Back to Archived Journals » Cell Health and Cytoskeleton » Volume 8
Desmosome assembly, homeostasis, and desmosomal disease
Authors Cirillo N
Received 13 October 2015
Accepted for publication 14 December 2015
Published 29 February 2016 Volume 2016:8 Pages 9—23
DOI https://doi.org/10.2147/CHC.S59510
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Denis Wirtz
Nicola Cirillo1,2
1Melbourne Dental School and Oral Health CRC, The University of Melbourne, Melbourne, VIC, Australia; 2Research Unit, Centre for Innovation, Research, Education, and Health (IRIS), Caposele, Italy
Abstract: Cell–cell adhesion is involved in all aspects of tissue behavior in multicellular organisms, from tissue morphogenesis (regulation of cell shape, apoptosis, cell movement, and development of complex structures) to aging and disease. A major player in the dynamic regulation of intercellular contacts is the desmosome. Knowledge of the desmosome has evolved over 150 years from the notion of a static, punctuate, adhesive barrier structure to one of the finely tuned multifunctional complexes involved in the regulation of numerous and diverse aspects of keratinocyte physiology and disease. In this context, nondesmosomal regulatory molecules have been acquiring increasing importance in the study of desmosome homeostasis and have become part of the extended desmosomal interactome named "desmo-adhesome". Among these associated molecules, kinases are the prominent regulators of both desmosome remodeling and acquisition of hyperadhesion, two novel concepts in cell–cell adhesion. Spatiotemporal changes in the expression and regulation of desmosomal proteins also underlie a number of genetic, infectious, autoimmune, and malignant conditions. In addition to offering a systems-level view of the molecular composition of desmosomes, we also discuss the mechanisms that regulate, and disrupt, desmosome homeostasis.
Keywords: cell adhesion, desmo-adhesome, pemphigus, cancer
Introduction
When Bizzozero described small dense nodules at the contact points between adjacent cells in 1864,1 the young Italian pathologist arguably did not imagine that >6,000 articles would be published about this organelle in the following 150 years. These adhesive cell–cell contact points were named desmosome by Josef Shaffer in 1920, from the Greek words “desmos” meaning “bond” and “soma” meaning “body”.2,3 Later, electron microscopy (EM) allowed the characterization of these “spot welds” as pairs of electron-dense attachment plaques, one for each adjacent cell. In the 1970s, the development of procedures to isolate intact desmosomes from tissues followed by the generation of specific antibodies against such desmosomal proteins led to the biochemical characterization and immunolocalization of the major protein components of the desmosomes.4 In the last 3 decades, cDNA cloning techniques and the advances in cell and molecular biology have clarified the structure and deduced the amino acid sequence of major desmosomal proteins.
However, it was not until recently that desmosomes have started to be regarded as highly dynamic structures involved in all aspects of epidermal physiology and pathophysiology, from tissue morphogenesis to aging and disease.5–7 Further insight into the understanding of desmosome function and regulation has come from the study of desmosome-targeting diseases of genetic, autoimmune, and infectious nature.8
Structure–function relationship
Intercellular cohesion is important at all levels in the human body but becomes an essential requirement in tissues subjected to mechanical stress, such as the epidermis.
Desmosomes are the principal intercellular junctions of stratified epithelia that ensure epithelial tissue homeostasis.9
Freeze-fracture EM reveals that desmosomes are disk-shaped structures of 0.2–0.5 μm in diameter consisting of a compact aggregation of intramembranous protein particles.10 These membrane-bound, electron-dense structures link intermediate filaments (IFs) to the plasma membrane region between cells.11
Ultrathin section electron microscopic observations demonstrate that desmosomes consist of a central electron-dense midline between two plasma membranes of adjacent cells (desmoglea) and dense plaques, ie, the outer and inner dense plaques, forming the symmetrical desmosomal structure (Figure 1A and B). Large bundles of IFs extend from the nuclear surface and cell’s interior out toward the plasma membrane, where they attach to desmosomes by interweaving with the cytoplasmic plaque of the adhesive complex.12 Thus, the cytoskeletal IF network is attached to the plasma membrane at the desmosomal plaque,13 and this multilevel structure is termed as desmosome/IF complex.
 | Figure 1 Schematic representation of desmosome and its components. |
With regard to size, the desmoglea encompass ~28 nm between the adjacent plasma membranes of adjacent cells, which appear as a pair of parallel black lines with a clear white gap of 3.5 nm. The outer dense plaque, which attaches to the inside of the plasma membrane with a slightly reduced electron-dense appearance, is ~20 nm in thickness.10 Forming a lesser electron-dense gap between the outer dense plaque is the inner dense plaques, also ~20 nm in thickness, and attached to the inside of the outer dense plaque (Figure 1).
The overall adhesive function of the desmosome is dependent upon the tethering of IFs to the desmosomal plaque, highlighting the integrated functions of adhesion and cytoskeletal elements. Thus, desmosomes are modular structures comprising adhesion molecules that bolt cells together, cytoskeletal cables that disperse forces, and linking molecules at the cytoplasmic plaque of the desmosome that carry mechanical load from the adhesion molecules to the IF cytoskeleton.12
Principal molecular constituents of the desmosome
In electron microscopical images, the center of the desmosomal junction is composed of the extracellular regions of cadherin family members, and a dense midline of their interleaved N termini runs through this. Just inside the plasma membrane is the outer dense plaque, which contains the armadillo-repeat protein family members, namely plakoglobin (Pg) and plakophilins (Pkps). The intracellular domains of the cadherins – namely desmogleins (Dsgs) and desmocollins (Dscs) – contribute to the outer dense plaque, as Pg and Pkps do. Beyond this lies a translucent zone and a further inner dense plaque, ie, composed largely of the plakin family members, including desmoplakin (Dsp), plectin, envoplakin (EVP), and periplakin (PPL) (Figure 1). Together, these proteins provide a highly organized supermolecular assembly that mediates stable yet adaptable mechanical coupling between the points of cell–cell adhesion and the cytoskeleton.14,15 Knockout studies in mice indicate that Dsp is the only component essential for the formation of desmosome-like structures.16–21
The intercellular components: desmosomal cadherins
Desmosomal cadherins are single-pass transmembrane adhesion molecules at their extracellular N-terminal domains. There are at least four different Dsg isoforms (Dsg1–4) and three different Dscs (Dsc1–3).22 The seven genes for human desmosomal cadherins are clustered on chromosome 18q12.1.23–25 In contrast to Dsgs, alternative splice variants of the Dscs affecting their C-terminal tails have been observed resulting in a long form (Dsc1–3a) and a shortened variant (Dsc1–3b). With respect to Dsgs, the expression of Dsg1 and Dsg3 isoforms is restricted to stratified squamous epithelia, whereas the Dsg2 isoform is expressed in all cells that make desmosomes.26 In the epidermis, Dsg1 (160 kDa) is expressed in the upper cell layers, while Dsg3 (130 kDa) is present in the lower cell layers.27 Dsg4 is expressed in the more highly differentiated layers of the epidermis, and specifically in the hair shaft cortex, the lower hair cuticle, and the upper inner root sheath cuticle.28 Similar to Dsgs, Dscs are expressed in tissue-specific and differentiation-dependent patterns in adult epithelial tissues.29
The overall structures of Dsgs and Dscs are highly homologous and related to E-cadherin, the classical prototype and ancestor of cadherins. The processed extracellular domain of desmosomal cadherins is composed of five extracellular cadherin (EC1–5)-repeat domains with three Ca2+-binding sites in between these EC repeats (Figure 1C). The fifth and more divergent membrane proximal repeat (EC5) sometimes is also referred to as extracellular anchor domain. A single transmembrane domain links the extracellular domain with a cytoplasmic tail assembling a specific set of desmosomal plaque proteins important for the correct assembly and organization of desmosomes, for the association with IFs, and for signaling. The major difference between Dsgs and Dscs resides in their cytoplasmic domains. A membrane proximal to the intracellular anchor domain and an intracellular cadherin segment (ICS) domain are common to both Dsgs and Dscs (except for the short Dscb splice variants). Dsg intracellular domains are extended by an intracellular proline-rich linker region, a repeated unit domain containing different numbers of 29±1 amino acid repeats, and a glycine-rich Dsg-terminal domain (DTD) at the C terminus30,31 (Figure 1C).
The desmosomal plaque components
In electron microscopical images, IFs attach to desmosomes at electron-dense cytoplasmic plaques of multiple proteins. The major plaque proteins can be assigned to two families, the armadillo-repeat protein family32,33 and the plakin protein family.34,35 Members of both families contain multiple protein–protein interaction motifs mediating their complex assembly at the desmosomal plaque. In addition, some of the proteins appear to be involved in signaling processes. The desmosomal armadillo-repeat proteins include Pg, Pkp1, -2, and -3, and p0071,36,37 occasionally designated as Pkp4 (Figure 1). Both Pg and p0071 localize to adherens junctions and desmosomes38 and have been proposed to be involved in the cross talk between these junctions. Four plakin family members, namely Dsp, plectin, EVP, and PPL, have been found at desmosomes.
Many of the junctional armadillo-repeat protein family members have been localized to cell–cell contacts and in the nucleus39 and, therefore, are assumed to have dual functions as best studied for β-catenin and its Drosophila homolog armadillo. A more detailed discussion of the signaling function of Pg is provided in a comprehensive review by Yin and Green.40 Pg (86 kDa) directly binds to the desmosomal cadherins except to Dscb variants, which only include part of the ICS domain without the Pg interaction site.41,42 At desmosomes, Pg acts as an adaptor to assemble further plaque proteins, including Dsp,43,44 Pkp2 and -3,45,46 and p0071,38 and functions as a bond to make robust lateral aggregations of desmosomal cadherins. The analysis of a Pg construct with deleted C-terminal domain suggests that Pg somehow participates in the regulation of desmosomal size.47 With respect to its signaling function, initial studies suggested that Pg similar to β-catenin activates transcription in association with LEF-1/TCF and Pg is also an important component of adherens junctions, where it links E-cadherin to actin.48 Since Pg is a component common to both desmosomes and adherens junctions, it may play a critical role in the cross talk between these two junctional structures in the regulation of cell–cell contacts.49
Pkps are the members of a subfamily of armadillo proteins that were named according to the founder member (p120-catenin) of the p120 family of armadillo proteins. This family comprises seven members, four of which are found primarily in adherens junctions (p120-catenin, NPRAP/δ-catenin, armadillo-repeat gene deleted in velo-cardio-facial syndrome, and p0071/Pkp4), whereas Pkp1–3 localize primarily at desmosomes.50 Pkp1 (75 kDa) is a major desmosomal component expressed in the suprabasal cells of the epidermis, binds to Dsp1 and KIF in vitro,51 and also takes part in the lateral interactions of Dsp and desmosomal cadherins.52 As for desmosome regulation, it should be noted that desmosomes of patients with skin fragility syndrome, which completely lack Pkp1, are characterized by widened intercellular spaces and smaller desmosome size, associated with KIF detachment from desmosomes and minor trauma-induced blistering.53 It is also of interest to note that Pkp1 interferes with Pg binding to Dsp, yet together with Pg, promotes clustering of desmosomal plaque complexes at cell borders.54 Pkp1 contains two functionally distinct domains, ie, the head domain, which could play a role in organizing the desmosomal plaque, and the armadillo-repeat domain, which might be involved in regulating the actin dynamics.50 Recently, in a cell culture system (A431DE cells) that expresses all proteins necessary for desmosomal assembly, except Pkp1, exogenous expression of Pkp1 allowed the desmosomes to be assembled. In addition, deletion mutagenesis experiments revealed that amino acids 686–726 in the carboxyl terminus of Pkp1 were required for its localization to the plasma membrane, while amino acids 1–34 in the amino terminus were necessary for the subsequent recruitment of Dsp to the membrane and desmosome assembly.55 Moreover, loss of Pkp1 causes a reduction in size and number of desmosomes, resulting in ectodermal dysplasia/skin fragility syndrome.53 Thus, it has been suggested that Pkp1 plays an important role in desmosome stability and/or assembly.
Plakins of keratinocyte desmosomes, ie, Dsps, EVP, and PPL, are the multidomain proteins that link the desmosomal cadherin–Pg complex to KIF.34,56 Dsps include at least two isoforms, namely Dsp1 and Dsp2, with molecular weights 250 and 220 kDa, respectively. The N terminus of the Dsps is embedded in the outer dense plaque and reaches the inner dense plaques of desmosomes. This N-terminal globular domain, namely the plakin domain, binds to desmosomal cadherins through Pg and consists of two pairs of spectrin repeats separated by a Src homology 3 domain.57 The globular C terminus of Dsp binds to KIF, and the N terminus binds to Pg, Pkps, and Dsc; they form a parallel dimer through the central coiled-coil domain.57 In this regard, epidermal-specific Dsp-knockout mice reveal that Dsp is essential in epidermal sheet formation.21 In these animals, the number of desmosomes is similar to those in wild type, but these desmosomes lack KIF binding (whereas it is reduced in the cultured conditions).
Desmosome-associated molecules and the desmo-adhesome
The evolution of the notion of desmosomes from static, punctuate, adhesive barrier structures to finely regulated multifunctional complexes resulted from the realization that regulatory molecules play a fundamental role in the homeostasis of cell adhesion structures,58–60 including desmosomes.61 An insight into the mechanisms that regulate the dynamics of keratinocyte adhesion, therefore, requires not only an understanding of desmosomal structure and function but also an understanding of the regulation of its component parts and their responses to perturbations.
In our studies describing for the first time the desmosomal network,61 we distinguished between intrinsic nodes, or proteins, (ie, structural components that have been reported to physically reside within the desmosome) and accessory proteins (eg, kinases and regulatory proteins), which interact directly with the intrinsic components in a junctional context (Figure 2).
 | Figure 2 Accessory components of the desmo-adhesome. |
Through data mining, we first used information derived from databases and published studies61 to address the molecular architecture, structure, and signaling of the desmo-adhesome at multiple levels. Dissection of the network into distinct, yet mutually interacting, families highlighted the functional role of certain components and their regulations. Importantly, we distinguished between binding interactions, which are always nondirectional, and signaling interactions, which are directional, either activating or inhibiting. This regulatory subnet was formed by membrane and adaptor proteins together with enzymes. This subnet included 28 accessory proteins, including kinases, phosphatases, and caspases.61,62 Many of these molecules, such as PKC, Src, MAPK, epidermal growth factor receptor (EGFR), and caspase 2/3, are now known to be involved in the assembly and disassembly of desmosomes.8,12,63,64 It is interesting to note that Pg, one of the main organizers of the desmosome that is crucial for adhesiveness, was highly connected to both protein kinases (n=5) and phosphatases (n=4), and this provided an in silico demonstration that the dynamic regulation of the desmosome may be orchestrated by this protein.61
At the time that the desmo-adhesome was published in 2009, it was well accepted that adherens junctions cooperated with desmosomes in providing tight intercellular adhesion. However, the exact molecular correlation and cross talk between these two adhesive structures were yet to be demonstrated. The inclusion of the E-cadherin-binding protein β-catenin and Src in the desmosomal interactome was therefore regarded with skepticism by some researchers. Now we know that a molecular cross talk does exist and that, for example, E-cadherin and Src associate with extradesmosomal Dsg3 and modulate desmosome assembly and adhesion.65 Thus, the desmosome-associated regulatory network should be fully regarded as a constituent part of the desmosome.
The dynamic desmosome: hyperadhesion and remodeling
Switching the calcium concentration from low (<0.1 mM) to high (~2 mM) in culture media has long been known to regulate desmosome assembly in vitro and convert, or perhaps differentiate, basal-like keratinocytes into suprabasal-like squames. In such model systems, desmosomes have been detected 5–15 minutes after the addition of physiological concentrations of calcium to the medium of various human keratinocyte cell lines (eg, HEKa cells) and MDCK (keratinocyte-like cells) cell cultures.66–69
Early biochemical studies revealed that the desmosomal cadherins and Pg cofractionate, while Dsp is a part of distinct multiprotein complexes that separately traffic to cell–cell contacts.12,69
Assembly of desmosomal membrane molecules occurs in several steps. In this process, transport of Pg to the cell border is required for the initiation of desmosome assembly both in vitro and in vivo. The first step is the transport of a population of Dsc2-enriched vesicles to the plasma membrane to initiate assembly. Pkp2 is required for kinesin-2-dependent trafficking of Dsc2, coordinating its fast transport and accumulation at the membrane. A population of vesicles enriched in Dsg2 traffic to the membrane a short time later, and their delivery to the plasma membrane likely requires kinesin-1.70
With regard to desmosomal plaque, cell–cell contact initiates desmosomal plaque assembly through three phases. In Phase I, Dsp accumulates at newly forming cell contacts beginning approximately 5 minutes, after E-cadherin, but around the time Dsg2 appears.71 Within 15–20 minutes, nonmembrane-bound Dsp-containing particles appear in the cortical region of the cell associated with the IF cytoskeleton; however, what signals the formation of these particles is unknown. Lastly, these precursors subsequently translocate to cell–cell contacts in Phase III to bolster the plaque in an MT-independent manner.72 Pkps play a key role in the temporal regulation of these steps.73
Disruption of desmosomes also occurs in an orderly fashion and in early stages may be induced either artificially by calcium chelation or during certain diseases, such as the autoimmune disease pemphigus. However, after maturation, the dismantling of desmosomes becomes more difficult. The term “hyperadhesion” was coined to describe this stronger adhesive state exhibited by desmosomes both in vitro and in vivo. Hyperadhesion functions to maintain tissue integrity and plays an important role in development, wound healing, and skin disease.74–76 No detectable changes in desmosome composition have been associated with hyperadhesive desmosomes. Comparison of the ultrastructure of desmosomes in normal and wound-edge epidermis showed that those of normal epidermis that were hyperadhesive exhibited very prominent midlines, whereas those at the wound edge, many of which were calcium dependent generally lacked midlines, and the intercellular space appeared amorphous.77,78
Upon wounding, desmosomes lose their hyperadhesive state in response to recruitment and activation of PKC to the wound edge.78 Protein phosphorylation both positively and negatively regulates desmosome assembly. For instance, following calcium-induced assembly, Dsc3 binds Pg and subsequently becomes serine phosphorylated by a currently unknown kinase(s). This is followed by Dsc3 interaction with Dsg3, thereby promoting desmosome formation.79 Activation of Src has recently been reported to be crucial for desmosome formation.65 Activation of PKCα has been previously reported to stimulate desmosome formation in low-calcium conditions or in the absence of adherens junctions. EGFR expression levels and tyrosine phosphorylation status of the cadherin tails and Pg constitute another mechanism to modulate desmosome assembly and stability.73
In summary, regulated desmosome remodeling by differential expression and postsynthetic modification of desmosome components is critical for normal epidermal structural integrity, especially that of cell–cell junctions, and epidermal differentiation. Regulation of desmosomal assembly and disassembly, ie, desmosome remodeling, appears to include both inside-out and outside-in signaling mechanisms that cross talk with adherens junctions. Impairment of desmosome remodeling is now thought to generate a number of cutaneous diseases, including the different forms of pemphigus, as well as Hailey–Hailey and Darier’s diseases. Even molecular structural deficiencies due to mutations of desmosome component genes, which cause a variety of diskeratosis, such as palmoplantar keratosis, wooly hairs, skin fragile syndrome, and even cardiomyopathy, are thought to generate aberrant activation of signaling pathways, which may then result in impairments of desmosome remodeling.64,80 Hence, a review of the diseases caused by desmosome dysfunction warranted to fully appreciate the importance of this organelle in humans (Figure 3).
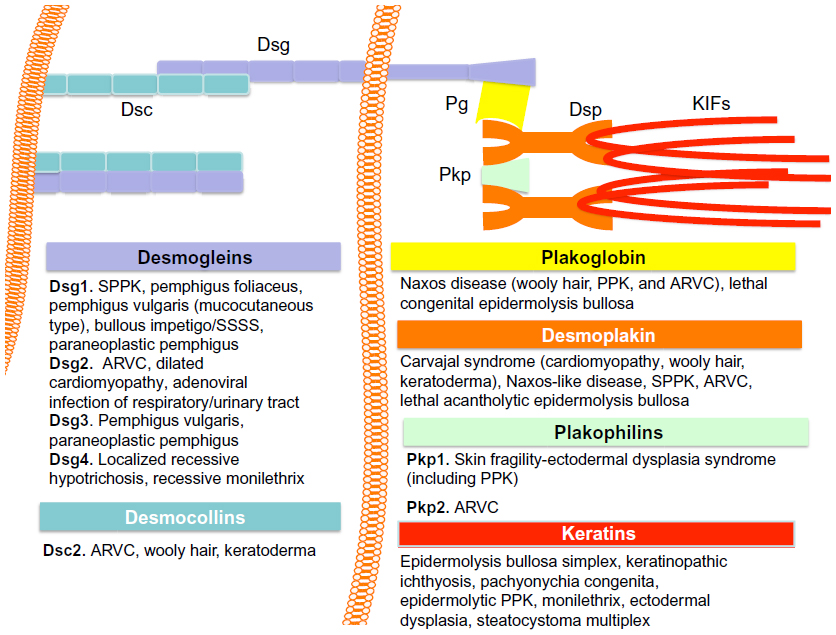 | Figure 3 Desmosome and disease. |
Desmosome and disease
Genetic disease of the desmosome
Over the past 15–20 years, inherited disease has revealed the fundamental principles of desmosome biology and continues to provide laboratory-based scientists with the lines of enquiry that provide greater insight into the mechanisms of these fascinating structures.
Plakophilin 1
Immunohistochemical analysis of skin biopsies from a patient displaying skin fragility coupled with an ectodermal dysplasia phenotype showed a complete absence of Pkp1, disruption of the keratin IF network, and distribution of perturbed Dsp.53 Mutation analysis from this patient’s genomic DNA revealed compound heterozygous premature termination codons within the Pkp1 gene. Further cases of this disorder, known as ectodermal dysplasia–skin fragility syndrome, have been reported, all of which are associated with recessively inherited mutations in the Pkp1 gene.82–84 At least eleven different mutations in this gene are currently known, all leading to similar clinical features of the affected individuals. The most common characteristics of this disorder are widespread skin fragility (with trauma-induced erosions and blistering), alopecia, palmoplantar keratoderma (PPK), nail dystrophy, and in some cases hypohidrosis. Desmosomes are still formed and retain several ultrastructural characteristics,53,85,86 and there is some, albeit compromised, intercellular adhesion throughout the epidermis without Pkp1. However, the fact that the skin of these patients cannot withstand minor trauma illustrates the important role of Pkp1 in cell adhesion.
Plakophilin 2
Pkp2 exists in two alternatively spliced variants (2a and 2b) and is expressed in all tissues that have desmosomes and also in some cell types lacking desmosomes. Pkp2 is the only Pkp isoform expressed in the myocardium.87 Gerull et al88 identified 25 different dominant Pkp2 mutations in 32 out of the 120 unrelated probands with arrhythmogenic right ventricular cardiomyopathy (ARVC). ARVC is a genetic disorder that results in fibro-fatty replacement of right ventricular myocytes and consequent ventricular arrhythmias,89 not infrequently leading to unexpected sudden death in the young, particularly athletes.90 Subsequent reports have confirmed the high prevalence, with reduced penetrance and variable expressivity, of dominant mutations in Pkp2 in a large series of ARVC probands.91–93 At present, 49 dominant mutations have been identified.
Whereas human Pkp1 mutations manifest in the skin, Pkp2 mutations only were seen to cause ARVC. Mutations in the Pkp1 gene lead to impaired association of keratin IFs with the membrane in keratinocytes and disrupted Dsp localization. In parallel, lack of Pkp2 in mouse cardiomyocytes causes IFs to retract from the membrane and Dsp to form cytoplasmic aggregates. These observations underscore the essential roles of both Pkp1 and Pkp2 in mediating Dsp association with desmosomes in the epidermis and heart, respectively.
Plakophilin 3
Although Pkp3 is a ubiquitous protein, no human mutations have yet been identified in the Pkp3 gene. Mice deficient in Pkp3 show morphological abnormalities of several subsets of hair follicles (HFs) and are prone to dermatitis.94 In these Pkp3-deficient mice, desmosomes are absent from the basal layer of the outer root sheath of HFs and from the matrix cells that are in contact with dermal papillae. In the basal layer of Pkp3-null epidermis, densities of desmosomes and adherens junctions were remarkably altered. Compensatory changes in several junctional proteins were observed. Pkp3-null mice housed in conventional facilities were prone to dermatitis.
Plakoglobin
Transgenic mice homozygous for a null mutation in the Pg gene die from embryonic day E10.5 onward, due to severe heart defects. Hearts of mice lacking Pg have no detectable desmosomes, as opposed to embryonic epithelia. Thus, Pg seems to be an essential component of cardiac but not of epithelial desmosomes.95 Consistent with the important roles of Pg revealed by mouse-knockout models, was the finding that the autosomal recessive Naxos disease mapped to 17q21.2, a locus that was known to include the Pg gene.96 The clinical features of this disorder comprise wooly hair, PPK, and ARVC. Recently, it was reported that the lack of Pg can also lead to a novel clinico-genetic entity belonging to a group of lethal congenital epidermolysis bullosa.
Desmoplakin
Human Dsp mutations can be inherited in both dominant and recessive modes of transmission. The resultant disorders range from relatively mild skin conditions caused by Dsp haploinsufficiency to severe fragility and morphogenetic defects in the integument of a neonatal patient or ARVC. In some cases, wooly hair is also seen. The first reported Dsp mutations were both autosomal-dominant nonsense mutations leading to haploinsufficiency.97,98 These mutations caused a striate form of palmoplantar keratoderma (SPPK), rather than skin fragility or ectodermal dysplasia syndrome, but the clinical features were also exacerbated by mechanical trauma. Histology and ultrastructural examination of the affected palmoplantar skin revealed a reduction in the number of desmosomes, accompanied by widening of intercellular spaces between suprabasal keratinocytes, and retracted KIFs.99
A subsequent report described another patient with compound heterozygosity for two mutations (6079C>T; R1934X/6370delTT; X2058) in the C terminus of Dsp, resulting in expression of a truncated protein that lacked the entire IF-binding domain. These mutations caused a lethal condition named “lethal acantholytic epidermolysis bullosa”.100 The patient presented with complete alopecia, neonatal teeth, and nail loss. This individual died shortly after birth from massive transcutaneous fluid loss as a result of severe fragility of skin and mucous membranes. EM revealed the presence of relatively normal desmosomes, but keratin IFs were retracted toward the nucleus, and desmosomes were often torn out of adjacent cells due to the lack of cytoskeletal attachment to the plaque. These observations indicate that the Dsp N terminus is sufficient for desmosome morphology and tissue development, allowing embryonic survival.
The first human autosomal recessive mutation in the Dp gene leading to a cardiocutaneous phenotype was described by Norgett et al.101 The phenotype was a combination of SPPK, wooly hair, and dilated left ventricular cardiomyopathy, resulting in heart failure early in life. Following an earlier clinical description of the same family,102 this phenotype is now referred to as the Carvajal syndrome. Another C-terminal missense mutation (G2375R) leads to defects in skin, hair, and heart (Naxos-like disease), when inherited in a homozygous fashion.103
All known Dsp mutations leading to ARVC without skin or hair involvement are inherited in a dominant fashion.104
Desmoglein 1
Nine dominant mutations in the Dsg1 gene leading to SPPK have been identified so far.105–107 Milingou et al108 reported a dominantly inherited heterozygous single base insertion in exon 3 of Dsg1 (121insT) leading to a premature termination codon. Interestingly, the clinical features of patients harboring this mutation are a focal nonstriated form of PPK associated with discrete keratinization at sites exposed to mechanical trauma, such as the knees, ankles, and finger knuckles, and with mild nail dystrophy. In addition, Keren et al109 reported a family affected with a diffuse nonstriated form of PPK that harbored a recurrent mutation in Dsg1, previously associated with cases of SPPK. These studies reveal that mutations in Dsg1 are not exclusively associated with SPPK.
Desmoglein 2 and Desmocollin 2
The identification of mutations in Pg, Dsp, and Pkp2 genes, which result in ARVC led researchers to believe that ARVC might be a disease of the desmosome. Therefore, Dsg2 and Dsc2, which code for the only isoforms of Dsg/Dsc known to be expressed in cardiac myocytes,110 were plausible candidate genes for this disorder. In fact, mutation screening of Dsg2 and Dsc2 in patients with ARVC revealed the presence of several mutations in these genes.111,112
Twenty-two dominant mutations in the Dsg2 gene, including missense, insertion–deletion, nonsense, and splice site, were identified in patients with ARVC.111–113 All mutations were predicted to disrupt functionally important parts of Dsg2. The majority of these mutations were spread throughout the EC N-terminal domain. EM observations of endomyocardial biopsy samples of some of these patients showed, in agreement, a decreased number of desmosomes as well as a widening of the intercalated disk gap.113 In addition, histological observations of patients’ biopsies revealed myocardial atrophy and fibro-fatty replacement.112
Desmoglein 4
The observation that mutations in Pkp1, Dp, and Pg caused hair defects raised the question as to whether cadherin gene mutations could also result in an abnormal inherited hair phenotype. In 2003, Kljuic et al identified a new desmosomal cadherin – Dsg4 – which is expressed in the suprabasal epidermis and HF.24 They also associated a mutation in Dsg4 with a hair disease phenotype. Dsg4 shares 41% identity with Dsg1, 37% identity with Dsg2, and 50% identity with Dsg3.25 Dsg4 is expressed only in the salivary gland, testis, prostate, and skin. In the HF, Dsg4 is the primary desmosomal cadherin present and is extensively expressed throughout the matrix, precortex, and inner root sheath.24
Through genetic linkage analysis, this group first linked a disease, namely localized hypotrichosis, to chromosome 18q12 and then identified a homozygous 5 kb intragenic deletion in Dgs4 in two families. This disorder is characterized by hypotrichosis restricted to the scalp, chest, arms, and legs. Histology of scalp skin revealed thin and atrophic HFs and hair shafts that often coil up within the skin due to their inability to penetrate the epidermis. There is also a marked swelling of the precortical region resulting in the formation of a bulbous “bleb” within the base of the hair shaft, similar to the shape of a lance, hence the name lanceolate.24
This and other mutations in Dsg4 have subsequently been found.116 Additionally, other mutations have been identified in patients who have monilethrix hairs as a part of their phenotypic presentation – monilethrix-like congenital hypotrichosis.110,118
Infectious diseases targeting the desmosome
Recent research has shown that the desmosome can be targeted during infection. Specifically, the discovery that staphylococcal exfoliative toxins (ETs) can cleave a desmosomal cadherin has shed light on the pathophysiology of a number of blistering diseases with common infective etiology. ETA and ETB are the first two Staphylococcus aureus ETs described.119 In 1994, a third potential ET (dominated ETC) was described by Sato et al.120 More recently, a new potential staphylococcal exfoliative toxin D (ETD) was identified from an isolate taken from a wound site. After recombinant expression, ETD showed specific protease activity in the neonatal mouse model and cleaved recombinant mouse Dsg1.121 The ETs are host specific, affecting humans, monkeys, mice, and hamsters but not rats, rabbits, dogs, guinea pig, chicken, and frogs.121,122
Staphylococcal scalded skin syndrome (SSSS) is a generalized exfoliative dermatitis typically occurring in newborns (Ritter’s disease) and in children younger than 5 years.123 The clinical features of SSSS vary from localized blisters to severe exfoliation affecting over 90% of the entire body surface.124 Characteristic fragile, thin-roofed, flaccid bullae are formed which rupture easily, resulting in red, denuded skin resembling a burn. Friction applied to the skin causes the epidermis to wrinkle and separate (Nikolsky’s sign). Widespread involvement of the entire skin surface can occur, but the mucous membranes are usually speared. ET-producing S. aureus can be recovered from the nasopharynx rather than from the skin.
SSSS adult cases are extremely rare.123,125 This has been attributed to the ability of adults to metabolize and excrete exfoliatin rapidly or to acquired immunity.126
Localized bullous impetigo occurs in neonates and children. In neonates, lesions are found mostly on the perineum, periumbilical area, or both, while in older children, they are found most often on the extremities.127 Small flaccid bullae are formed and filled with fluid that varies from clear, cloudy, opaque, or purulent to white or yellow pus.128 These bullae rupture and heal, leaving a yellow–brown crust and a nontender lesion.129 Fluid obtained from these bullae is found to contain S. aureus. This is the mildest form of the staphylococcal exfoliating disease as the surrounding skin remains normal, and there are no systemic symptoms or signs.
Staphylococcal scarlet fever, also called scarlatiniform erythroderma, was until recently considered to be a milder or abortive form of SSSS.121,130 Patients usually develop fever and generalized erythroderma involving flushing of the skin and late skin desquamation. Unlike generalized SSSS, the scarlatiniform eruption is not associated with the formation of bullae. The clinical feature is very similar to those of other infectious erythrodermal causes, such as toxic shock syndrome and streptococcal scarlet fever.131
Strains from patients with generalized exfoliative syndrome and bullous impetigo are shown to produce either ETA or ETB, whereas those from patients with staphylococcal scarlet fever most frequently produce TSST-1 or staphylococcal enterotoxin A to staphylococcal enterotoxin D and very rarely an ET.132 Therefore, staphylococcal scarlet fever should not be classified as a form of SSSS but as an abortive form of toxic shock syndrome or as a separate syndrome.
Autoimmune disease and desmosomes
The paradigm of autoimmunity against desmosome is represented by pemphigus, a group of autoimmune-blistering diseases that are characterized by the loss of cell–cell adhesion between keratinocytes (acantholysis) in the epidermis, and autoantibodies against Dsg3133 and/or Dsg1,134 in addition to a variety of other desmosomal, nondesmosomal, and nonepithelial IgGs.135–137 Classically, pemphigus can be classified into two major groups, ie, pemphigus vulgaris (PV), which is characterized by suprabasal acantholysis and by autoantibodies against Dsg3 or both Dsg3 and Dsg1, and pemphigus foliaceus, which is characterized by superficial acantholysis in the granular cell layer of the epidermis and autoantibodies against Dsg1. Additional desmosomal autoantigens include Dscs, such Dsc3,138,139 as well as Pg,140 Dsp,141 and Pkp3.142
Insights into pathomechanisms underlying the generation of acantholysis in PV after autoantibodies bind to desmosomal and nondesmosomal antigens on the keratinocyte surface have lead to two conflicting, yet complementary hypotheses. The first hypothesis proposes that anti-Dsg3 antibody interferes with intercellular adhesive function(s) of Dsg3 either by steric hindrance or by outside-in signaling, leading directly to desmosomal dissociation.143 The second hypothesis proposes that several PV-IgG-induced intracellular signaling events could lead to desmosomal dissociation via intracellular and inside-out pathways.144–146 Research suggests that PV-IgG does not directly inhibit desmosome formation, even though antibodies in PV-IgG may also cause steric hindrance between homophilic Dsg3 interactions and heterophilic Dsc3 interactions. However, longer PV-IgG incubation ultimately leads to cell–cell detachment, suggesting that impairment of desmosome remodeling may be involved in the PV acantholysis.64 The signaling-related events thought to be involved in the pathomechanisms of PV include not only phosphorylation of Dsg3 and a Ca2+/PKC pathway but also apoptosis signaling, as well as modulations of Pg, p38MAPK, heat shock protein 27, cdk2, Src, RhoA, and others.147–149
Overall, research published so far shows that, while desmosome disassembling is a key pathogenic event in pemphigus, the mechanisms by which the splitting occurs involve intracellular signaling pathways possibly triggered by nondesmosomal molecules.
The desmosome in cancer
Despite the lack of a cause–effect relationship, current data provide substantial evidence that desmosomes or their protein components are involved in different processes during the development and progression of cancer.
The seminal paper demonstrating a strict correlation between desmosome and cancer showed that expression of desmosomal cadherins inhibited the invasive behavior of nonadhesive fibroblasts.150 Since then, a multitude of articles have shown direct and indirect roles of desmosomal components in malignancy.
Reduced expression of desmosomal cadherins, which may be responsible for the weakening of cell–cell adhesion observed in cancer cells, was observed in different tumors, such as skin,151 head and neck,152 lung,153 breast,154 and a variety of other epithelial malignancies,155 including gastric156 and colon157 cancers. In one example, knockdown of Pkp3 in colon cancer cells promoted anchorage-independent growth and tumor growth in immunocompromised mice.158 However, there are instances where desmosomal molecules are overexpressed,159 although this could represent a compensatory mechanism for the loss of adhesion strength. Alternatively, the desmosomal cadherins may serve as signaling molecules via their adaptor proteins, thus controlling cell cycle and apoptosis. The altered desmosomal cadherin expression seen in cancer occurs via three main mechanisms: transcriptional deregulation; impaired transport, targeting, and assembly into mature desmosomes; and inactivation by proteolytic cleavage.155
Another mechanism that defines the cancer phenotype is escaped from programed cell death or apoptosis. While it was long known that E-cadherin-mediated cell–cell adhesion is crucial for escaping cell death in the absence of ECM attachment (aka anoikis),160 a role for desmosomal cadherin had not been elucidated until recently. Research shows that the overexpression of Dsg2 increases anchorage-independent cell growth in an EGFR and nuclear factor kappa B-dependent pathway, resulting in upregulation of c-myc and antiapoptotic Bcl-XL.161 Knockdown of Dsg1 in keratinocytes is associated with decreased rates of apoptosis in response to ultraviolet (UV) irradiation.162 Expression of high levels of Pg inhibits apoptosis and correlates with an increased expression of the antiapoptotic factor Bcl-2.163 Loss of Rnd3/RhoE leads to an enhanced expression of desmosomal proteins and protects keratinocytes from cisplatin-induced apoptosis in a Pg-dependent manner.164 A recent study suggested that Pkp2 is also involved in the regulation of apoptosis.165 Furthermore, reexpression of Dsp in cells with an epigenetic inactivation of the Dsp gene exhibited an increased sensitivity to apoptosis by the upregulation of Pg and the concomitant inhibition of β-catenin –TCF/LEF-dependent transcription.166
Abnormal proliferative signaling of cancer cells may also be partially controlled by desmosomes via their accessory components (Figure 2), including tyrosine kinases, which are frequently upregulated in cancer. Keratinocytes from Pg-null mice exhibit an increased Src activity correlating with enhanced cell motility. Both the Src inhibitor PP2 and a dominant negative Src kinase attenuated cell migration in Pg-negative cells.167 Furthermore, Src is involved in a cross talk between Dsg3 and E-cadherin.168 In these researches, Pg was identified as a target of Src, Fyn, and Fer kinases, and this is consistent with the predictions of the desmosomal interactome.61
Desmosomes are related to central regulators of cell cycle and apoptosis, such as the retinoblastoma proteins Rb and p53.169 A link between p53 and its close relative p63 to desmosomal structure and function is certain. Both directly control the expression of not only desmosomal genes, including Dsc3, Dsg1, and Dsp,155 but also p53 apoptosis effector related to peripheral myelin protein-22. Knockout of peripheral myelin protein-22 in mice results in a loss of desmosomes, impaired wound healing, and enhanced tumorigenesis,170,171 thus further emphasizing the role of desmosomes in cancer progression.
Conclusion
The crucial role of the desmosome in maintaining epidermal integrity has widely been demonstrated by the large number of studies on diseases that occur when the function of one or more desmosomal constituents is impaired (Figure 2). At the same time, the study of disease pathophysiology has allowed more insights into the basic elements of desmosome structure and function to be gained. Thus, the desmosome best exemplifies how basic, translational, and clinical research integrate to nurture and advance scientific knowledge.
Disclosure
The author reports no conflicts of interest in this work.
References
Bizzozero G. Delle cellule cigliate, del reticolo Malpighiano dell’epidermide. Ann Univ Med. 1864;190:110–118. | |
Sacchi S. Observations on histochemistry of so-called nodes of Ranvier, Bizzozero or desmosomes of Schaffer. Dermatologica. 1952;105:158–162. | |
Delva E, Tucker DK, Kowalczyk AP. The desmosome. Cold Spring Harb Perspect Biol. 2009;1:a002543. | |
Skerrow CJ, Matoltsy AG. Isolation of epidermal desmosomes. J Cell Biol. 1974;63:515–523. | |
Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol. 2004;5:271–281. | |
Stahley SN, Kowalczyk AP. Desmosomes in acquired disease. Cell Tissue Res. 2015;360:439–456. | |
Harmon RM, Green KJ. Structural and functional diversity of desmosomes. Cell Commun Adhes. 2013;20:171–187. | |
Cirillo N. 150th anniversary series: desmosomes in physiology and disease. Cell Commun Adhes. 2014;21:85–88. | |
Bazzi H, Christiano AM. Broken hearts, woolly hair, and tattered skin: when desmosomal adhesion goes awry. Curr Opin Cell Biol. 2007;19:515–520. | |
He W, Cowin P, Stokes DL. Untangling desmosomal knots with electron tomography. Science. 2003;302:109–113. | |
Ihrie RA, Marques MR, Nguyen BT, et al. Perp is a p63-regulated gene essential for epithelial integrity. Cell. 2005;120:843–856. | |
Kowalczyk AP, Green KJ. Structure, function, and regulation of desmosomes. Prog Mol Biol Transl Sci. 2013;116:95–118. | |
North AJ, Bardsley WG, Hyam J, et al. Molecular map of the desmosomal plaque. J Cell Sci. 1999;112:4325–4336. | |
Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. 2008;1778:572–587. | |
Al-Jassar C, Bikker H, Overduin M, et al. Mechanistic basis of desmosome-targeted diseases. J Mol Biol. 2013;425:4006–4022. | |
Andrä K, Lassmann H, Bitter R, et al. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 1997;11:3143–3156. | |
Gallicano GI, Bauer C, Fuchs E. Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development. 2001;128:929–941. | |
Aho S, Li K, Ryoo Y, et al. Periplakin gene targeting reveals a constituent of the cornified cell envelope dispensable for normal mouse development. Mol Cell Biol. 2004;24:6410–6418. | |
Määttä A, DiColandrea T, Groot K, et al. Gene targeting of envoplakin, a cytoskeletal linker protein and precursor of the epidermal cornified envelope. Mol Cell Biol. 2001;21:7047–7053. | |
Vasioukhin V, Bowers E, Bauer C, et al. Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol. 2001;3:1076–1085. | |
Dusek RL, Godsel LM, Green KJ. Discriminating roles of desmosomal cadherins: beyond desmosomal adhesion. J Dermatol Sci. 2007;45:7–21. | |
Nollet F, Kools P, van Roy F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J Mol Biol. 2000;299:551–572. | |
Kljuic A, Bazzi H, Sundberg JP, et al. Desmoglein 4 in hair follicle differentiation and epidermal adhesion: evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell. 2003;113:249–260. | |
Whittock NV, Bower C. Genetic evidence for a novel human desmosomal cadherin, desmoglein 4. J Invest Dermatol. 2003;120:523–530. | |
Collins JE, Legan PK, Kenny TP, et al. Cloning and sequence analysis of desmosomal glycoproteins 2 and 3 (desmocollins): cadherin-like desmosomal adhesion molecules with heterogeneous cytoplasmic domains. J Cell Biol. 1991;113:381–391. | |
Legan PK, Yue KK, Chidgey MA, et al. The bovine desmocollin family: a new gene and expression patterns reflecting epithelial cell proliferation and differentiation. J Cell Biol. 1994;126:507–518. | |
Bazzi H, Getz A, Mahoney MG, et al. Desmoglein 4 is expressed in highly differentiated keratinocytes and trichocytes in human epidermis and hair follicle. Differentiation. 2006;74:129–140. | |
King IA, Angst BD, Hunt DM, et al. Hierarchical expression of desmosomal cadherins during stratified epithelial morphogenesis in the mouse. Differentiation. 1997;62:83–96. | |
Kowalczyk AP, Stappenbeck TS, Parry DA, et al. Structure and function of desmosomal transmembrane core and plaque molecules. Biophys Chem. 1994;50:97–112. | |
Weiske J, Schöneberg T, Schröder W, et al. The fate of desmosomal proteins in apoptotic cells. J Biol Chem. 2001;276:41175–41181. | |
Hatzfeld M. The armadillo family of structural proteins. Int Rev Cytol. 1999;186:179–224. | |
Coates JC. Armadillo repeat proteins: beyond the animal kingdom. Trends Cell Biol. 2003;12:463–471. | |
Leung CL, Green KJ, Liem RKH. Plakins: a family of versatile cytolinker proteins. Trends Cell Biol. 2002;12:37–45. | |
Sonnenberg A, Liem RK. Plakins in development and disease. Exp Cell Res. 2007;313:2189–2203. | |
Hatzfeld M. Plakophilins: multifunctional proteins or just regulators of desmosomal adhesion? Biochim Biophys Acta. 2007;1773:69–77. | |
Schmidt A, Jäger S. Plakophilins – hard work in the desmosome, recreation in the nucleus? Eur J Cell Biol. 2005;84:189–204. | |
Hatzfeld M, Green KJ, Sauter H. Targeting of p0071 to desmosomes and adherens junctions is mediated by different protein domains. J Cell Sci. 2003;116:1219–1233. | |
Brembeck FH, Rosario M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev. 2006;16:51–59. | |
Yin T, Green KJ. Regulation of desmosome assembly and adhesion. Semin Cell Dev Biol. 2004;15:665–677. | |
Troyanovsky SM, Troyanovsky RB, Eshkind LG, et al. Identification of the plakoglobin-binding domain in desmoglein and its role in plaque assembly and intermediate filament anchorage. J Cell Biol. 1994;127:151–160. | |
Troyanovsky SM, Troyanovsky RB, Eshkind LG, et al. Identification of amino acid sequence motifs in desmocollin, a desmosomal glycoprotein, that are required for plakoglobin binding and plaque formation. Proc Natl Acad Sci U S A. 1994;91:10790–10794. | |
Hofmann I, Mertens C, Brettel M, et al. Interaction of plakophilins with desmoplakin and intermediate filament proteins: an in vitro analysis. J Cell Sci. 2000;113:2471–2483. | |
Smith EA, Fuchs E. Defining the interactions between intermediate filaments and desmosomes. J Cell Biol. 1998;141:1229–1241. | |
Bonné S, Gilbert B, Hatzfeld M, et al. Defining desmosomal plakophilin-3 interactions. J Cell Biol. 2003;161:403–416. | |
Chen X, Bonné S, Hatzfeld M, et al. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta-catenin signaling. J Biol Chem. 2002;277:10512–10522. | |
Palka HL, Green KJ. Roles of plakoglobin end domains in desmosome assembly. J Cell Sci. 1997;110:2359–2371. | |
Cowin P, Kapprell HP, Franke WW, et al. Plakoglobin: a protein common to different kinds of intercellular adhering junctions. Cell. 1986;46:1063–1073. | |
Lewis JE, Wahl JK, Sass KM, et al. Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J Cell Biol. 1997;136:919–934. | |
Hatzfeld M, Wolf A, Keil R. Plakophilins in desmosomal adhesion and signaling. Cell Commun Adhes. 2014;21(1):25–42. | |
Hatzfeld M, Haffner C, Schulze K, et al. The function of plakophilin 1 in desmosome assembly and actin filament organization. J Cell Biol. 2000;149:209–222. | |
Kowalczyk AP, Hatzfeld M, Bornslaeger EA, et al. The head domain of plakophilin-1 binds to desmoplakin and enhances its recruitment to desmosomes. Implications for cutaneous disease. J Biol Chem. 1999;274:18145–18148. | |
McGrath JA, McMillan JR, Shemanko CS, et al. Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet. 1997;17:240–244. | |
Bornslaeger EA, Godsel LM, Corcoran CM, et al. Plakophilin 1 interferes with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell-cell borders. J Cell Sci. 2001;114:727–738. | |
Sobolik-Delmaire T, Katafiasz D, Wahl JK 3rd. Carboxyl terminus of plakophilin-1 recruits it to plasma membrane, whereas amino terminus recruits desmoplakin and promotes desmosome assembly. J Biol Chem. 2006;281:16962–16970. | |
Ruhrberg C, Watt FM. The plakin family: versatile organizers of cytoskeletal architecture. Curr Opin Genet Dev. 1997;7:392–397. | |
Jefferson JJ, Ciatto C, Shapiro L, et al. Structural analysis of the plakin domain of bullous pemphigoid antigen1 (BPAG1) suggests that plakins are members of the spectrin superfamily. J Mol Biol. 2007;366:244–257. | |
Paris L, Bazzoni G. The protein interaction network of the epithelial junctional complex: a system-level analysis. Mol Biol Cell. 2008;19(12):5409–5421. | |
Winograd-Katz SE, Fässler R, Geiger B, et al. The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol. 2014;15(4):273–288. | |
Robertson J, Jacquemet G, Byron A, et al. Defining the phospho-adhesome through the phosphoproteomic analysis of integrin signalling. Nat Commun. 2015;6:6265. | |
Cirillo N, Prime SS. Desmosomal interactome in keratinocytes: a systems biology approach leading to an understanding of the pathogenesis of skin disease. Cell Mol Life Sci. 2009;66(21):3517–3533. | |
Cirillo N. Merging experimental data and in silico analysis: a systems-level approach to autoimmune disease and cancer. Expert Rev Clin Immunol. 2012;8(4):361–372. | |
Green KJ, Simpson CL. Desmosomes: new perspectives on a classic. J Invest Dermatol. 2007;127:2499–2515. | |
Kitajima Y. 150(th) anniversary series: desmosomes and autoimmune disease, perspective of dynamic desmosome remodeling and its impairments in pemphigus. Cell Commun Adhes. 2014;21:269–280. | |
Rötzer V, Hartlieb E, Vielmuth F, et al. E-cadherin and Src associate with extradesmosomal Dsg3 and modulate desmosome assembly and adhesion. Cell Mol Life Sci. 2015;72(24):4885–4897. | |
Hennings H, Michael D, Cheng C, et al. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell. 1980;19:245–254. | |
Hennings H, Holbrookm KA. Calcium regulation of cell-cell contact and differentiation of epidermal cells in culture. An ultrastructural study. Exp Cell Res. 1983;143:127–142. | |
Sheu HM, Kitajama Y, Yaoita H. Involvement of protein kinase C in translocation of desmoplakins from cytosol to plasma membrane during desmosome formation in human squamous cell carcinoma cells grown in low to normal calcium concentration. Exp Cell Res. 1989;185:176–190. | |
Pasdar M, Krzeminski KA, Nelson WJ. Regulation of desmosome assembly in MDCK epithelial cells: coordination of membrane core and cytoplasmic plaque domain assembly at the plasma membrane. J Cell Biol. 1991;113:645–655. | |
Nekrasova OE, Amargo EV, Smith WO, et al. Desmosomal cadherins utilize distinct kinesins for assembly into desmosomes. J Cell Biol. 2011;195:1185–1203. | |
Godsel LM, Dubash AD, Bass-Zubek AE, et al. Plakophilin 2 couples actomyosin remodeling to desmosomal plaque assembly via RhoA. Mol Biol Cell. 2010;21:2844–2859. | |
Godsel LM, Hsieh SN, Amargo EV, et al. Desmoplakin assembly dynamics in four dimensions: multiple phases differentially regulated by intermediate filaments and actin. J Cell Biol. 2005;171:1045–1059. | |
Nekrasova O, Green KJ. Desmosome assembly and dynamics. Trends Cell Biol. 2013;23(11):537–546. | |
Berika M, Garrod D. Desmosomal adhesion in vivo. Cell Commun Adhes. 2014;21:65–75. | |
Garrod D, Kimura TE. Hyper-adhesion: a new concept in cell-cell adhesion. Biochem Soc Trans. 2008;36(pt 2):195–201. | |
Cirillo N, Lanza A, Prime SS. Induction of hyper-adhesion attenuates autoimmune-induced keratinocyte cell-cell detachment and processing of adhesion molecules via mechanisms that involve PKC. Exp Cell Res. 2010;316(4):580–592. | |
Garrod DR, Berika MY, Bardsley WF, et al. Hyper-adhesion in desmosomes: its regulation in wound healing and possible relationship to cadherin crystal structure. J Cell Sci. 2005;118(pt 24):5743–5754. | |
Thomason HA, Cooper NH, Ansell DM, et al. Direct evidence that PKCα positively regulates wound re-epithelialization: correlation with changes in desmosomal adhesiveness. J Pathol. 2012;227(3):346–356. | |
Aoyama Y, Yamamoto Y, Yamaguchi F, et al. Low to high Ca2+-switch causes phosphorylation and association of desmocollin 3 with plakoglobin and desmoglein 3 in cultured keratinocytes. Exp Dermatol. 2009;18:404–408. | |
Kitajima Y. New insights into desmosome regulation and pemphigus blistering as a desmosome-remodeling disease. Kaohsiung J Med Sci. 2013;29(1):1–13. | |
McGrath JA, Hoeger PH, Christiano AM, et al. Skin fragility and hypohidrotic ectodermal dysplasia resulting from ablation of plakophilin 1. Br J Dermatol. 1999;140:297–307. | |
Sprecher E, Molho-Pessach V, Ingber A, et al. Homozygous splice site mutations in PKP1 result in loss of epidermal plakophilin 1 expression and underlie ectodermal dysplasia/skin fragility syndrome in two consanguineous families. J Invest Dermatol. 2004;122:647–651. | |
Steijlen PM, van Steensel MA, Jansen BJ, et al. Cryptic splicing at a non-consensus splice-donor in a patient with a novel mutation in the plakophilin-1 gene. J Invest Dermatol. 2004;122:1321–1324. | |
South AP, Wan H, Stone MG, et al. Lack of plakophilin 1 increases keratinocyte migration and reduces desmosome stability. J Cell Sci. 2003;116:3303–3314. | |
McMillan JR, Haftek M, Akiyama M, et al. Alterations in desmosome size and number coincide with the loss of keratinocyte cohesion in skin with homozygous and heterozygous defects in the desmosomal protein plakophilin 1. J Invest Dermatol. 2003;121:96–103. | |
Chen X, Bonne S, Hatzfeld M, et al. Protein binding and functional characterization of plakophilin 2: evidence for its diverse roles in desmosomes and β-catenin signaling. J Biol Chem. 2002;277:10512–10522. | |
Gerull B, Heuser A, Wichter T, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. | |
Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. | |
Thiene G, Nava A, Corrado D, et al. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. | |
Syrris P, Ward D, Asimaki A, et al. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:356–364. | |
Nagaoka I, Matsui K, Ueyama T, et al. Novel mutation of plakophilin-2 associated with arrhythmogenic right ventricular cardiomyopathy. Circ J. 2006;70:933–935. | |
Hall C, Li S, Li H, et al. Arrhythmogenic right ventricular cardiomyopathy plakophilin-2 mutations disrupt desmosome assembly and stability. Cell Commun Adhes. 2009;16:15–27. | |
Sklyarova T, Bonné S, D’Hooge P, et al. Plakophilin-3-deficient mice develop hair coat abnormalities and are prone to cutaneous inflammation. J Invest Dermatol. 2008;128(6):1375–1385. | |
Ruiz P, Brinkmann V, Ledermann B, et al. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 1996;135:215–225. | |
Coonar AS, Protonotarios N, Tsatsopoulou A, et al. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation. 1998;97:2049–2058. | |
Armstrong DK, McKenna KE, Purkis PE, et al. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet. 1999;8:143–148. | |
Whittock NV, Ashton GH, Dopping-Hepenstal PJ, et al. Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatol. 1999;113:940–946. | |
Whittock NV, Wan H, Morley SM, et al. Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol. 2002;118:232–238. | |
Jonkman MF, Pasmooij AM, Pasmans SG, et al. Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet. 2005;77:653–660. | |
Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766. | |
Carvajal-Huerta L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol. 1998;39:418–421. | |
Alcalai R, Metzger S, Rosenheck S, et al. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol. 2003;42:319–327. | |
Rampazzo A, Nava A, Malacrida S, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:120020–120026. | |
Rickman L, Simrak D, Stevens HP, et al. N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum Mol Genet. 1999;8:971–976. | |
Kljuic A, Gilead L, Martinez-Mir A, et al. A nonsense mutation in the desmoglein 1 gene underlies striate keratoderma. Exp Dermatol. 2003;12:523–527. | |
Barber AG, Wajid M, Columbo M, et al. Striate palmoplantar keratoderma resulting from a frameshift mutation in the desmoglein 1 gene. J Dermatol Sci. 2007;45:161–166. | |
Milingou M, Wood P, Masouye I, et al. Focal palmoplantar keratoderma caused by an autosomal dominant inherited mutation in the desmoglein 1 gene. Dermatology. 2006;212:117–122. | |
Keren H, Bergman R, Mizrachi M, et al. Diffuse nonepidermolytic palmoplantar keratoderma caused by a recurrent nonsense mutation in DSG1. Arch Dermatol. 2005;141:625–628. | |
Schaffer JV, Bazzi H, Vitebsky A, et al. Mutations in the desmoglein 4 gene underlie localized autosomal recessive hypotrichosis with monilethrix hairs and congenital scalp erosions. J Invest Dermatol. 2006;126:1286–1291. | |
Awad MM, Dalal D, Cho E, et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet. 2006;79:136–142. | |
Syrris P, Ward D, Asimaki A, et al. Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: a genotype-phenotype characterization of familial disease. Eur Heart J. 2007;28:581–588. | |
Pilichou K, Nava A, Basso C, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. | |
Moss C, Martinez-Mir A, Lam H, et al. A recurrent intragenic deletion in the desmoglein 4 gene underlies localized autosomal recessive hypotrichosis. J Invest Dermatol. 2004;123:607–610. | |
Schweizer J. More than one gene involved in monilethrix: intracellular but also extracellular players. J Invest Dermatol. 2006;126:1216–1219. | |
Wiley BB, Rogolsky M. Molecular and serological differentiation of staphylococcal exfoliative toxin synthesized under chromosomal and plasmid control. Infect Immun. 1977;18:487–494. | |
Sato H, Matsumori Y, Tanabe T, et al. A new type of staphylococcal exfoliative toxin from a Staphylococcus aureus strain isolated from a horse with phlegmon. Infect Immun. 1994;62:3780–3785. | |
Yamaguchi T, Nishifuji K, Sasaki M, et al. Identification of the Staphylococcus aureus etd pathogenicity island which encodes a novel exfoliative toxin, ETD, and EDIN-B. Infect Immun. 2002;70:5835–5845. | |
Bailey CJ, Lockhart BP, Redpath MB, et al. The epidermolytic (exfoliative) toxins of Staphylococcus aureus. Med Microbiol Immunol. 1995;184:53–61. | |
Gemmell CG. Staphylococcal scalded skin syndrome. J Med Microbiol. 1995;43:318–327. | |
Ladhani S, Evans RW. Staphylococcal scalded skin syndrome. Arch Dis Child. 1998;78:85–88. | |
Cribier B, Piemont Y, Grosshans E. Staphylococcal scalded skin syndrome in adults. A clinical review illustrated with a new case. J Am Acad Dermatol. 1994;30:319–324. | |
Ladhani S, Joannou CL, Lochrie DP, et al. Clinical, microbial, and biochemical aspects of the exfoliative toxins causing staphylococcal scalded-skin syndrome. Clin Microbiol Rev. 1999;12:224–242. | |
Melish ME. Staphylococci, streptococci and the skin: review of impetigo and staphylococcal scalded skin syndrome. Semin Dermatol. 1982;1:101–109. | |
Ginsburg CM. Staphylococcal toxin syndromes. Pediatr Infect Dis. 1983;2:S23–S27. | |
Scales JW, Fleischer AB Jr, Krowchuk DP. Bullous impetigo. Arch Pediatr Adolesc Med. 1997;151:1168–1169. | |
Melish ME, Glasgow LA. The staphylococcal scalded-skin syndrome. N Engl J Med. 1970;282:1114–1119. | |
Todd JK. Staphylococcal toxin syndromes. Annu Rev Med. 1985;36:337–347. | |
Lina G, Gillet Y, Vandenesch F, et al. Toxin involvement in staphylococcal scalded skin syndrome. Clin Infect Dis. 1997;25:1369–1373. | |
Eyre RW, Stanley JR. Identification of pemphigus vulgaris antigen extracted from normal human epidermis and comparison with pemphigus foliaceus antigen. J Clin Invest. 1988;81:807–812. | |
Koulu L, Kusurni A, Steinberg MS, et al. Human autoantibodies against a desmosomal core protein in pemphigus foliaceus. J Exp Med. 1984;160:1509–1518. | |
Cirillo N, Gombos F, Lanza A. Pemphigus vulgaris immunoglobulin G can recognize a 130 000 MW antigen other than desmoglein 3 on peripheral blood mononuclear cell surface. Immunology. 2007;121(3):377–382. | |
Nguyen VT, Ndoye A, Shultz LD, et al. Antibodies against keratinocyte antigens other than desmogleins 1 and 3 can induce pemphigus vulgaris-like lesions. J Clin Invest. 2000;106:1467–1479. | |
Cirillo N, Cozzani E, Carrozzo M, et al. Urban legends: pemphigus vulgaris. Oral Dis. 2012;18(5):442–458. | |
Spindler V, Heupel WM, Efthymiadis A, et al. Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in pemphigus. J Biol Chem. 2009;284(44):30556–30564. | |
Mao X, Nagler AR, Farber SA, et al. Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol. 2010;177(6):2724–2730. | |
Korman NJ, Eyre RW, Klaus-Kovtun V, et al. Demonstration of an adhering-junction molecule (plakoglobin) in the autoantigens of pemphigus foliaceus and pemphigus vulgaris. N Engl J Med. 1989;321(10):631–635. | |
Mimouni D, Foedinger D, Kouba DJ, et al. Mucosal dominant pemphigus vulgaris with anti-desmoplakin autoantibodies. J Am Acad Dermatol. 2004;51:62–67. | |
Kalantari-Dehaghi M, Anhalt GJ, Camilleri MJ, et al. Pemphigus vulgaris autoantibody profiling by proteomic technique. PLoS One. 2013;8(3):e57587. | |
Koga H, Tsuruta D, Ohyama B, et al. Desmoglein 3, its pathogenecity and a possibility for therapeutic target in pemphigus vulgaris. Expert Opin Ther Targets. 2013;17(3):293–306. | |
Chernyavsky AI, Arredondo J, Kitajima Y, et al. Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J Biol Chem. 2007;282(18):13804–13812. | |
Marchenko S, Chernyavsky AI, Arredondo J, et al. Antimitochondrial autoantibodies in pemphigus vulgaris: a missing link in disease pathophysiology. J Biol Chem. 2010;285(6):3695–3704. | |
Grando SA, Bystryn JC, Chernyavsky AI, et al. Apoptolysis: a novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp Dermatol. 2009;18(9):764–770. | |
Spindler V, Waschke J. Desmosomal cadherins and signaling: lessons from autoimmune disease. Cell Commun Adhes. 2014;21(1):77–84. | |
Cirillo N, AlShwaimi E, McCullough M, et al. Pemphigus vulgaris autoimmune globulin induces Src-dependent tyrosine-phosphorylation of plakophilin 3 and its detachment from desmoglein 3. Autoimmunity. 2014;47(2):134–140. | |
Berkowitz P, Hu P, Warren S, et al. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A. 2006;103(34):12855–12860. | |
Tselepis C, Chidgey M, North A, et al. Desmosomal adhesion inhibits invasive behavior. Proc Natl Acad Sci U S A. 1998;95:8064–8069. | |
Xin Z, Yamaguchi A, Sakamoto K. Aberrant expression and altered cellular localization of desmosomal and hemidesmosomal proteins are associated with aggressive clinicopathological features of oral squamous cell carcinoma. Virchows Arch. 2014;465:35–47. | |
Shinohara M, Hiraki A, Ikebe T, et al. Immunohistochemical study of desmosomes in oral squamous cell carcinoma: correlation with cytokeratin and E-cadherin staining, and with tumour behaviour. J Pathol. 1998;184:369–381. | |
Cui T, Chen Y, Yang L, et al. DSC3 expression is regulated by p53, and methylation of DSC3 DNA is a prognostic marker in human colorectal cancer. Br J Cancer. 2011;104:1013–1019. | |
Oshiro MM, Kim CJ, Wozniak RJ, et al. Epigenetic silencing of Dsc3 is a common event in human breast cancer. Breast Cancer Res. 2005;7:R669–R680. | |
Huber O, Petersen I. 150th anniversary series: desmosomes and the hallmarks of cancer. Cell Commun Adhes. 2015;22(1):15–28. | |
Yashiro M, Nishioka N, Hirakawa K. Decreased expression of the adhesion molecule desmoglein-2 is associated with diffuse-type gastric carcinoma. Eur J Cancer. 2006;42:2397–2403. | |
Kamekura R, Kolegraff KN, Nava P, et al. Loss of the desmosomal cadherin desmoglein-2 suppresses colon cancer cell proliferation through EGFR signaling. Oncogene. 2013;33:4531–4536. | |
Kundu ST, Gosavi P, Khapare N, et al. Plakophilin3 downregulation leads to a decrease in cell adhesion and promotes metastasis. Int J Cancer. 2008;123:2303–2314. | |
Brennan D, Mahoney MG. Increased expression of Dsg2 in malignant skin carcinomas: a tissue-microarray based study. Cell Adh Migr. 2009;3:148–154. | |
Grossmann J. Molecular mechanisms of “detachment-induced apoptosis – anoikis”. Apoptosis. 2002;7(3):247–260. | |
Brennan D, Hu Y, Choi YW, et al. Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J Cell Sci. 2007;120:758–771. | |
Dusek RL, Getsios S, Chen F, et al. The differentiation-dependent desmosomal cadherin desmoglein 1 is a novel caspase-3 target that regulates apoptosis in keratinocytes. J Biol Chem. 2006;281:3614–3624. | |
Hakimelahi S, Parker HR, Gilchrist AJ, et al. Plakoglobin regulates the expression of the anti-apoptotic protein Bcl-2. J Biol Chem. 2000;275:10905–10911. | |
Ryan KR, Lock FE, Heath JK, et al. Plakoglobin dependent regulation of keratinocyte apoptosis by Rnd3. J Cell Sci. 2012;125:3202–3209. | |
Kim C, Wong J, Wen J, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–110. | |
Yang L, Chen Y, Cui T, et al. Desmoplakin acts as a tumor suppressor by inhibition of the Wnt/β-catenin signaling pathway in human lung cancer. Carcinogenesis. 2012;33:1863–1870. | |
Yin T, Getsios S, Caldelari R, et al. Plakoglobin suppresses keratinocyte motility through both cell-cell adhesiondependent and -independent mechanisms. Proc Natl Acad Sci U S A. 2004;102:5420–5425. | |
Tsang SM, Brown L, Lin K, et al. Non-junctional human desmoglein 3 acts as an upstream regulator of Src in E-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J Pathol. 2012;227:81–93. | |
Sherr CJ, McCormick F. The Rb and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. | |
Beaudry VG, Jiang D, Dusek RL, et al. Loss of the p53/p63 regulated desmosomal protein Perp promotes tumorigenesis. PLoS Genet. 2010;6:e1001168. | |
Dusek RL, Bascom JL, Vogel H, et al. Defi ciency of the p53/p63 target Perp alters mammary gland homeostasis and promotes cancer. Breast Cancer Res. 2012;14:R65. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.