, and atmosphere.
DLS Parameters
The assessment of sample size and surface charge was executed using a Cordouan Technology system (located in Pessac, France) that comprised a size detector (Vasco analyzer) and a charge measurement module (Wallis). The following parameters were established to determine the particle size and its distribution: temperature (23.0±0.5 °C), time interval (21±2 μs), number of channels (440±10), laser power (80±5%), continuous acquisition, and Pade-Laplace analysis. The parameters set to detect surface charge were as follows: the same temperature, automatic applied field, medium resolution, 5 measures/sequence, laser power (70%), and Henry function (Smoluchowski).
SEM
The morphology of the sample was examined utilizing a TESCAN 3 VEGA scanning electron microscope secondary electron detector (Brno, Czech Republic). The following settings were employed: the accelerating voltage at 20.0 kV and the magnifications to 500x and 1000x.
Small-Angle Neutron Scattering
Small-angle neutron scattering (SANS) experiments were conducted on the samples as dry powders that were placed into 2mm-thick quartz cuvettes from Hellma (Müllheim, Germany) and measured without any pre-treatment. Neutron scattering experiments were conducted on the Yellow Submarine instrument at the Budapest Neutron Centre, which is a pin-hole type instrument with a two-dimensional neutron detector. The instrument was used with two sample-to-detector distances (1.2m and 5.3m) and two wavelengths (4.95Å and 9.70Å). The beam diameter was 7mm. The samples were measured for 60–120 minutes at each setting, at room temperature. By changing the wavelengths and sample-to-detector distances, a Q range of 0.006–0.400 Å−1 was covered. The Q (scattering vector) was calculated using the Equation 2:
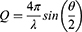
where λ is the wavelength of the monochromatic neutron beam, and 2θ is the scattering angle.
After accounting for background scattering, detector sensitivity, empty cell scattering, and sample transmission, the observed scattering intensity was averaged radially. The values of neutron intensity are expressed in arbitrary units since the amount of in-beam material was overlooked.
Cells Proliferation
For the experiment, human dermal fibroblast (HDFa) cells were utilized, which were seeded in a medium supplemented with FBS and penicillin-streptomycin mix. The cells were grown under standard conditions in a specific environment that was previously established.28 The cells were cultivated in such a manner that the medium was modified every 2–3 days in order to promote optimal growth. The proliferation of the cells was assessed using the MTT colorimetric technique. To analyze the cytotoxicity, HDFa (2x10^4 cells/well) were seeded in 100 µL of medium in 96-well cell culture plates and allowed to adhere for 24 hours. After 24 hours, the growth medium was replaced with samples diluted in different ratios: 90 µL medium + 10 µL sample, 95 µL medium + 5 µL sample, and 99 µL medium + 1 µL sample. Control cell batches were treated with DMSO at equivalent concentrations to the test samples. All tests, performed in triplicate, involved samples suspended in DMSO. Cell proliferation was assessed at 24, 48, and 72 hours post-treatment using the MTT assay, and absorbance at 590 nm was measured with a Bio-Tek Synergy H1 spectrophotometer. The proliferation of cells was calculated using Equation 3:28

where At is the absorbance for the test solutions and Ac is the absorbance for the control. The calculation was based on the average of three successive measurements.
Skin Irritation
A comprehensive pharmaco-toxicological evaluation was carried out on newly synthesized compounds using specially-sensitive mice, which were divided into five groups. The first two groups served as controls, one consisting of mice treated only with the solvent, while the other received a 2.0% SDS solution, a known skin irritant. The hair on the back of CB17SCID mice was shaved at the beginning of the experiment and each subsequent week, providing a surface area of 3–4 cm2. The samples under investigation (40±3 µL each) were applied to the back skin of the mice every third day for a period of 15 days. Measurements of various parameters were taken 30 minutes later by the same operator under standardized conditions of temperature and humidity. Erythema was assessed using a Mexameter® MX 18 probe, while skin hydration levels were determined using a Corneometer® CM 825 probe. Both devices were connected to a Multiprobe Adapter System (MPA5) from Courage-Khazaka (Koln, Germany).
Statistics
The IBM SPSS Software version 27.0.0.0 (Armonk, NY, USA) was utilized for data analysis. The mean and standard deviation of mean were used to present the results for continuous variables. To assess the normality of the dataset, a Kolmogorov–Smirnov (K-S) test was conducted; Student’s t-test, ANOVA, Kruskal–Wallis, and multiple comparison Tukey’s post-hoc test was also used. A p-value of less than 0.05 was considered to be statistically significant (# indicate p > 0.05, * indicate p < 0.05, ** indicate p < 0.01, and *** indicate p < 0.001). The charts were created in Excel, which is part of the Microsoft® Office Professional Plus 2019 software package from Microsoft Corp. (Washington, USA).
Animals: Before Irritation Testing
The present study was conducted in full compliance with the ethical principles outlined in the Declaration of Helsinki; prior to commencement, the study protocol underwent evaluation and received approval from the Ethical Committee at “Victor Babes” University of Medicine and Pharmacy Timisoara, Romania (approval no. 42 from October 29, 2021). The experimental methodology adhered to the guidelines established by the National Institute of Animal Health; animal subjects were housed under controlled environmental conditions, including a 12-hour light-dark cycle, unrestricted access to food and water, ambient temperature of 25±1 °C, and relative humidity exceeding 55%.
Results
Physicochemical Properties
The results of the pH evaluation demonstrated that the samples were neutral and safe, with the following values obtained: 6.77±0.19 (sample PU_0) and 6.75±0.14 (sample PU_1). Both samples had pH values that fell within the acceptable range for pharmaceutical products. Moreover, the refractive index values were found to be 1.60 for both samples, which is an important measure for characterizing the samples. The refractive index analysis can be used to verify the purity and isotropic nature of the samples.
Encapsulation Efficiency and in vitro Release
The quantity of free DNA was used to calculate the loaded amount. A graph of absorbance versus calibration was created using the absorbance of nine standard solutions with varying concentrations. The average encapsulation efficiency for samples PU_1 was 72.5±2.1% based on the amount of free DNA reported as a percentage of the total quantity added to synthesis.
The cumulated DNA release is depicted in Figure 2, which was collected under nearly ideal sink conditions, as previously described in a study.29 To more precisely characterize the observed pattern of release, we conducted an analysis of the data using the principal kinetic models, including zero-order, first-order, Korsmeyer-Peppas, Hickson-Crowell, and Higuchi models; the highest R2 value was recorded at 0.9886 in the case of Higuchi model.
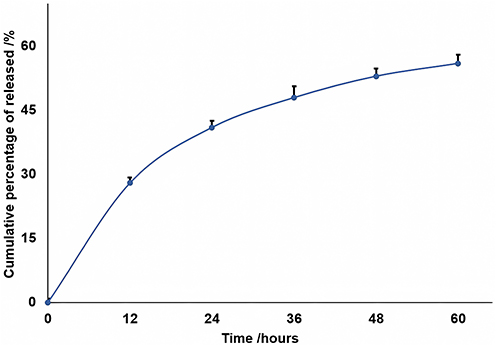 |
Figure 2 The evolution of the cumulated DNA release. |
Penetrability Rate
The release of DNA to a target can be significantly influenced by its carrier ability to penetrate various membranes, which is a crucial rate-limiting parameter. The permeation of PU particles from both samples through an artificial membrane was remarkably similar (Figure 3), with around 40% of the particles penetrating within the first ten hours and approximately 65% passing through in the first 24 hours.
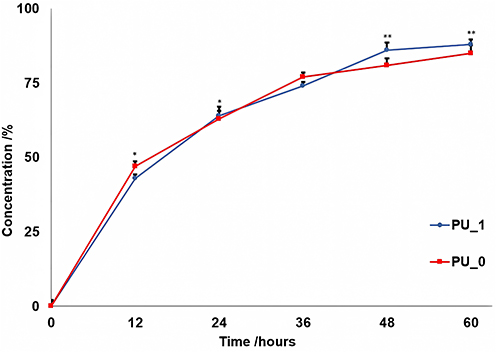 |
Figure 3 The penetrability rates. The significant difference between the samples was indicated by *p < 0.05 and **p < 0.01 from t-test at the same time. |
FTIR-UATR Spectroscopy
FTIR-UATR analysis was used to comparatively determine the functional groups present in the obtained samples, which is often used in materials science, chemical analysis, and biomedical research to determine the composition of a sample and gain insight into its properties and behavior. Figure 4 shows the FTIR spectra of the analyzed samples.
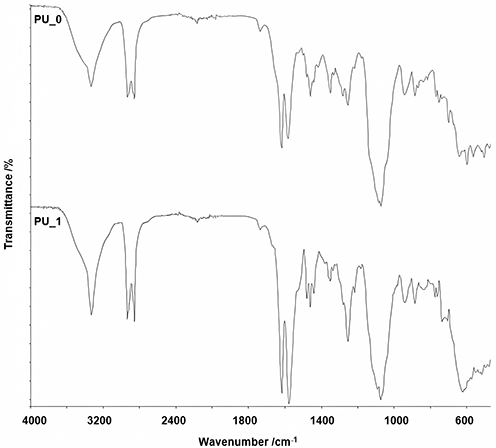 |
Figure 4 FTIR spectra of samples. |
The following absorption bands were identified: -NH- stretching (3340 cm−1 for PU_0 and 3334 cm−1 for PU_1), -CH2- stretching (sharp peaks between 2851 and 2928 cm−1); other vibrations associated with the carbonyl absorption bands between 1738 and 1624 cm−1, while -CH2- were identified by the bands located at 1452, 1347, and 1256 cm−1 for PU_0 and at 1466, 1347, and 1247 cm−1 for PU_1. The characteristic group of vibrations of the -NH- bond is very intense at 611 cm−1 and 1576 cm−1 for PU_1 and at 1586 cm−1 for PU_0. The presence of bands in the range of 1079–1069 cm−1 suggests the existence of a C-O-C bond, while the bands at 935 cm−1 are characteristic of aliphatic alcohols, respectively 888–720 cm−1 for mono-substituted alcohols. The two spectra have similar characteristics in terms of composition and functional group presence. Both the samples displayed peaks at identical wavelengths, albeit with minor variations in intensity.
Raman Spectroscopy
The Raman spectra of samples (Figure 5) show bands at 235.5 cm−1, 272.5 cm−1, and 339.9 cm−1 (PU_0), at 211.3 cm−1 and 280.2 cm−1 (PU_1) that correspond to the cleavage of C-C aliphatic bonds, a band at 442.9 cm−1 (PU_0) characteristic for alcohols,30 and bands between 536.3 cm−1 (PU_1) and 544.7 cm−1 (PU_0) attributed to the stretch of C-C alicyclic and aliphatic bonds. The C-O-C bond is indicated by the strong bands occurring at 859.6 cm−1 (PU_1) and between 744.9 and 964.8 cm−1 (PU_0). The bands between 1226.2 and 1494.9 cm−1 (PU_0), between 1108.4 and 1441.8 cm−1 (PU_1) are associated with the stretch and asymmetric cleavage of -CH3 and -CH2- bonds. The presence of moderate bands between 1585.1 and 1747.8 cm−1 suggests the presence of C=O bonds, while the observation of bands around 1971.9 cm−1 indicates the presence of C-O-O bonds. The moderate band at 2141.1 cm−1 (PU_0) is associated with C=C bonds, while the signal of C=N bonds appears between 2238.2 and 2245.1 cm−1. The strong band at 2885.8 cm−1 (PU_1) is specific to C-CH3 groups, while moderate bands around 3150 cm−1 can be attributed to amines.
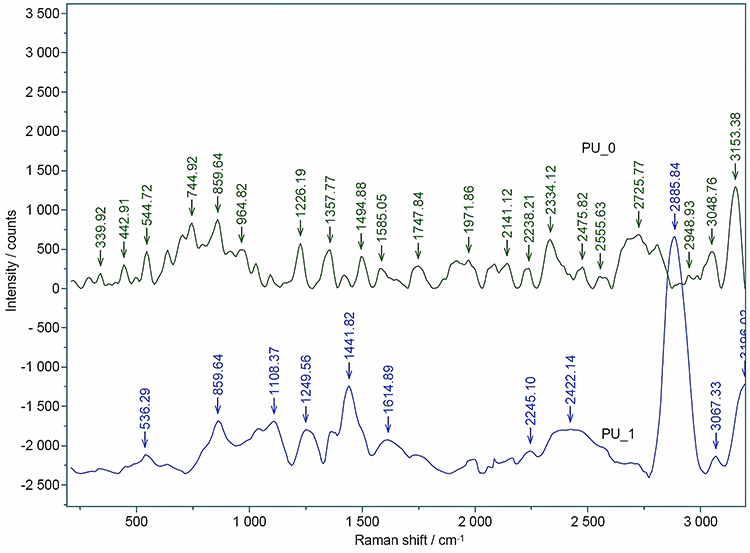 |
Figure 5 Raman spectra of samples. |
Thermal Analyses
Figures 6–8 illustrate the results of the thermal analyses, including TG/DTG and DSC. The thermal degradation of PU samples can be attributed to multiple degradation steps and varying stability, which depends on the composition and the presence of various raw materials. Up to 100 °C, the decomposition of the macromolecular chains is generally considered to be minimal, but the evaporation of acetone and water can be observed. The most intense degradation step, with a DTG peak maximum around 200°C, is ascribed to thermolysis processes that disrupt the weakest bonds. This is followed by two temperature ranges of combustion, whose maximums of the decomposition rates approximately distribute in 300–350°C and in 370–450°C, respectively. The first stage represents the decomposition of PU and the release of volatile compounds, while the second stage corresponds to the oxidation process of the residues.
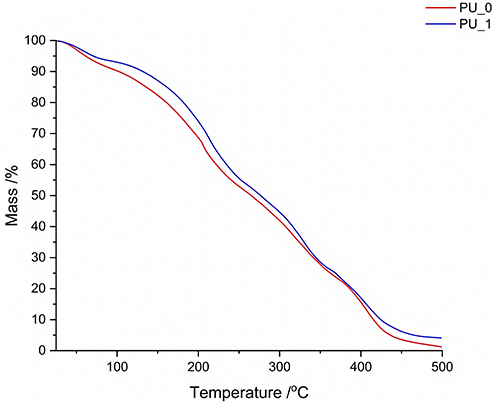 |
Figure 6 The TG curves of the synthesized samples. |
 |
Figure 7 The DTG curves of samples. |
 |
Figure 8 The DSC curves of the tested samples. |
DLS Parameters
The necessity of determining the size distribution and Zeta potential via dynamic light scattering before deploying any delivery system is generally recognized and considered indispensable. These methods have been extensively employed in numerous research domains such as the stability of nanoparticles, bio-colloids, and polymer science. Table 1 lists the values obtained from Zetasizer evaluations.
 |
Table 1 DLS Parameters of the Developed Samples |
SEM
The morphological aspects of the PU_0 (Figure 9) and PU_1 (Figure 10) samples were investigated at two different magnifications: 500x (a) and 1000x (b). After a comparison of the figures for DNA loaded and bare PU capsules, no major differences were observed.
 |
Figure 9 SEM image of the PU_0 sample: (a) 500x; (b) 1000x. |
 |
Figure 10 SEM image of the PU_1 sample: (a) 500x; (b) 1000x. |
SANS
In Figure 11, the neutron intensity is represented with respect to Q momentum transfer. The curves were modelled using a power-law model (Equation 4):
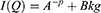
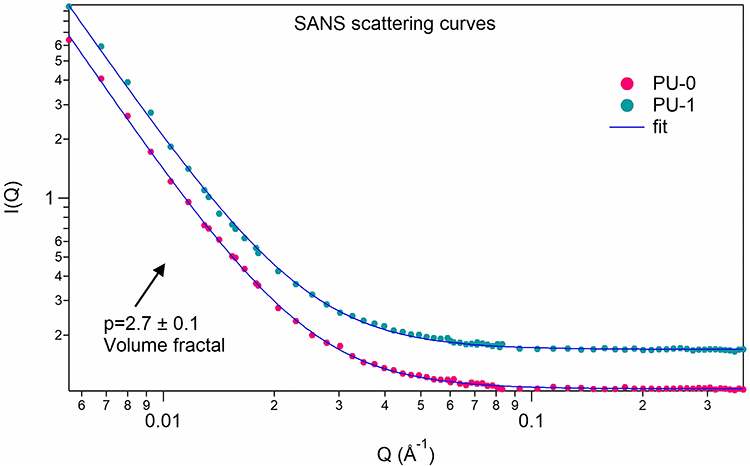 |
Figure 11 SANS intensity versus scattering vector curves. |
where A contains the instrument-related scaling factor and neutron contrast, p is the power value related to the fractal exponent, and Bkg is the incoherent background scattering.
Cell Viability
The viability of HDFa cells (Figure 12) was determined 24, 48, and 72 hours after stimulation with the samples at concentrations of 1%, 5%, and 10% to determine their cytotoxicity. At 24 hours, concentration-dependent increases in cell viability were observed for both samples. The highest values were observed at the concentration of 10% (160% for PU_0 and 167% for PU_1).
 |
Figure 12 Relative cells viability after (a) 24, (b) 48, and (c) 72 hours. #p> 0.05, *p< 0.05, **p< 0.01, ***p< 0.001 vs control. Bars represent mean ± standard deviation of the mean. |
A similar trend was observed in terms of cell viability after 48 hours. Thus, the maximum concentration tested for samples increased cell viability to 194% (PU_0) and 196% (PU_1) compared to the control. At 72 hours of incubation, PU_0 increased cell viability compared to the control by 227%, while PU_1 exhibited the highest values at the studied concentrations (270%).
Skin Irritation
Two mouse strains were used in this study owing to their different skin sensitivities. As shown in Figures 13 and 14, slight variations in the examined skin parameters (erythema and hydration) were apparent when comparing the consequences observed in the tested PU samples to those found in SDS.
 |
Figure 13 Comparative evolution of erythema for (a) CB17SCID mice and (b) Balb-c. #p> 0.05, *p < 0.05, **p < 0.01, ***p < 0.001. Bars represent mean ± standard deviation of the mean (n= 30). |
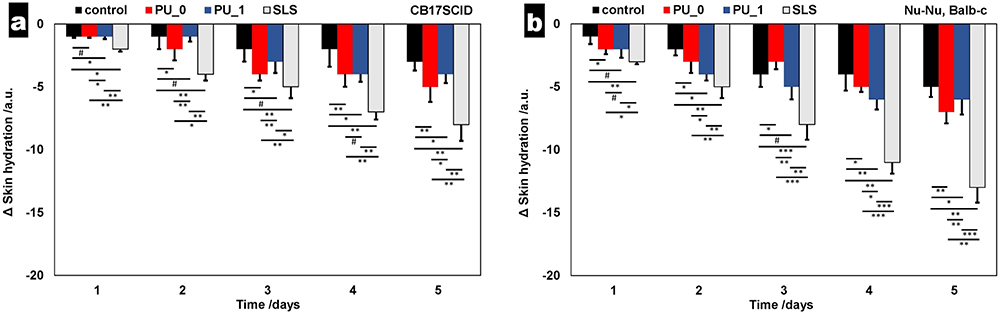 |
Figure 14 Comparative evolution of skin hydration for (a) CB17SCID mice and (b) Balb-c. #p> 0.05, *p < 0.05, **p < 0.01, ***p < 0.001. Bars represent mean ± standard deviation of the mean (n= 30). |
Discussion
To deliver therapeutic nucleic acids into patient cells either to compensate for genetic abnormalities or to introduce new genetic material capable of resisting disease processes, DNA delivery is an essential part of the treatment.31 The potential of DNA delivery to alter the course of severe and sometimes fatal genetic illnesses, while providing previously unattainable effective therapies emphasizes the significance of this technology.32 Although viral vectors have demonstrated excellent transport efficiency, concerns about their safety have raised interest in non-viral vectors, such as polymer-based carriers and cationic liposomes. These non-viral vectors offer advantages such as ease of production and low immunogenicity.33
Two samples of a polyurethane carrier, one with DNA and one without, were obtained and comparatively characterized using physicochemical, in vitro, and in vivo evaluations. These analyses confirmed the spectral properties, stability, size, charge, and shape of the newly synthesized products. The synthesis process involved combining an aqueous with an organic phase. PEG400, used in the aqueous phase, served to form soft segments of the macromolecular chains and control their hydrophilicity and release profile. PEG400 is also commonly used as a pore-forming agent. PCL was included to enhance hydrolysis-induced degradation, while HD functioned as a chain extender. The ratio of these raw materials was determined based on our previous research to achieve a pro-longed release profile and a specific range of PU structure sizes.16–21 On the other hand, MDEA was used to introduce cationic groups. IPDI, dissolved in acetone was chosen for the organic phase due to its non-carcinogenic properties. Banana DNA has been chosen due to its advantages for scientific investigations, especially in the genetic diversity analysis and breeding programs.34
The pH evaluation revealed that both samples were neutral and safe, with pH values within the falling within the acceptable range for the topical application of pharmaceutical products. Previous studies have shown that the refractive index is a critical parameter for assessing the purity and isotropic nature of samples. The obtained refractive index value of 1.60 provides valuable information into the safety and purity of samples, which is essential for their use in pharmaceutical applications.
Encapsulation efficacy has crucial potential for improving therapeutic outcomes through increased bioavailability and controlled release of the loaded active agent, which can lead to more effective treatment and reduced side effects. On the other side, the encapsulation efficiency is an important parameter in reducing drug loss during formulation and manufacturing. A result of 72.5% was obtained by reporting the quantity of free DNA from the washing mixture to the total amount of DNA added to the synthesis; although the value is acceptable, existing literature indicates that PEI and lipid nanoparticles exhibit higher values.35,36 Farshbaf et al37 describe that cationic polymers present very good values for the encapsulation efficacy and their potential for therapeutic use, particularly in nucleic acids delivery for gene therapy, is heightened by this aspect, making them a compelling candidate for these applications.
The discharge of active agents relies heavily on their release profile, which is a crucial factor in the delivery process. Typically, manipulation of polymer degradation rates and drug diffusion from polymer particles is used to control the release profile. The study presented a rapid burst release due to the discharge of surface-bound molecules in the first 12 hours (Figure 2),38 followed by a slow release that occurred simultaneously with the degradation of the polymer and diffusion of the active agent through the pores of the polymer particle over the next 24–36 hours. Finally, there was a stable “plateau” phase that determined the release profile. On the other side, the penetrability rate is an important rate-limiting parameter that influence the release of the active agent to the target. From our measurements, it was observed that the penetrability of the particles through the PVDF membrane was approximately 45% in the first ten hours and it is worth mentioning that almost the entire amount passed through the artificial membrane within the next 50 hours.
The data obtained by FTIR and Raman spectroscopy can be used to optimize the design and performance of carriers, ensuring that they are safe and effective for use in medical applications. When discussing the safety aspects of polyurethane products, it is essential to acknowledge the significant toxicity associated with isocyanates, a key raw material used in the synthesis.39 Figure 5 shows a minor peak for the DNA phosphate at 1108 cm−1 for PU_1, which is consistent with the literature;40 however, the absence of other signals at 779, 1334, 1484, 1574, and 1665 cm−1 might be due to the effective encapsulation, which may limit the vibrational motions required for IR detection. This incorporation frequently causes the masking or shifting of distinctive infrared peaks because of the interactions between the drug and carrier.
The synthesized polyurethane particles exhibited excellent thermal stability according to the findings of this study. The ability to maintain structural integrity and functional properties at a range of temperatures is critical because many active agents are sensitive to high or low temperatures, which can compromise their potency and efficacy. The current carrier demonstrates significant thermal stability up to 200 °C. Although assessment above this temperature is not relevant for topical biomaterials, it provides valuable insights into the macromolecular chains, allowing for the prediction of the half-life of pharmaceutical formulations and the assessment of compatibility with various excipients.41
In contrast to our previously generated PU particles,16–21,28 the DNA carrier we developed a larger particle size distribution and particle size because of the incorporation of a higher amount of chain extender (HD) during the synthesis process. The benefits of these changes include enhanced encapsulation effectiveness and capacity to sustain DNA release, thereby guaranteeing therapeutic levels over an extended duration. The SEM images and DLS parameters (Table 1) demonstrated the emergence of dispersed systems comprising two distinct particle populations. Previous research has indicated that carriers comprising dispersed particles typically ensure an extended release,42 as a result of their differing degradation rates.
Polyurethane is known to be prone to phase separation and microphase segregation, which can lead to inhomogeneities in the material. The presence of DNA or other additives can further influence the material’s nanostructure, potentially leading to inhomogeneities. Understanding how DNA affects the nanostructure of this material is essential for developing biomaterials with specific properties. One of the advantages of SANS is that it provides average information over the whole volume of the sample (approximately 25 mm3), whereas microscopic methods are limited to surface-level observations. Therefore, a nanophase separation with or without DNA would clearly alter the power-law behavior of the SANS curves.
The SANS data in Figure 11 reveal typical power-law behavior, confirming the homogeneity of the material in the size range of approximately 20–200 nm. The observed volume or mass fractal-like behavior is characterized by a p-exponent value of 2.7 ± 0.1. This type of fractal behavior is typically associated with structures containing branching and crosslinking, forming three-dimensional networks. Notably, both the PU_0 and PU_1 samples exhibit similar SANS curves, indicating that the addition of DNA does not compromise the polyurethane skeleton.43
Polyurethane biomaterials have already demonstrated the ability to promote cell adhesion and growth at specific concentrations, as demonstrated by a study conducted by Zanetta et al.44 A study by González-García et al45 also examined the cytocompatibility of two PU-based biomaterials designed for bone tissue regeneration and their results revealed excellent cell adhesion to the material’s surface, which was completely covered by cells, and good cell proliferation. Additionally, in a previous study conducted by our research team, we investigated the impact of a PU carrier containing ursolic and oleanolic acids on skin tumors and found that cell viability was slightly higher in the sample with encapsulated acids than in the sample with free acids.46 The data presented in Figure 12 (HDFa cell viability) provided strong evidence for the non-cytotoxic profile of PU particles as a delivery system for nucleic acids. After subjecting the cells to 24, 48, or 72 hours of stimulation, the cell viability increased in a time- and dose-dependent manner. The highest level of cell proliferation (270%) was recorded for PU_1 when tested at the highest concentration (10%) after 72 hours.
Skin assessment in mouse models is important because it allows researchers to study skin biology and disease in a controlled and reproducible manner. Furthermore, skin assessments in mouse models can provide insights into the efficacy and safety of topical treatments. Overall, the use of mouse models is critical for the development of new therapies.47 Our in vivo noninvasive and quantitative evaluation of erythema and skin hydration was used to investigate the irritative effects of the PU samples. Figure 13 (erythema differences in a 5-day assessment on two mice strains) presents the comparative evolution of this skin parameter; it can be noticed that erythema values increase between 15–40 arb. units in the control group of CB17SCID mice and 20–60 arb. units for Nu-Nu Balb-c mice because of the higher sensitivity of hairless mice. The increase in erythema values is often associated with irritation; however, the amplitude of this increase is very important; for SDS, the difference is equal to 126 arb. units (CB17SCID), and 182 arb. units (Nu-Nu Balb-c). Similarly, the decrease in skin hydration indicates an irritation potential of a chemical compound of pharmaceutical formulation, but it can be noticed that the differences found for mice treated with PU_0 and PU_1 are much closer to the differences recorded for the control group than for the SDS group.
Physicochemical, in vitro, and in vivo evaluations of the synthesized carrier revealed that the development of polyurethane particles for the delivery of nucleic acids represents a novel approach to this important research area, and the results of this study provide valuable insights into the potential of this new technology. However, although the results of this study are promising, further clinical trials are needed to fully evaluate the safety and efficacy of these particles as delivery systems for DNA.
The small sample size of this study is its primary drawback; undoubtedly, the quantity of MDEA used plays a significant role in determining the efficiency of DNA delivery. Therefore, it is essential to examine a series of PU carriers with varying MDEA concentration. The absence of investigations into the temporal stability of the formulation and the effects of samples on irritated skin should be recognized as limitations of this study. A comparative study exploring the efficacy of PU and polyethyleneimine as DNA carriers could yield valuable information. To comprehensively characterize these samples, it is essential to conduct a long-term cytotoxicity assay to assess carrier biocompatibility, evaluate prolonged DNA release, and examine skin irritation over extended periods, with a minimum duration of one month.
Conclusion
This study demonstrates the successful fabrication of cationic polyurethane (PU) particles as potential non-viral carriers for topical DNA delivery. Particles synthesized from aliphatic non-carcinogenic raw materials showed favorable physicochemical properties, efficient DNA encapsulation, controlled release, and good membrane penetration. Analytical techniques confirmed that DNA encapsulation did not alter the structural integrity of the carrier, while thermal analysis indicated stability up to 200 °C. In vitro and in vivo assessments revealed dose- and time-dependent cell proliferation and minimal skin irritation, thereby supporting the biocompatibility of the system. The small sample size and lack of temporal stability are the main limitations of this study. Comparing PU and polyethyleneimine carriers would be valuable. Future studies should assess carrier biocompatibility, DNA release, and skin irritation over one month.
Acknowledgments
This research was funded by “Victor Babes” University of Medicine and Pharmacy Timisoara, grant number 5EXP/1244/30.01.2020. The authors would like to acknowledge “Victor Babes” University of Medicine and Pharmacy Timisoara for their support in covering the costs of publication for this research paper.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Al-Dosari MS, Gao X. Nonviral gene delivery: principle, limitations, and recent progress. AAPS J. 2009;11(4):671–681. doi:10.1208/s12248-009-9143-y
2. Nayerossadat N, Maedeh T, Ali PA. Viral and nonviral delivery systems for gene delivery. Adv Biomed Res. 2012;1:27. doi:10.4103/2277-9175.98152
3. Piperno A, Sciortino MT, Giusto E, et al. Recent advances and challenges in gene delivery mediated by polyester-based nanoparticles. Int J Nanomed. 2021;16:5981–6002. doi:10.2147/IJN.S321329
4. Gigante A, Li M, Junghänel S, et al. Non-viral transfection vectors: are hybrid materials the way forward? Medchemcomm. 2019;10(10):1692–1718. doi:10.1039/c9md00275h
5. Zu H, Gao D. Non-viral vectors in gene therapy: recent development, challenges, and prospects. AAPS J. 2021;23:78. doi:10.1208/s12248-021-00608-7
6. Yang TF, Chin WK, Cherng JY, et al. Synthesis of novel biodegradable cationic polymer: N,N-Diethylethylenediamine polyurethane as a gene carrier. Biomacromolecules. 2004;5(5):1926–1932. doi:10.1021/bm049763v
7. Rolińska K, Mazurek-Budzyńska M, Parzuchowski PG, et al. Synthesis of shape-memory polyurethanes: combined experimental and simulation studies. Int J Mol Sci. 2022;23(13):7064. doi:10.3390/ijms23137064
8. Wang J, Dai D, Xie H, et al. Biological effects, applications and design strategies of medical polyurethanes modified by nanomaterials. Int J Nanomed. 2022;17:6791–6819. doi:10.2147/IJN.S393207
9. Niesiobędzka J, Datta J. Challenges and recent advances in bio-based isocyanate production. Green Chem. 2023;25:2482–2504. doi:10.1039/D2GC04644J
10. Bouchemal K, Briançon S, Perrier E, et al. Synthesis and characterization of polyurethane and poly(ether urethane) nanocapsules using a new technique of interfacial polycondensation combined to spontaneous emulsification. Int J Pharm. 2004;269(1):89–100. doi:10.1016/j.ijpharm.2003.09.025
11. Bouchemal K, Briançon S, Perrier E, et al. Nano-emulsion formulation using spontaneous emulsification: solvent, oil and surfactant optimisation. Int J Pharm. 2004;280(1–2):241–251. doi:10.1016/j.ijpharm.2004.05.016
12. Bouchemal K, Briançon S, Couenne F, et al. Stability studies on colloidal suspensions of polyurethane nanocapsules. J Nanosci Nanotechnol. 2006;6(9–10):3187–3192. doi:10.1166/jnn.2006.468
13. El‐Sayed A, Kantouch F, Kantouch A. Preparation of cationic polyurethane and its application to acrylic fabrics. J Appl Polym Sci. 2011;121:777–783. doi:10.1002/app.33558
14. Palencia M, Lerma TA, Arrieta AA. Antibacterial and non-hemolytic cationic polyurethanes with N-carboxymethyl-N,N,N-triethylammonium groups for bacteremia-control in biomedical-using materials. Mat Today Comm. 2020;22:100708. doi:10.1016/j.mtcomm.2019.100708
15. Namviriyachote N, Lipipun V, Akkhawattanangkul Y, et al. Development of polyurethane foam dressing containing silver and asiaticoside for healing of dermal wound. Asian J Pharm Sci. 2019;14(1):63–77. doi:10.1016/j.ajps.2018.09.001
16. Munteanu MF, Ardelean A, Borcan F, et al. Mistletoe and garlic extracts as polyurethane carriers—a possible remedy for choroidal melanoma. Curr Drug Deliv. 2017;14:1178–1188. doi:10.2174/1567201814666170126113231
17. Borcan L-C, Dudas Z, Len A, et al. Synthesis and characterization of a polyurethane carrier used for a prolonged transmembrane transfer of a chili pepper extract. Int J Nanomed. 2018;13:7155–7166. doi:10.2147/IJN.S181667
18. Borcan F, Chirita-Emandi A, Andreescu NI, et al. Synthesis and preliminary characterization of polyurethane nanoparticles with ginger extract as a possible cardiovascular protector. Int J Nanomed. 2019;14:3691–3703. doi:10.2147/IJN.S202049
19. Gălușcan A, Jumanca D, Borcan F, et al. Comparative study on polyurethane and cyclodextrin carrier for triclosan. Rev Chim Bucharest. 2014;65(2):190–193.
20. Borcan F, Len A, Bordejevic DA, et al. Obtaining and characterization of a polydisperse system used as a transmembrane carrier for isosorbide derivatives. Front Chem. 2020;8:492. doi:10.3389/fchem.2020.00492
21. Borcan F, Len A, Dehelean CA, et al. Design and assessment of a polyurethane carrier used for the transmembrane transfer of acyclovir. Nanomaterials. 2021;11(1):51. doi:10.3390/nano11010051
22. Omara D, Barugahare B. Protocol Optimization of DNA Extraction from Banana Fruits. J Adv Biol Biotechnol. 2022;25:1–5. doi:10.9734/JABB/2022/v25i430274
23. Khare P, Raj V, Chandra S, et al. Quantitative and qualitative assessment of DNA extracted from saliva for its use in forensic identification. J Forensic Dent Sci. 2014;6(2):81–85. doi:10.4103/0975-1475.132529
24. Kokubo T, Takadama H. Simulated Body Fluid (SBF) as a standard tool to test the bioactivity of implants. In: Bäuerlein E, editor. Handbook of Biomineralization: Biological Aspects and Structure Formation. Weinheim (Germany): Wiley-VCH Verlag GmbH & Co. KGaA; 2007.
25. Jerca FA, Jerca VV, Hoogenboom R. In vitro assessment of the hydrolytic stability of Poly(2-isopropenyl-2-oxazoline). Biomacromolecules. 2021;22(12):5020–5032. doi:10.1021/acs.biomac.1c00994
26. Rehman F, Guo J, Arshad M, et al. Surface engineered mesoporous silica carriers for the controlled delivery of anticancer drug 5-fluorouracil: computational approach for the drug-carrier interactions using density functional theory. Front Pharmacol. 2023;14:1146562. doi:10.3389/fphar.2023.1146562
27. Yu Z, Zhou Y, Grote JG, et al. Photoluminescence and lasing from deoxyribonucleic acid (DNA) thin films doped with sulforhodamine. Appl Opt. 2007;46(9):1507. doi:10.1364/ao.46.001507
28. Borcan F, Vlase T, Vlase G, et al. The Influence of an isocyanate structure on a polyurethane delivery system for 2′-Deoxycytidine-5′-monophosphate. J Funct Biomater. 2023;14:526. doi:10.3390/jfb14100526
29. Zeng L, An L, Wu X. Modeling drug-carrier interaction in the drug release from nanocarriers. J Drug Deliv. 2011;2011:370308. doi:10.1155/2011/370308
30. Lin-Vien D, Colthup NB, Fateley WG, et al. Alcohols and Phenols. In: Lin-Vien D, Colthup NB, Fateley WG, et al. editors. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules. Amsterdam, The Netherland: Academic Press Inc.; 1991:125–141.
31. Jiao Y, Xia ZL, Ze LJ, et al. Research Progress of nucleic acid delivery vectors for gene therapy. Biomed Microdevices. 2020;22(1):16. doi:10.1007/s10544-020-0469-7
32. Brunetti-Pierri N. 30 - Gene therapy and gene editing. In: Dhar SU, Nagamani SCS, Eble TN, editors. Handbook of Clinical Adult Genetics and Genomics. Cambridge, MA: Academic Press; 2020:463–477.
33. Shui M, Chen Z, Chen Y, et al. Engineering polyphenol-based carriers for nucleic acid delivery. Theranostics. 2023;13(10):3204–3223. doi:10.7150/thno.81604
34. Slameto S. Genetic diversity and molecular analysis using RAPD markers of banana cultivars in the five regions of East Java, Indonesia. Biodiversitas. 2023;24(9):5035–5043. doi:10.13057/biodiv/d240947
35. Tiyaboonchai W, Woiszwillo J, Middaugh CR. Formulation and characterization of DNA-polyethylenimine-dextran sulfate nanoparticles. Eur J Pharm Sci. 2003;19(4):191–202. doi:10.1016/s0928-0987(03)00102-7
36. Mendonça MCP, Kont A, Kowalski PS, et al. Design of lipid-based nanoparticles for delivery of therapeutic nucleic acids. Drug Discov Today. 2023;28(3):103505. doi:10.1016/j.drudis.2023.103505
37. Farshbaf M, Davaran S, Zarebkohan A, et al. Significant role of cationic polymers in drug delivery systems. Art Cells Nanomed Biotechnol. 2018;46(8):1872–1891. doi:10.1080/21691401.2017.1395344
38. Lin CH, Chen CH, Lin ZC, et al. Recent advances in oral delivery of drugs and bioactive natural products using solid lipid nanoparticles as the carriers. J Food Drug Analysis. 2017;25(2):219–234. doi:10.1016/j.jfda.2017.02.001
39. Nakashima K, Takeshita T, Morimoto K. Review of the occupational exposure to isocyanates: mechanisms of action. Environ Health Prev Med. 2002;7(1):1–6. doi:10.1007/BF02898058
40. Chandra GK, Eklouh-Molinier C, Fere M, et al. Probing in vitro ribose induced DNA-glycation using raman microspectroscopy. Anal Chem. 2015;87(5):2655–2664. doi:10.1021/acs.analchem.5b00182
41. Tita B, Fulias A, Marian E, et al. Thermal Stability and Decomposition Kinetics Under Non-isothermal Conditions of Sodium Diclofenac. Rev Chim Bucharest. 2009;60:524–528.
42. Danaei M, Dehghankhold M, Ataei S, et al. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics. 2018;10(2):57. doi:10.3390/pharmaceutics10020057
43. Almásy L. New measurement control software on the Yellow Submarine SANS instrument at the Budapest Neutron Centre. J Surf Investig. 2021;15:527–531. doi:10.1134/S1027451021030046
44. Zanetta M, Quirici N, Demarosi F, et al. Ability of polyurethane foams to support cell pro-liferation and the differentiation of MSCs into osteoblasts. Acta Biomater. 2009;5(4):1126–1136. doi:10.1016/j.actbio.2008.12.003
45. González-García DM, Marcos-Fernández Á, Rodríguez-Lorenzo LM, et al. Synthesis and in vitro cytocompatibility of segmented Poly(Ester-Urethane)s and Poly(Ester-Urea-Urethane)s for bone tissue engineering. Polymers. 2018;10(9):991. doi:10.3390/polym10090991
46. Oprean C, Borcan F, Pavel I, et al. In vivo biological evaluation of polyurethane nanostructures with ursolic and oleanolic acids on chemically-induced skin carcinogenesis. In Vivo. 2016;30(5):633–638.
47. Nowotarski SL, Feith DJ, Shantz LM. Skin carcinogenesis studies using mouse models with altered polyamines. Cancer Growth Metastasis. 2015;8(Suppl 1):17–27. doi:10.4137/CGM.S21219
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.