Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Developing genomic knowledge bases and databases to support clinical management: current perspectives
Authors Huser V, Sincan M, Cimino J
Received 17 May 2014
Accepted for publication 10 July 2014
Published 9 September 2014 Volume 2014:7 Pages 275—283
DOI https://doi.org/10.2147/PGPM.S49904
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Video abstract presented by Vojtech Huser
Views: 242
Vojtech Huser,1 Murat Sincan,2,3 James J Cimino1,4
1Laboratory for Informatics Development, National Institutes of Health Clinical Center, Bethesda, MD, USA; 2Undiagnosed Diseases Program, 3Office of the Clinical Director, National Human Genome Research Institute, National Institutes of Health, MD, USA; 4National Library of Medicine, National Institutes of Health, MD, USA
Abstract: Personalized medicine, the ability to tailor diagnostic and treatment decisions for individual patients, is seen as the evolution of modern medicine. We characterize here the informatics resources available today or envisioned in the near future that can support clinical interpretation of genomic test results. We assume a clinical sequencing scenario (germline whole-exome sequencing) in which a clinical specialist, such as an endocrinologist, needs to tailor patient management decisions within his or her specialty (targeted findings) but relies on a genetic counselor to interpret off-target incidental findings. We characterize the genomic input data and list various types of knowledge bases that provide genomic knowledge for generating clinical decision support. We highlight the need for patient-level databases with detailed lifelong phenotype content in addition to genotype data and provide a list of recommendations for personalized medicine knowledge bases and databases. We conclude that no single knowledge base can currently support all aspects of personalized recommendations and that consolidation of several current resources into larger, more dynamic and collaborative knowledge bases may offer a future path forward.
Keywords: personalized medicine, knowledge bases, databases, clinical decision support, clinical informatics
Personalized medicine
Personalized medicine, also referred to as precision or individualized medicine, can be defined as a combination of state-of-the-art molecular profiling (-omics methods) and traditional methods, such as family history, to create diagnostic and therapeutic strategies precisely tailored to individual patients. Although there are many molecular profiling methods available in research settings, only genomic analyses such as genotyping or exome sequencing are in significant use for patient care. This article is intended to complement a previous perspective paper on pharmacogenetic testing in primary care settings1 by discussing genomic databases supporting current personalized medicine practice that uses genomic data, particularly the use of germline (as opposed to somatic) DNA.
Tailoring patient care to an individual, based on genetic traits, is not a novel phenomenon in medicine. Historical elements, such as family history, have long been used to screen for risk assessment. Laboratory studies such as sickle cell preparations and hemoglobin electrophoresis are commonplace methods for indirectly assessing mutations by characterizing gene products. Other examples are genetic tests for von Hippel-Lindau disease or breast cancer. In November 2013, the US Food and Drug Administration granted marketing authorization for Illumina’s MiSeqDx Universal Kit (Illumina, San Diego, CA, USA), which provides the opportunity for clinical laboratories to create their own genetic tests.2
The availability of more comprehensive genetic testing greatly increases the problem space. As a result, the practice of personalized medicine is dominated by, and for all intents and purposes synonymous with, modern genetic testing.
The challenge now is no longer the performance of the tests but the decisions around when to perform them and how to interpret their results.3 As the number and complexity of genetic findings far exceed human mental capacity, knowledge bases are needed that can assist with personalized medical decision making. Current knowledge bases are, in turn, derived from sets of individual genomic data residing in large databases.4 This article describes the interplay of input data with knowledge bases, with a case study to illustrate how genomic and nongenomic input data are interpreted with the help of knowledge bases to produce personalized medicine recommendations.
Genomic input data
There are many types of genomic data that can be collected into data sets, such as single nucleotide polymorphism (SNP) data produced by genotyping DNA arrays of various sizes, Sanger sequencing data of individual genes, next-generation targeted sequencing of sets of genes, and nontargeted sequencing of either whole exomes or whole genomes. To focus our perspective paper on a single, well-defined scenario, we chose to consider nontargeted, germline, whole-exome sequencing, as we believe this scenario will be the most prevalent and economical in the clinical context in the coming years.
Although the exome makes up only 1.7% of the whole human genome, it is still large, with approximately 60,000,000 data points (60 MB), depending on the capture kit used (see the McInerney-Leo study5 for a kits comparison). Exome sequencing is popular for a number of reasons. First, the exome is considered the most important part of the genome, as any change in this part has the potential to directly affect protein coding and downstream gene products. Second, it is much easier, quicker, and cheaper to sequence only 1.7% of the genome. Third, downstream data management tasks including storage, annotation, and analysis of variants are simpler and made even easier by storing only the data points that represent variants from some standard sequence. Therefore, unless there is a clear clinical or scientific reason to sequence the whole genome, most institutions offering clinical sequencing confine themselves to exome sequencing. We focus on germline genomic data because, in our view, their acquisition is most mature with respect to clinical use. We acknowledge that somatic genomics are rapidly progressing and are potentially an important future part of personalized medicine.
For our model scenario, we assume that personalized medicine is delivered by an interdisciplinary team that includes a specialist physician (eg, an outpatient endocrinologist), a genetic counselor, and a molecular geneticist.3 We further limit the context to clinical settings, not research studies, and assume that the cost of the exome sequencing was paid by the patient or the health plan (eg, because of a diagnostic odyssey or family relative having a genetic disease warranting exome sequencing of close relatives). We also expect that the patient has several channels by which genetic reports may be communicated. The most significant constraint in our scenario is a review of genetic findings relevant to a specialist, using a specialty-specific genetic report (see Figure 1). Other channels include consultation visits with a genetic counselor covering incidental (off-target) findings outside the specialty in question, as well as a patient-interfacing Web-application for on-demand, self-guided inspection of genomic findings (see Partners Healthcare and Personal Genome Project6–8 for sample genetic reports). In addition, patients may receive notifications if new scientific findings modify their exome interpretation such that a reconsultation visit is required (eg, when genomic knowledge bases receive new updates that trigger new findings reportable to the patient).
Knowledge bases
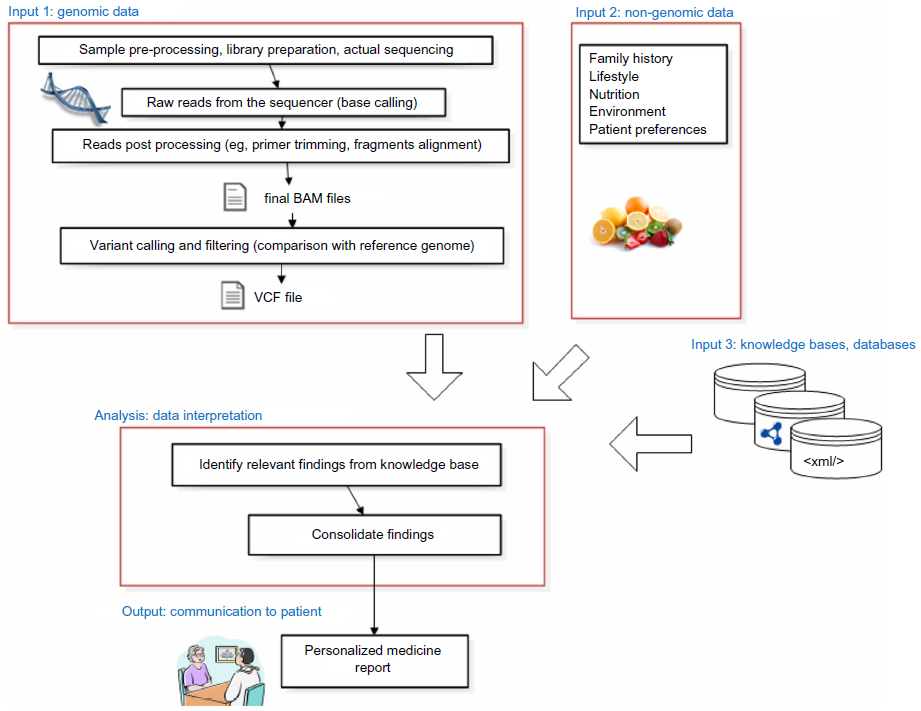 | Figure 1 Data flow in personalized medicine. |
To provide personalized medicine recommendations, knowledge relevant to individual patient data must be obtained. We use the term knowledge bases to characterize resources that include information about the interpretation and implication of specific genomic findings. Knowledge bases typically contain aggregate knowledge and no patient-level data.
Table 1 presents an overview of selected knowledge bases available today, ranging from commercial products such as the Human Gene Mutation Database (HGMD) to freely available resources such as ClinVar. These knowledge bases differ in their level of expert curation and orientation toward practicing clinicians versus molecular geneticists. The ClinGen knowledge base, announced in September 2013, is the most recently created resource specifically targeted for clinical sequencing applications (http://www.iccg.org/about-the-iccg/clingen/).
 | Table 1 Overview of selected genomic knowledge bases and resources |
In terms of content or internal organization, knowledge bases can be primarily structured by publication (PubMed), disease (Online Mendelian Inheritance in Man), gene (Gene Ontology), protein product (Human Protein Atlas), or individual gene position (ClinVar). In many cases, however, knowledge on various levels is related, and multiple organizing principles are needed, as well as links among knowledge bases.
Some knowledge bases serve as a primary source of data about human reference sequences, variants, and functional implications, whereas other resources, such as the University of California Santa Cruz Genome Browser and Ensembl, bring a set of several resources together and present them in an integrated way. This allows users to obtain information from multiple sources within a single platform. A special type of knowledge base is the reference human genome build itself (currently at build 38; http://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26), which provides knowledge on gene and exon/intron boundaries.
In terms of format, knowledge bases differ in how they can eventually be consumed by teams interpreting genomic variants. For example, the Single Nucleotide Polymorphism Database (dbSNP)9 is a relational database that consists of several interconnected tables, including an allele table listing unique alleles across several organisms, an allele frequency table for different subpopulations, and a publication table with PubMed articles linked to a given dbSNP record. Another popular format is eXtensible Markup Language, which is not restricted by relational or spreadsheet paradigms. ClinVar,10 for example, is available as a single file (905 MB) that contains a single hierarchically arranged record per variant that subsumes all related information for that variant.
In terms of function, knowledge bases help reduce the large set of about 20,000 variants that are typically present in an average exome variant call file for a single patient to a smaller set of variants that are clinically important. Unfortunately, no single database currently provides a complete set of variants coupled with a reliable classification of clinical effect.11
Peterson and colleagues recently examined the differences in information covered by several knowledge bases including ClinVar, the Online Mendelian Inheritance in Man, the Universal Protein Resource (http://www.uniprot.org/), and HGMD. They found, for example, that although 619 variants were found in all four knowledge bases, HGMD (the professional version) contained 35,920 variants that were not found in any of the other knowledge bases.12 In another knowledge base comparison analysis, Johnston and Biesecker note that HGMD contains the largest number of records, but they also mention that it requires a license fee and that only ClinVar offers a mechanism for ongoing collection of variant annotation information.11
The ideal knowledge base should offer best-available variant frequency data and consistent curation mechanisms with external dynamic community inputs, including disease-specific teams of practicing clinicians. During the previous 15 years, hundreds of locus-specific knowledge bases (LSKBs) have been created, but many are no longer maintained. The most comprehensive list (at http://www.hgvs.org/dblist/glsdb.html) was last updated in May 2011 and contains entries on 1,550 genes maintained by 314 distinct curating teams. In the future, we expect consolidation of many LSKBs into larger knowledge bases, such as ClinVar or ClinGen,13 with more well-developed maintenance approaches. Multiplicity of knowledge sources and conflicting recommendations can be demonstrated in the ClinVar June 2014 release, which supports retrieval of ClinVar’s variants in the 56 genes listed in the American College of Medical Genetics and Genomics (ACMG) incidental findings guideline.14 Of the total 24,160 ClinVar’s applicable variants, 1,441 of those (6%) come from multiple submitters, and 525 of those (36%) have conflicting recommendations (query http://www.ncbi.nlm.nih.gov/clinvar?term=“gene acmg incidental 2013” with filters for “review status” and “clinical significance”).
The published literature (as represented in PubMed) can be viewed as the ultimate knowledge base. PubMed is often used for final variant curation before interpretation of genomic test results are reported to the patient. However, given a known publication bias, only a fraction of variants will have sufficient data to warrant publication.13
The ability to annotate variants related to polygenic common diseases remains an unsolved challenge. Many experts rightly justify first tackling annotation of highly penetrant variants linked with Mendelian diseases. Such annotation is potentially relevant to as many as 30 million US individuals.13 However, although less feasible, building knowledge base infrastructure and consensus for polygenic diseases or whole-exome pharmacogenomics is potentially relevant to a much larger pool of patients.
Knowledge base companion tools
In addition to creating knowledge bases, additional software tools used in conjunction with knowledge bases are important in the interpretation process. The genome coordinate system used by a particular knowledge base plays an important role in the efficient use of the knowledge for annotating patient variant call files. To illustrate the coordinate system, consider a missense variation in PCSK9 gene on chromosome 1 (rs11591147, G>T). In human build 38, it is the 55,039,974th base, whereas in build 37(patch13), it is located 465,673 bases downstream (on position 55,505,647). With each new human genome build, either every knowledge base must adapt to the new reference sequence or the original genomic data must be converted to match the knowledge base.
Companion tools, such as the Genome Remapping Service15 from the National Center for Biotechnology Information, are crucial for informatics annotation processes dealing with data using past human genome builds.
Other companion software tools, such as the National Center for Biotechnology Information’s Variation Reporter16 or ANNOVAR (which stands for “annotate variation”),17 provide reference (or exemplary) implementation of one or multiple knowledge bases. These tools make knowledge bases readily accessible to genetic counselors and patients because they are self-standing and do not require programming expertise or custom integration. They typically instruct the user to upload a single variant call file and output a list of filtered significant variants based on user-set or some default thresholds.
Patient level databases
In addition to resources that capture medical knowledge, further development of personalized medicine will also require accumulation of long-term outcomes. Large, prospectively collected cohorts of national scale have been proposed.18 Patient level databases, in contrast to knowledge bases, are not limited to aggregated data and contain individual patient genomic data; in addition, in some cases, they also contain limited or lifetime deidentified clinical data. However, genomic data can be considered to be effective personal identifiers. Analyses of SNP data have demonstrated that 30–80 SNP data points can uniquely identify a single person in a theoretical population of 10 billion.19 Because genomic data are highly identifiable, cross-institutional sharing of data sets that have complete patient genotype and deidentified phenotype data remains a legal challenge. Existing patient-level databases, such as the Database of Genotypes and Phenotypes (http://www.ncbi.nlm.nih.gov/gap), limit access to qualified investigators, require study-based approval, and provide only a select subset of phenotypic variables, rather than complete, potentially reidentifiable electronic health record (EHR) data.
However, sharing of patient-level data does exist today at the level of single genes. For example, the Leiden Open Variation Database (http://www.lovd.nl/3.0/home) contains data on 2.2 million variants from 170,000 individuals.13 The BRCA1 and BRCA2 gene variants database from Myriad Genetics is another example, notable for its highly proprietary nature.20 The size of the patient-level database supporting the aggregated, non-patient-level knowledge base greatly determines the ratio of variants of unknown significance ultimately reported to patients. Recent emergence of molecular pathological epidemiology21 as a field that calls for extending traditional epidemiology studies with data on molecular heterogeneity of a given disease also implies the need for large patient-level databases.
Patient-level databases are usually limited to a single institution, such as Vanderbilt University’s Synthetic Derivative22 or UK Biobank.23 However, federated databases are necessary for optimal genomic data annotation. For example, consider an interpretation of an autosomal-dominant variant that has incomplete penetrance and variable expressivity. Without a large database that could provide data on penetrance and severity for a significant population of past carriers of this variant, it is difficult to provide a clear clinical message to a patient who possesses the variant. Repositories of genomic data of phenotypically defined individuals (eg, healthy adults aged 25–35 years or verified individuals without the phenotype in question) can provide much-needed insights into variant penetrance.
An example of an altruistic, patient-powered data set containing phenotypic data combined with genomic data (including facial photographs in some cases) comes from the Personal Genomes Project,24 which to date has released more than 600 exomes with some phenotypic data. Other examples of large patient-level databases include the Veterans Administration’s Million Veteran program25 and the United Kingdom’s 100,000 Genome Project.26 The largest database is the Exome Sequencing Project, funded by the National Heart, Lung, and Blood Institute, which has variant information on more than 7,500 patients. Aggregated data organized by genetic variant are available publically via the Exome Variant Server (http://evs.gs.washington.edu/EVS/). Complete data (subject to request approval) are available via the Database of Genotypes and Phenotypes (see study dbGaP:phs000281.v5.p3 for example).
In terms of classification, there are essentially two kinds of patient level databases: databases that enable public access and greatly limit genotype and phenotype data and databases with project-based or user-based access control that use data use agreements to govern access to more detailed (and sensitive) data. Federated exchanges of genotype–phenotype data sets remain in the proposal stage.27 In the future, we can expect increased pressure to provide nonresearch users with access to patient-level databases, new innovative approaches for access control and user credentialing, and new patient consent policies.
In terms of function, person-level databases with lifetime patient follow-up are important for determining frequency, penetrance, and other parameters of the knowledge bases13 and for long-term validation of the recommendations generated by the knowledge bases (eg, did the predicted outcome truly occur?). The use of larger databases to validate and update existing knowledge bases is a constant iteration cycle.
In terms of format, patient-level databases differ widely in their format eventually exposed to investigators. In addition, because of complex privacy issues, there has been a much smaller scale of use. Predominant use is by researchers, rather than in the context of a single clinical encounter. Because of that, there is limited standardization of phenotypic data. For example, there is only limited standardization of phenotypic variables across different studies within the Database of Genotypes and Phenotypes.
Case study with incidental findings
To understand the use of some of the knowledge bases mentioned earlier, consider a recent exome sequencing research project carried out by the Undiagnosed Diseases Program (UDP) at the National Institutes of Health. Within this project, a total of 543 patients from 159 families underwent exome sequencing. Although it was done within a research context focusing on undiagnosed rare diseases, the clinical implications of off-target incidental findings (limited to the 56 genes identified by the ACMG guideline14) were returned to the attending physician. A detailed report by Lawrence and colleagues on the UDP team’s experience is published separately.28 The UDP team started with an examination of 56 genes in 543 exomes and found a total of 5,948 variants. This number was reduced to 996 mutations after filtering out mutations that were not present in any database or that had been documented to have no known significant biologic consequence.
For 250 of the 996 variants (25.1%), the UDP team performed manual curation (in the form of a literature search) and found that existing knowledge bases generally do not provide complete information. They primarily used HGMD (DM type of mutation), dbSNP, ClinVar, and locus-specific databases registered in the Leiden Open Variation Database. For unregistered locus-specific databases, annotations were manually collected from the individual databases and used to annotate the variants on the basis of matching Human Genome Variation Society nomenclature.28 Although the ACMG recommendation clearly enumerates 56 genes associated with addressable diseases, it does not provide information at the level of the variants and does not unambiguously identify which variants within the 56 genes need to be reported to patients. Variants of unknown significance, that is, those not found in any database, are especially troublesome.
The UDP established diagnoses in approximately 24% of the 160 cases seen during the first 2 years. In addition to diagnosing several common disorders on clinical grounds, the UDP defined two new disorders and identified 21 rare diagnoses in 23 individuals on molecular or biochemical bases. Whole-exome sequencing proved critical for the diagnosis of six different disorders.29 The UDP sequencing study did not address the value of the sequencing for the patient, but a recent review of clinical sequencing30 concluded that clinical exome or genome sequencing (with a cost between $4,000–$15,000) is only two to four times more expensive than a single-gene sequencing and that it can be the most efficient test. They also point out that even if sequencing test results do not change prognosis or clinical management, they may at least allow termination of a potentially expensive and invasive diagnostic odyssey.
We acknowledge that the selected case study does not fully satisfy our model scenario (nonresearch sequencing funding source and genetic counselor visit with long-term updates provided to patients); however, it does represent direct experience with genomic interpretation performed locally at our institution and partially demonstrates the points discussed earlier. This perspective paper also draws on our experience with delivering off-target findings as part of the new 2014 Genomics Opportunity project at the National Institutes of Health Clinical Center. The aim of that project is to recruit additional staff (eg, genetics counselors) and enhance bioinformatics infrastructure devoted to clinical sequencing.31
The informatics of future knowledge bases and databases
We see several factors shaping the future of personalized medicine knowledge bases and databases. This section provides an overview of trends or change requirements we can expect in the near future across all three areas discussed earlier.
Knowledge bases
Consolidation of multiple knowledge bases
Although each knowledge base may cover a unique aspect of personalized medicine, for institutions establishing their personalized medicine clinics, the explosion in their overall number is not optimal. With a large number of LSKBs seeking a new knowledge base coordinator (eg, the research laboratory that initiated the knowledge base no longer exists), we expect the consolidation of LSKBs into larger knowledge bases.
Clinical bioinformatics focus
Many current knowledge bases were created with a molecular biologist as a target user. Some current knowledge bases are suitable for use by a genetic counselor, but no current platform directly targets a single specialty clinician as the system user. We see a middle ground in both increasing genetics training for today’s physicians and other clinical staff and developing better user interfaces and modifications in knowledge base structures that make existing platforms more clinician-friendly.
Patient-level databases
Growth and innovations in patient level databases
The interpretation of proper genomic results benefits greatly from databases that contain genotype and long-term phenotype information. Changes to patient consent for transparent enrollment into such databases and rigorous user-access credentialing will be required, as well as a tight semantic integration of existing knowledge bases with patient-level databases.
General
The reference sequence is still a moving target
Each new human genome build improves biologic accuracy but also necessitates significant maintenance of knowledge bases and databases. Emerging genomic remapping tools can mitigate this problem. From 2001–2004, the average lifespan of the reference build was 4.7 months. Since March 2006 (release of build GRCh36.1), the lifespan has increased considerably, to an average of 37 months. The current build (GRCh38, released in December 2013) came 57 months after the previous build. Projecting the trend seen since 2006, we can expect GRCh39 to be released sometime in 2020. This change in the frame of reference requires realignment of old genomic data as well as knowledge bases. Variant identification used by dbSNP (rs identifiers), however, transcends human genome build changes, as it is not identified by genomic position but, rather, by a separate numbering system.
Storage of genomic data within EHRs and personal health record systems
Despite some standardization, such as the Health Level 7 genetic testing report32 or new codes in the Logical Observation Identifiers Names and Codes terminology supporting structured genetic testing reports,33 storage of genomic data and existing genomic reports within today’s EHR systems continues to be a challenge.34 Institutions participating in the Electronic Medical Records and Genomics network provide the best examples of the EHR modifications needed for the provision of point-of-care advice and tracking patient outcomes.35
Conclusion
Practical personalized medicine requires an amount of knowledge that exceeds the capacity of an individual physician’s memory. Developing databases and knowledge bases that facilitate the process of delivering personalized medicine recommendations is inevitable. At this time, several such databases exist, and we see early experience with those. Exome sequencing is increasingly being used as the tool of choice for the identification of deleterious variants responsible for Mendelian diseases.
Existing knowledge bases, with exception of ClinVar and ClinGen, often lack detailed fields on a variant’s clinical effect in the context of a patient presenting with a given variant. Geneticists building existing knowledge bases and databases could collaborate with clinical informatics experts experienced with maintaining long-term phenotypic records.
Transition from a research context (such as clinical trials) to a clinical context (actual patient care) has not yet occurred for a sufficient quantity of patients. Many of the existing databases are neither meant to be used by front-line clinicians nor directly target physicians as primary recipients of the resulting reports. Informatics standards as well as EHR systems still have gaps in matching the functionality of custom-built research systems. However, the newest initiatives, such as ClinGen funding reserved for expert curation of variants’ clinical relevancy and the second generation of larger genomic initiatives resulting in larger patient level databases, will undoubtedly increase the clinicians’ and patients’ awareness of personalized medicine and advance the field.
Disclosure
VH and JJC are supported by the Intramural Research Program of the National Institutes of Health Clinical Center and the National Library of Medicine. MS is supported by the Intramural Research Program of the National Human Genome Research Institute. The authors report no other conflicts of interest in this work.
References
Mills R, Voora D, Peyser B, Haga SB. Delivering pharmacogenetic testing in a primary care setting. Pharmgenomics Pers Med. 2013;6:105–112. | |
FDA-approved Next-Generation sequencing system could expand clinical genomic testing: experts predict MiSeqDx system will make genetic testing more affordable for smaller labs. Am J Med Genet A. 2014;164A(3):x–xi. | |
Biesecker LG, Burke W, Kohane I, Plon SE, Zimmern R. Next-generation sequencing in the clinic: are we ready? Nat Rev Genet. 2012;13(11):818–824. | |
Overby CL, Kohane I, Kannry JL, et al. Opportunities for genomic clinical decision support interventions. Genet Med. 2013;15(10):817–823. | |
McInerney-Leo AM, Marshall MS, Gardiner B, et al. Whole exome sequencing is an efficient, sensitive and specific method of mutation detection in osteogenesis imperfecta and Marfan syndrome. Bonekey Rep. 4, 2013;2:456. | |
Partners Healthcare. MedSeq project. 2014. Available from: http://www.genomes2people.org/wp-content/uploads/2013/09/GGR_sample.pdf. Accessed April 18, 2014. | |
Personal Genome Project. Exome Results & Raw Data Summary. 2014; Available from: https://my.pgp-hms.org/user_file/download/501. Accessed April 30, 2014. | |
Personal Genome Project. GET-Evidence variant report. 2014; Available from http://evidence.pgp-hms.org/genomes?display_genome_id=4a7464a2bb7bca0ca2e602994f228dc6889a5faa. Accessed April 29, 2014. | |
Sayers EW, Barrett T, Benson DA, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011;39(Database issue):D38–D51. | |
Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–D985. | |
Johnston JJ, Biesecker LG. Databases of genomic variation and phenotypes: existing resources and future needs. Hum Mol Genet. 2013;22(R1):R27–R31. | |
Peterson TA, Doughty E, Kann MG. Towards precision medicine: advances in computational approaches for the analysis of human variants. J Mol Biol. 2013;425(21):4047–4063. | |
Cutting GR. Annotating DNA variants is the next major goal for human genetics. Am J Hum Genet. 2014;94(1):5–10. | |
Green RC, Berg JS, Grody WW, et al; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–574. | |
National Center for Biotechnology Information. NCBI Genome Remapping Service. National Center for Biotechnology Information; 2014. http://www.ncbi.nlm.nih.gov/genome/tools/remap. Accessed May 4, 2014. | |
National Center for Biotechnology Information. Variation Reporter. National Center for Biotechnology Information; 2014. http://www.ncbi.nlm.nih.gov/variation/tools/reporter. Accessed May 6, 2014. | |
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. | |
Green ED, Guyer MS, National Human Genome Research I. Charting a course for genomic medicine from base pairs to bedside. Nature. 2011;470(7333):204–213. | |
Lin Z, Owen AB, Altman RB. Genetics. Genomic research and human subject privacy. Science. 2004;305(5681):183. | |
Cook-Deegan R, Conley JM, Evans JP, Vorhaus D. The next controversy in genetic testing: clinical data as trade secrets? Eur J Hum Genet. 2013;21(6):585–588. | |
Ogino S, Lochhead P, Giovannucci E, Meyerhardt JA, Fuchs CS, Chan AT. Discovery of colorectal cancer PIK3CA mutation as potential predictive biomarker: power and promise of molecular pathological epidemiology. Oncogene. 2014;33(23):2949–2955. | |
Roden DM, Pulley JM, Basford MA, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clinical pharmacology and therapeutics. 2008;84(3):362–369. | |
Genomics England. 100k Genome Project. 2014. Available from: http://www.genomicsengland.co.uk. Accessed May 7, 2014. | |
Ball MP, Bobe JR, Chou MF, et al. Harvard Personal Genome Project: lessons from participatory public research. Genome Med. 2014;6(2):10. | |
VA. Milion Veteran Program. 2014. Available from: http://www.research.va.gov/MVP. Accessed May 2, 2014. | |
UK Department of Health. The 100,000 Genomes Project. 2014. Available from: http://www.genomicsengland.co.uk/the-100000-genomes-project. Accessed May 7, 2014. | |
GlobalAliance. Creating a Global Alliance to Enable Responsible Sharing of Genomic and Clinical Data. 2013. Available from: http://www.broadinstitute.org/files/news/pdfs/GAWhitePaperJune3.pdf. Accessed April 24, 2014. | |
Lawrence L, Sincan M, Markello T, et al. The implications of familial incidental findings from exome sequencing: the NIH Undiagnosed Diseases Program experience. Genet Med. 2014. | |
Gahl WA, Markello TC, Toro C, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14(1):51–59. | |
Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014;370(25):2418–2425. | |
Clinical Center Genomics Opportunity (project overview). 2014; http://www.genome.gov/27557011. Accessed May 14, 2014. | |
Bosca D, Marco L, Burriel V, et al. Genetic testing information standardization in HL7 CDA and ISO13606. Studies in health technology and informatics. 2013;192:338–342. | |
HL7. HL7 Implementation Guide: Clinical Genomics: Fully LOINC-Qualified Genetic Variation Model, Release 2. 2013; http://www.hl7.org/documentcenter/private/standards/v251/V2IG_CG_ LOINCGENVAR_R2_INFORM_2013MAR.pdf. Accessed April 10, 2014. | |
Tarczy-Hornoch P, Amendola L, Aronson SJ, et al. A survey of informatics approaches to whole-exome and whole-genome clinical reporting in the electronic health record. Genetics in medicine: official journal of the American College of Medical Genetics. Oct 2013;15(10):824–832. | |
eMERGE Network Workshop (January 22, 2014): Future Directions for the eMERGE Network. http://www.genome.gov/Pages/About/OD/OPG/eMERGE2014/eMERGE_FullSummary.pdf. Accessed May 25, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.