Back to Journals » International Journal of Nanomedicine » Volume 13
Design of a transdermal formulation containing raloxifene nanoparticles for osteoporosis treatment
Authors Nagai N ![]() , Ogata F, Otake H, Nakazawa Y
, Ogata F, Otake H, Nakazawa Y ![]() , Kawasaki N
, Kawasaki N ![]()
Received 5 May 2018
Accepted for publication 3 July 2018
Published 6 September 2018 Volume 2018:13 Pages 5215—5229
DOI https://doi.org/10.2147/IJN.S173216
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Noriaki Nagai,1 Fumihiko Ogata,1 Hiroko Otake,1 Yosuke Nakazawa,2 Naohito Kawasaki1
1Faculty of Pharmacy, Kindai University, Higashi-Osaka, Osaka, Japan; 2Faculty of Pharmacy, Keio University, Minato-ku, Tokyo, Japan
Purpose: In the clinical setting, raloxifene, a second-generation selective estrogen receptor modulator, is administered orally; however, the bioavailability (BA) is only 2% because of its poor solubility in aqueous fluids and its extensive first-pass metabolism. Therefore, it is expected that the development of a transdermally delivered formulation may reduce the necessary dose without compromising its therapeutic efficacy. In this study, we designed transdermal formulations containing raloxifene nanoparticles and evaluated their usefulness for osteoporosis therapy.
Methods: Raloxifene was crushed with methylcellulose by the bead mill method, and the milled raloxifene was gelled with or without menthol (a permeation enhancer) by Carbopol® 934 (without menthol, Ral-NPs; with menthol, mRal-NPs). The drug release and transdermal penetration were measured using a Franz diffusion cell, and the therapeutic evaluation of osteoporosis was determined in an ovariectomized rat model.
Results: The mean particle size of raloxifene in the transdermal formulation (Ral-NPs) was 173.7 nm. Although the raloxifene released from Ral-NPs remained in the nanoparticle state, the skin penetration of raloxifene nanoparticles was prevented by the stratum corneum in rat. On the other hand, inclusion of menthol in the formulation attenuated the barrier function of the stratum corneum and permitted the penetration of raloxifene nanoparticles through the skin. Moreover, macropinocytosis relates to the skin penetration of the formulation including menthol (mRal-NPs), since penetration was inhibited by treatment with 2 µM rottlerin, a macropinocytosis inhibitor. In addition, the application of 0.3% mRal-NPs (once a day) attenuated the decreases in calcium level and stiffness of the bones of ovariectomized rat.
Conclusion: We prepared raloxifene solid nanoparticles by a bead mill method and designed a novel transdermal formulation containing nanoparticles and permeation enhancers. These transdermal formulations overcome the barrier properties of the skin and show high drug penetration through the transdermal route (BA 8.5%). In addition, we found that raloxifene transdermal formulations are useful for the treatment of osteoporosis in ovariectomized rat.
Keywords: nanomedicine, transdermal delivery system, permeation enhancer, endocytosis, skin
Plain language summary
In the clinical setting, raloxifene, a second-generation selective estrogen receptor modulator, is administered orally; however, the bioavailability (BA) is only 2% because of its poor solubility in aqueous fluids and its extensive first-pass metabolism. In this study, we designed transdermal formulations containing raloxifene nanoparticles to improve the low BA by using a permeation enhancer (menthol) and bead mill method and evaluated their usefulness for osteoporosis therapy. Menthol attenuates the barrier function of the stratum corneum, allowing raloxifene particles ~100–450 nm in size to transit into the underlying epidermis. After that, raloxifene may dissolve and diffuse into the epidermis and dermis and finally be absorbed into the blood circulation (BA 8.5%). The enhanced levels of plasma raloxifene provide efficient and effective therapy for osteoporosis. It appears that macropinocytosis is related to the skin penetration of raloxifene nanoparticles into the epidermis or dermis layers of the skin.
Introduction
Osteoporosis is a common systemic skeletal condition that is related to low bone mineral density and pathological fractures.1 Osteoporosis results from insufficient postmenopausal estrogen levels and collapse of the balance between osteoclasts and osteoblasts.2 The main pharmacologic agents used for treatment are anti-resorptives that prevent the action and development of osteoclasts.3 Raloxifene hydrochloride (raloxifene), one such pharmacologic agent, has a molecular weight of 510.04 g/mol and an octanol/water partition coefficient of 3.81. Raloxifene is a benzothiophene derivative, classified pharmacologically as a selective estrogen receptor modulator (SERM), and is effective for the prevention and treatment of osteoporosis.4 It is mostly supplied as 60 mg tablets as the daily dose for patients and is approved for the prevention and treatment of postmenopausal osteoporosis. In addition, it serves as an estrogen substitute in long-term female hormone replacement therapy and reduces the risk of invasive breast cancer in postmenopausal women.5,6 Raloxifene is characterized by its high permeability, low solubility, and low BA7 and belongs to Biopharmaceutical Classification System class II drugs. Moreover, the absolute BA of raloxifene in oral administration is ~2% due to its poor water solubility and extensive first-pass metabolism via the liver.8 Therefore, it is hypothesized that raloxifene could be a more effective drug for the prevention and treatment of osteoporosis, for the prevention of breast cancer, and for hormone replacement therapy if its BA could be enhanced.9
A transdermal delivery system offers a way to avoid first-pass metabolism and drug–drug interactions. The steps needed for the percutaneous absorption of traditional transdermal formulations are as follows: 1) penetration into the stratum corneum under the influence of a concentration gradient, 2) subsequent diffusion through the stratum corneum → underlying epidermis → dermis, and 3) transition into the blood circulation. In this process, the transdermal route is limited by the barrier properties of the skin’s outer stratum corneum, and the amount that can be administered transdermally is quite low. Therefore, it is necessary to overcome the barrier properties of the skin if a drug is to be delivered via that route. The use of a micro-needle,10,11 therapeutic 1 MHz frequency sonophoresis,12,13 iontophoresis,14,15 patches,16,17 permeation enhancers, and liposomes18 have been demonstrated to be helpful for overcoming the barrier properties of the skin. Formulations containing nanoparticles have been investigated as nanomedicines.19–23 We also reported that transdermal formulations containing solid nanoparticles can enhance the percutaneous absorption of a drug,24 and it is expected that solid nanoparticles may provide a novel strategy for transdermal drug delivery systems.
In this study, we designed a transdermal formulation containing raloxifene nanoparticles (raloxifene transdermal formulation) and demonstrated the mechanism of skin penetration in the application of this raloxifene transdermal formulation. In addition, we investigated the therapeutic effect of the raloxifene transdermal formulation on postmenopausal osteoporosis using ovariectomized rats (OVX rat) as a model.
Materials and methods
Animals
Female 5-week-old Wistar rats were purchased from Shimizu Laboratory Supplies Co., Ltd. (Kyoto, Japan), and either sham operated (Sham rat) or ovariectomized (OVX rat), and used for the therapeutic evaluation of osteoporosis. In addition, female 7-week-old Wistar rats were used in experiments to evaluate the skin penetration of raloxifene transdermal formulations. All rats were provided with a normal diet (CE-2 formulation diet, http://www.clea-japan.com/Feed/ce2.html) and water (Clea Japan Inc., Tokyo, Japan), and housed at 25°C (light: 7:00 am to 7:00 pm). All experiments using rats were carried out in accordance with the Pharmacy Committee Guidelines for the Care and Use of Laboratory Animals in Kindai University and approved on April 1, 2013 (project identification code KAPS-25-002). In addition, all procedures were in accordance with the Guiding Principles approved by The Japanese Pharmacological Society and with the guidelines for animal experimentation of the International Association for the Study.
Chemicals
Conventional raloxifene powder (particle size, 4.91±0.31 μm, mean ± SD), l-menthol (menthol), isoflurane, methyl p-hydroxybenzoate, and cytochalasin D were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Carbopol® 934 (carbopol and carboxypolymethylene) and nystatin were provided from Serva (Heidelberg, Germany), and Sigma-Aldrich Japan (Tokyo, Japan) respectively. Methylcellulose (MC), obtained from Shin-Etsu Chemical Co., Ltd. (Tokyo, Japan), had an average viscosity of ~4 Pa·s at 20°C. Dynasore and rottlerin were purchased from Nacalai Tesque (Kyoto, Japan). All other chemicals were used with the highest purity commercially available.
Preparation of raloxifene transdermal formulations
Raloxifene powder and MC were mixed in distilled water and crushed at 3,000 rpm for 30 s ×30 times by the bead mill method using Bead Smash 12 (Wakenyaku Co. Ltd, Kyoto, Japan).24,25 In the preparation of raloxifene transdermal formulation containing menthol, menthol was added to the milled raloxifene dispersion, then stirred and sonicated. After that, the raloxifene dispersions with or without menthol were gelled by carbopol dissolved in distilled water (without menthol, Ral-NPs; with menthol, mRal-NPs). Transdermal formulations containing raloxifene microparticles were prepared by mixing raloxifene powder, MC, and/or menthol in carbopol gel (without menthol, Ral-MPs; with menthol, mRal-MPs). Table 1 shows the compositions of the transdermal formulations containing raloxifene micro- and nanoparticles.
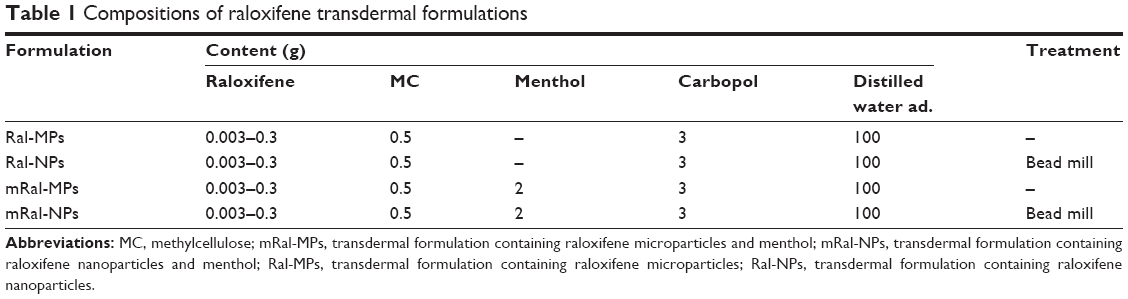 | Table 1 Compositions of raloxifene transdermal formulations |
Particle size and images of raloxifene in transdermal formulations
The particle size of raloxifene was measured by a nanoparticle size analyzer SALD-7100 (Shimadzu Corp., Kyoto, Japan) and NANOSIGHT LM10 (QuantumDesign Japan, Tokyo, Japan), and the number of raloxifene nanoparticles was also determined using the NANOSIGHT LM10. The complex refractive index was evaluated from the measurement data for raloxifene micro- and nanoparticles and set at 1.60–0.010i in the SALD-7100. The conditions for NANOSIGHT LM10 were as follows: measurement time, 60 s; wavelength, 405 nm (blue); viscosity of the suspension; 0.904–0.906 mPa·s. Particle images were obtained under an atomic force microscope using SPM-9700 (Shimadzu Corp.).
Dispersity of raloxifene particles in transdermal formulations
The experiment was performed according to our previous report.24 Raloxifene transdermal formulations were divided into 10 parts and kept at 22°C for 0–30 days. The raloxifene concentration in each part was measured by a LC-20AT system (HPLC; Shimadzu Corp.) with 1 μg/mL methyl p-hydroxybenzoate used as an internal standard. The mobile phase consisted of 50 mM phosphate buffer/acetonitrile (75/25, v/v) at a flow rate of 0.25 mL/min. A 2.1×50 mm Inertsil® ODS-3 column (GL Science Co., Inc., Tokyo, Japan) was used at 35°C, and the wavelength for detection was 287 nm. The particle size, number, and concentration of raloxifene in the samples were obtained by the NANOSIGHT LM10 and HPLC methods described above.
Release of raloxifene from transdermal formulations
The experiment was performed according to our previous report using a membrane filter and Franz diffusion cell.24 An O-ring flange (1.6 cm internal diameter) was placed on a 450 nm-pore size MF™-MEMBRANE FILTER (Merck Millipore, Tokyo, Japan), and 0.3 g of transdermal formulation was spread uniformly over the filter. The filter was set in a Franz diffusion cell (reservoir volume 12.2 mL) filled with 10 mM phosphate buffer (measurement of drug release) or distilled water (measurement of particle size frequency), and 100 μL of sample solution was withdrawn from the reservoir chamber.24 The Franz diffusion cell was thermoregulated at 37°C, and the particle size, number, and concentration of raloxifene in the samples were determined using the NANOSIGHT LM10 and HPLC methods described above. The area under the raloxifene concentration–time curve (AUCRelease) was estimated by the trapezoidal rule up to the last raloxifene measurement point (24 h).
Skin penetration of raloxifene transdermal formulations
The experiment was performed according to our previous report using rat skin and a Franz diffusion cell.24 The hair on the abdominal area of rats was removed on the day before the experiment. The abdominal skin with or without the stratum corneum was removed from the rats at 7 weeks of age, and 0.3% raloxifene transdermal formulation (0.3 g) was spread uniformly over the abdominal skin. The stratum corneum was removed by the tape stripping method as necessary. In the experiment using skin treated with menthol and endocytosis inhibitors, 2% menthol solution or endocytosis inhibitors were applied to the abdominal skin for 1 h prior to the application of the transdermal formulation. After that, the skin samples were set in the Franz diffusion cell filled with 10 mM phosphate buffer (in the experiment involving endocytosis inhibition, 10 mM phosphate buffer with endocytosis inhibitor was used). The experimental conditions were the same as for the Franz diffusion cell experiment described above. In this study, four endocytosis inhibitors dissolved in 0.5% dimethyl sulfoxide (DMSO; 54 μM nystatin,26 caveolae-dependent endocytosis inhibitor; 40 μM dynasore,27 clathrin-dependent endocytosis inhibitor; 2 μM rottlerin,28 macropinocytosis inhibitor; 10 μM cytochalasin D,26 phagocytosis inhibitor) were used. The areas under the penetrated raloxifene concentration–time curves (AUCPenetration) were measured by the AUC method described above.
Percutaneous absorption of raloxifene transdermal formulations
The experiment was performed according to our previous report.24 The hair on the abdominal area was removed on the day before the experiment. Raloxifene 0.3% transdermal formulations (0.3 g) were applied to the shaved abdominal skin of 7-week-old Wistar rats, and blood sample was collected from the right jugular vein. In the repetitive application study, the skin surface was washed with saline, and the transdermal formulation was reapplied every 24 h. The blood sample was centrifuged (800× g, 20 min, 4°C), and the plasma concentrations and the areas under the plasma raloxifene concentration–time curves (AUCPlasma) were measured by the HPLC and AUC methods described above. In addition, the raloxifene concentration was analyzed according to Equations 1–3.24
In a single intravenous injection, 300 μL of raloxifene in 1% DMSO (200 μg/kg) was injected into the femoral vein, and the concentration in the plasma was calculated according to Equation 1:
|
|
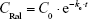
where CRal is raloxifene concentration in the plasma, C0 is the initial concentration in the plasma (40.8±3.9 ng/mL), ke is the elimination rate constant (2.0±0.1×10−2 h−1), and Vd is distribution volume (4.9 mL/g). These data were obtained from seven experiments and used to analyze the pharmacokinetic parameters for the percutaneous absorption by using Equations 2 and 3.
In a single transdermal administration, the percutaneous absorption of raloxifene was calculated according to Equation 2:
|
|
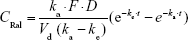
where CRal is the plasma raloxifene concentration, D is the dose (0.3% mRal-NPs, 0.3 g), ka is the absorption rate constant, t is time (0–24 hour), F is the fraction. Vd was obtained from the above data for a single intravenous injection.
The plasma raloxifene concentration data after repetitive transdermal administration (0.3 g/day, interval 24 h) were estimated by Equation 3:
|
|
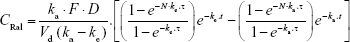
where N and τ are the frequency and interval (24 h), respectively. A nonlinear least-squares fitting program (MULTI Excel edition, Microsoft Office Excel 2016; Microsoft Corporation, Redmond, WA, USA) was employed for these calculation (Equations 1–3).24
Application of raloxifene transdermal formulations to OVX rats
The hair on the abdominal area was removed on the day before the experiment, and 0.003%–0.3% raloxifene transdermal formulations (0.3 g) were applied to the shaved abdominal skin once a day (2:00 pm) for 1 month (6- to 10-week-old OVX rats). After the first application, the hair on the abdominal area was carefully removed every 4 days.
Measurement of Ca content in the bones of OVX rats
The femurs of OVX rats were removed and boiled for 2 h and then calcined in a muffle furnace KDF S-80 (Sansyo Co., Ltd., Osaka, Japan) at 550°C for 48 h. The calcined femur was dissolved in 1% nitric acid and filtered through a 0.45 μm membrane filter. In this study, the calcium ion (Ca2+) content of the filtrated solution was measured using an inductively coupled plasma-atomic emission spectrometer ICPS-7500 (ICP-AES; Shimadzu Corp.) and represented as the calcium (Ca) content in the femur.
Measurement of bone stiffness in OVX rats
The femurs of OVX rats were removed and washed in saline. The stiffness of the washed femur was measured using a hardness tester (ICP-AES; Shimadzu Corp.).
Statistical analysis
A minimum P-value of 0.05 (P<0.05) was chosen as the significance level. Student’s or Aspin–Welch’s t-tests were used for two group comparisons, and one-way ANOVA followed by Dunnett’s multiple comparison was used for multiple group comparisons. Data from the SALD-7100 are expressed as the mean ± SD; other data are expressed as the mean ± standard error (SE) of the mean.
Results
Effect of menthol on the transdermal delivery of raloxifene nanoparticles in rats
Figure 1 shows the size frequency and an image of the raloxifene particles in the transdermal formulation. The raloxifene became meringue-like when subjected to the bead mill method using Ral-MC. On the other hand, the raloxifene particles remained in the nano size range produced by the combination of MC and bead mill treatment, and the particle size and number in 0.3% Ral-NPs were 173.7±7.1 nm and 67.6±1.88×109 particles/0.3 g, respectively (NANOSIGHT LM10). The size frequency and image show no differences between transdermal formulations with or without menthol, and the particle size and number in mRal-NPs were 167.2±6.8 nm and 70.1±1.53×109 particles/0.3 g, respectively. In addition, the levels of dissolved raloxifene were low in the 0.3% raloxifene transdermal formulations listed in Table 1 (Ral-MPs 8.15±0.51 μg/g, mRal-MPs 8.28±0.46 μg/g, Ral-NPs 10.45±0.54 μg/g, mRal-NPs 10.81±0.55 μg/g, n=5). Figure 2 shows the changes in particle size, form, and concentration after the preparation of the raloxifene transdermal formulations. No changes in form or concentration in either Ral-NPs or mRal-NPs were observed 30 days after the bead mill treatment, and the particle size (Ral-NPs 220.0±14.7 nm, mRal-NPs 218.6±14.5 nm) remained in the nanoparticle range. The number of raloxifene nanoparticles in mRal-NPs was also similar to that in Ral-NPs (Ral-NPs 73.4±2.35×109 particles/0.3 g, mRal-NPs 78.6±2.27×109 particles/0.3 g). Figure 3 shows the release of raloxifene from the transdermal formulations. Raloxifene release from Ral-MPs was lower than that from Ral-NPs, and the results show that the raloxifene microparticles were unable to penetrate the 0.45 μm membrane filter since the particle size of the Ral-MPs was 5.47±0.38 μm. Moreover, the addition of menthol had no effect on drug release from raloxifene transdermal formulations. Raloxifene nanoparticles were detected in the reservoir chamber of the Franz diffusion cell 24 h after the application of Ral-NPs, and the mean particle size was 195.5±9.3 nm. The raloxifene particle number in the reservoir chamber 24 h after the application of Ral-NPs was 88% of that in the transdermal formulation (Ral-NPs). In addition, the AUCRelease in the mRal-NPs was similar to Ral-NPs, and the particle size frequency and number of raloxifene molecules from mRal-NPs also showed no significant difference in the values of Ral-NPs. Figure 4 shows the skin penetration profiles of raloxifene following the application of Ral-NPs and mRal-NPs. The skin penetration when Ral-NPs were applied to rats was similar to that when Ral-MPs or mRal-MPs were applied, although mRal-NPs showed significantly increased skin penetration. In addition, the skin penetration of raloxifene nanoparticles was also enhanced by pretreatment with menthol. On the other hand, no raloxifene nanoparticles were detected in the reservoir chamber of the Franz diffusion cell 0–24 h after the application of transdermal formulations containing raloxifene micro- or nanoparticles (raloxifene nanoparticles were not detectable by NANOSIGHT LM10). Endocytosis is the major route by which nanomedicines are transported across the membrane. Therefore, we investigated the relationship between endocytosis and skin penetration of mRal-NPs using various types of endocytosis inhibitors. Figure 5 shows the relationship between endocytosis and skin penetration of mRal-NPs. Skin penetration in rats treated with nystatin (caveolae-dependent endocytosis inhibitor), dynasore (clathrin-dependent endocytosis inhibitor), and cytochalasin D (phagocytosis inhibitor) were similar to the vehicle groups. On the other hand, the skin penetration of mRal-NPs was significantly decreased by treatment with rottlerin (macropinocytosis inhibitor), and the AUCpenetration treated with rottlerin was only 30.7% of the vehicle.
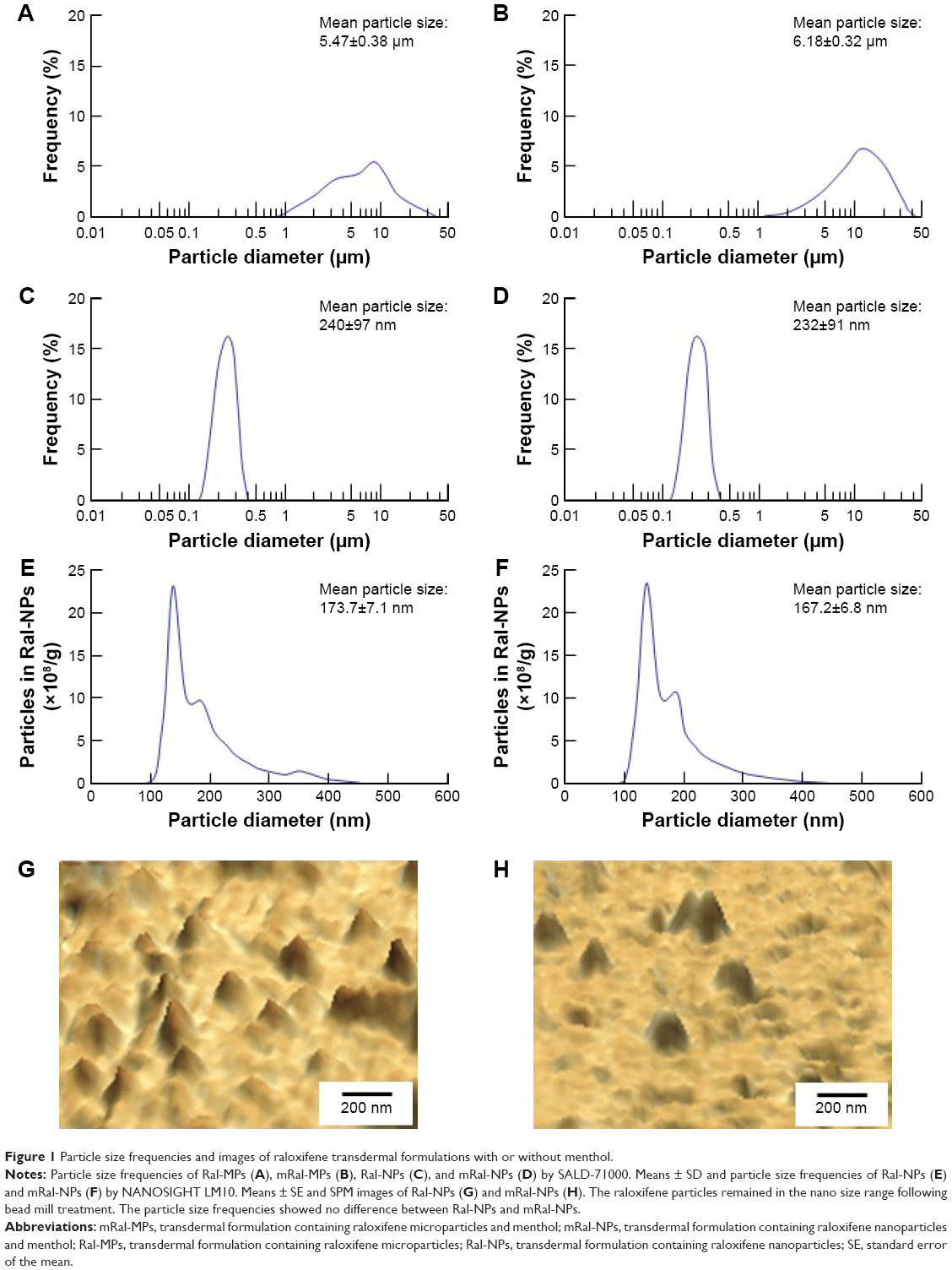 | Figure 1 Particle size frequencies and images of raloxifene transdermal formulations with or without menthol. |
 | Figure 2 Stability of raloxifene transdermal formulations 15 and 30 days after bead mill treatment. |
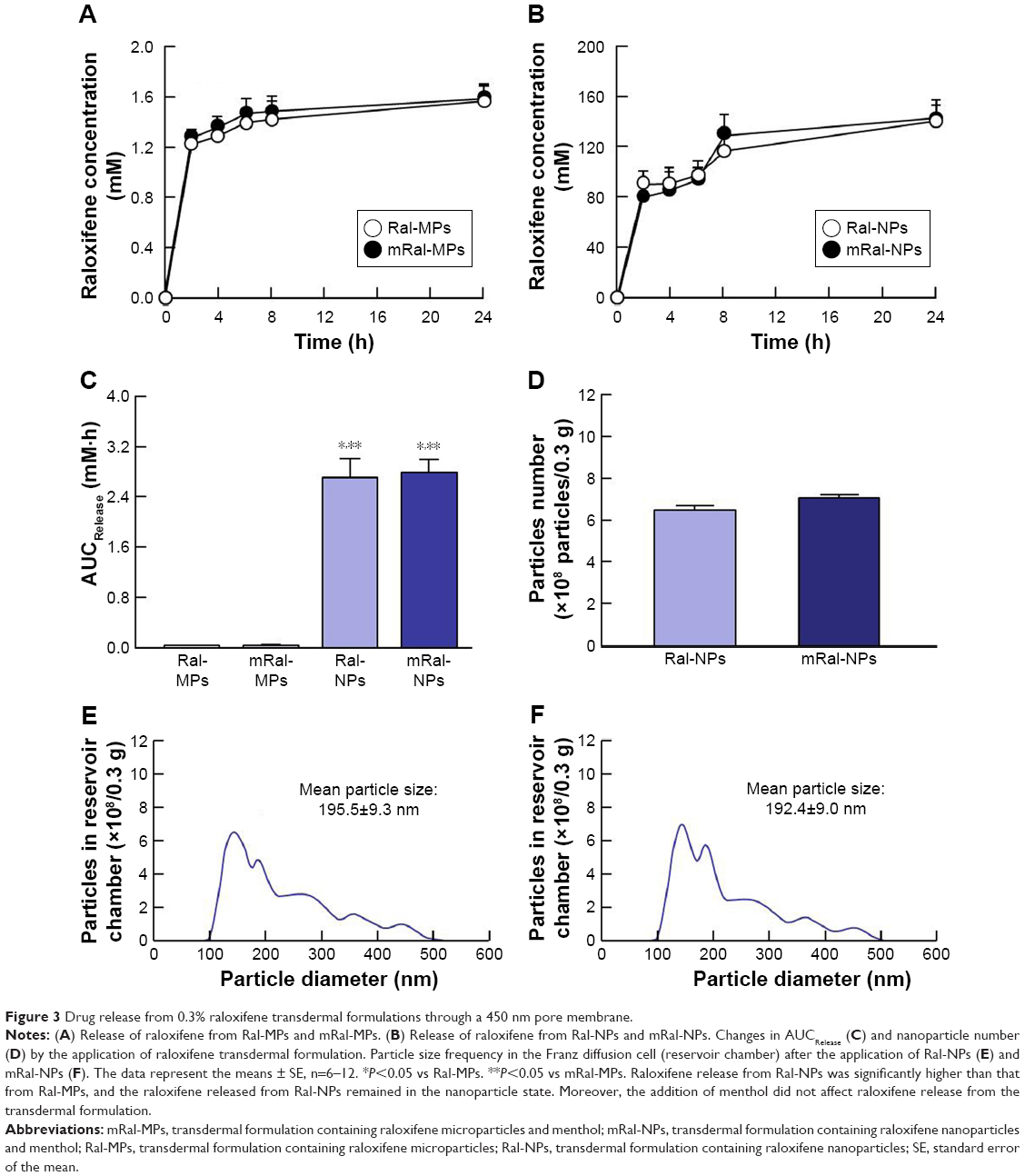 | Figure 3 Drug release from 0.3% raloxifene transdermal formulations through a 450 nm pore membrane. |
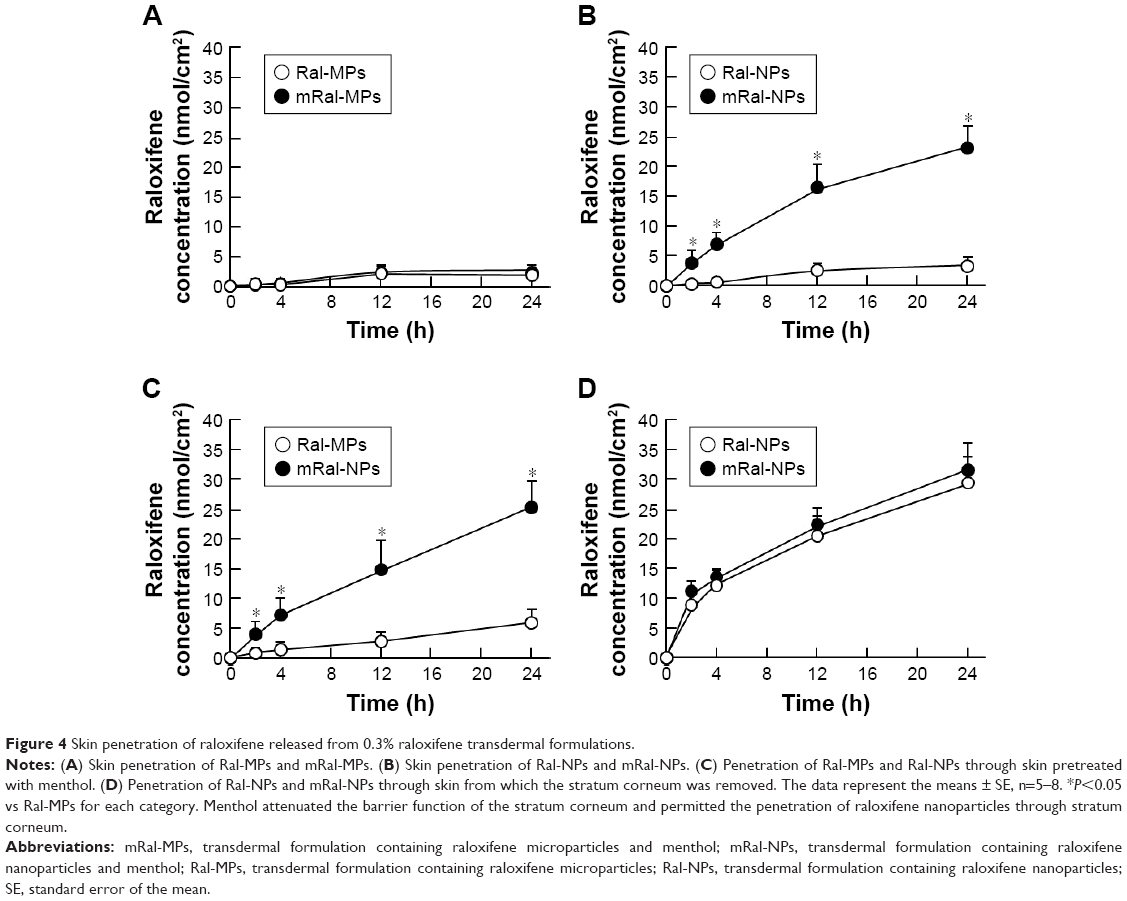 | Figure 4 Skin penetration of raloxifene released from 0.3% raloxifene transdermal formulations. |
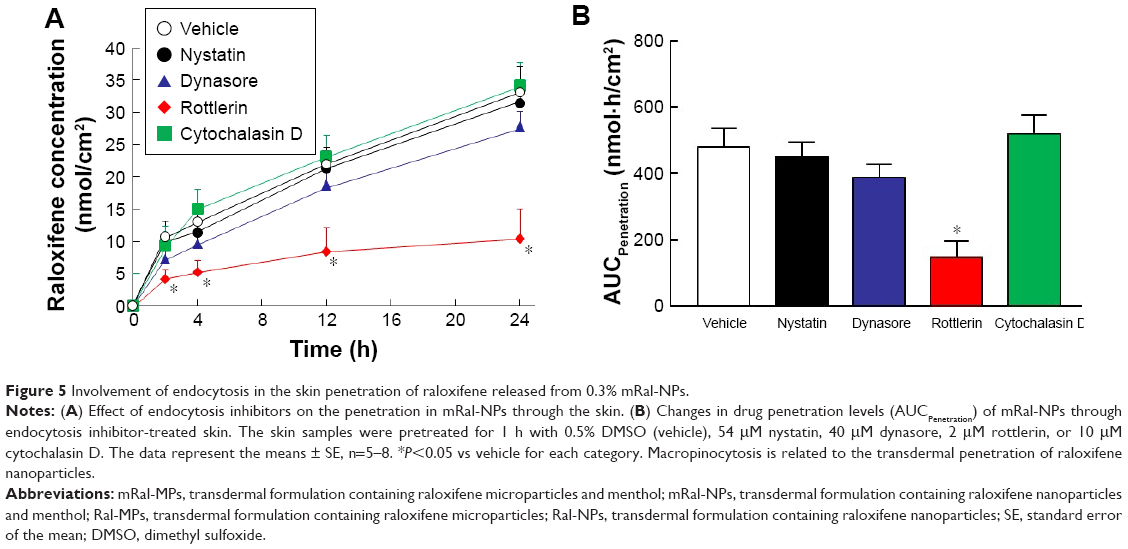 | Figure 5 Involvement of endocytosis in the skin penetration of raloxifene released from 0.3% mRal-NPs. |
Therapeutic effect of mRal-NPs on osteoporosis in OVX rats
Figure 6 shows the percutaneous absorption from raloxifene transdermal formulations, and Table 2 summarizes the pharmacokinetic parameters analyzed from the data for the percutaneous absorption of 0.3% mRal-NPs (Figure 6B). No differences were found between the percutaneous absorptions from Ral-MPs and Ral-NPs, and the Cmax of the plasma raloxifene concentration was 5.4 nM. The plasma concentration of raloxifene in rats treated with mRal-MPs was also similar to that of rats treated with Ral-MPs. On the other hand, the percutaneous absorption was enhanced by the combination of raloxifene nanoparticles and menthol, with the AUCPlasma in rats treated with mRal-NPs significantly higher than that in rats treated with Ral-NPs. The BA was 8.5%, and the steady state concentration (minimum–maximum) of raloxifene was 47.1–76.4 nM for the repetitive application of 0.3% Ral-NPs (0.03%, 3.9–7.8 nM; 0.003%, 0.39–0.79 nM). Figure 7A, C, and E shows changes in body weight (A), Ca content of the bone (C), and bone stiffness (E) of rats following ovariectomy. The increase in body weight in OVX rats was lower than that in sham rats. Twenty-eight days after ovariectomy, the Ca content and stiffness of the bones of OVX rats were 74.5% and 65.3% of those of sham rats, respectively. Figure 7B, D, and F shows the changes in body weight (B), Ca content of the bone (D), and bone stiffness (F) in OVX rats treated with 0.003%–0.3% mRal-NPs. The application of 0.3% Ral-MPs or 0.3% mRal-MPs had no effect on body weight (Ral-MPs 251±18 g, mRal-MPs 248±17 g, n=5), Ca content of the bone (Ral-MPs 366±21 mg/g, mRal-MPs 377±25 mg/g, n=5), or bone stiffness (Ral-MPs 19.2±1.9 kg, mRal-MPs 20.5±1.8 kg, n=5), and no therapeutic effects were observed in OVX rats treated with 0.3% Ral-NPs (body weight 259±15 g, Ca contents 378±23 mg/g, bone stiffness 20.8±1.6 kg, n=5). In contrast to the results of 0.3% Ral-MPs, mRal-MPs, and Ral-NPs, the decreases in body weight and the Ca content and stiffness of the bone of OVX rats were attenuated by the application of 0.3% mRal-NPs: body weight, Ca content, and stiffness of the bone were 1.16-, 1.15-, and 1.36-fold of those of vehicle-treated OVX rats, respectively.
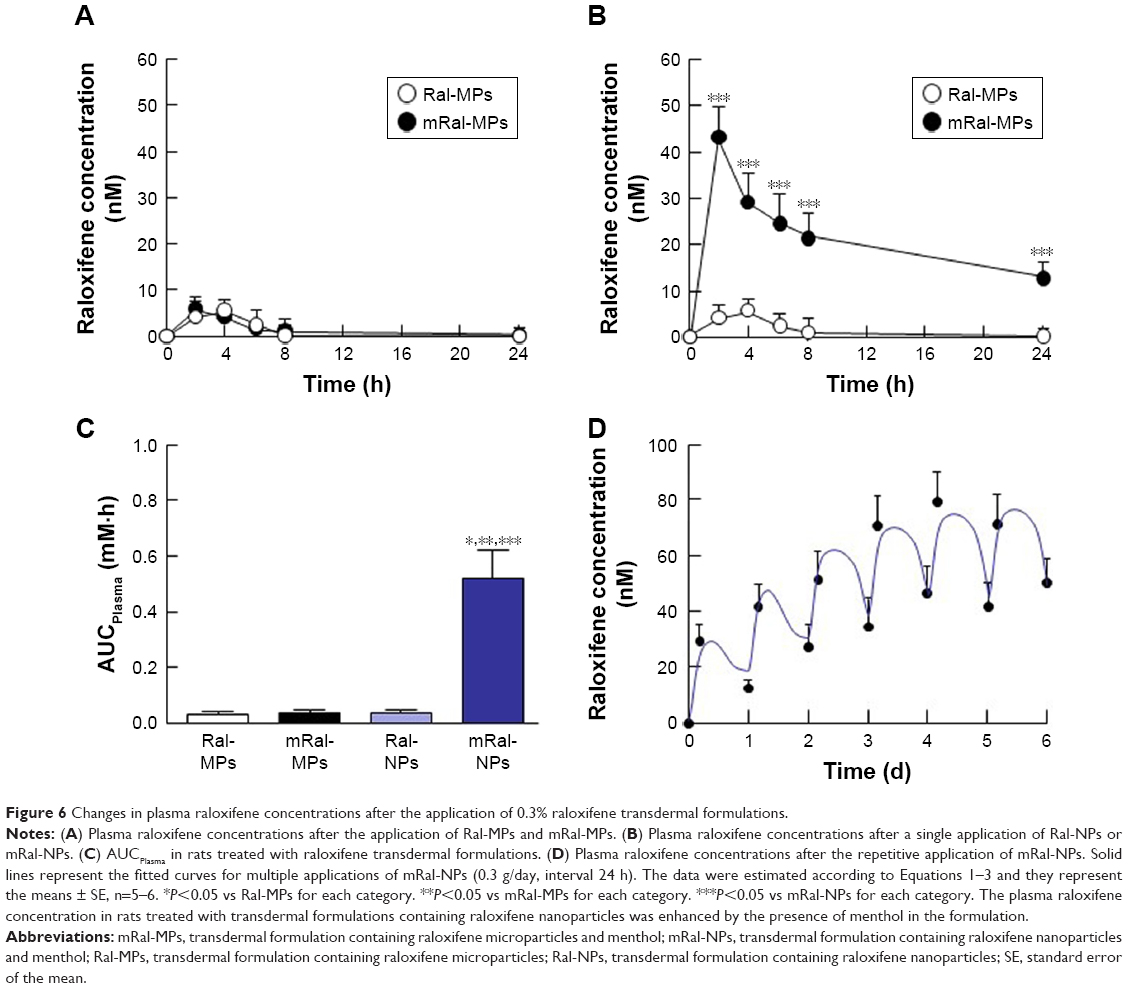 | Figure 6 Changes in plasma raloxifene concentrations after the application of 0.3% raloxifene transdermal formulations. |
 | Table 2 Pharmacokinetic parameters for the percutaneous absorption of mRal-NPs |
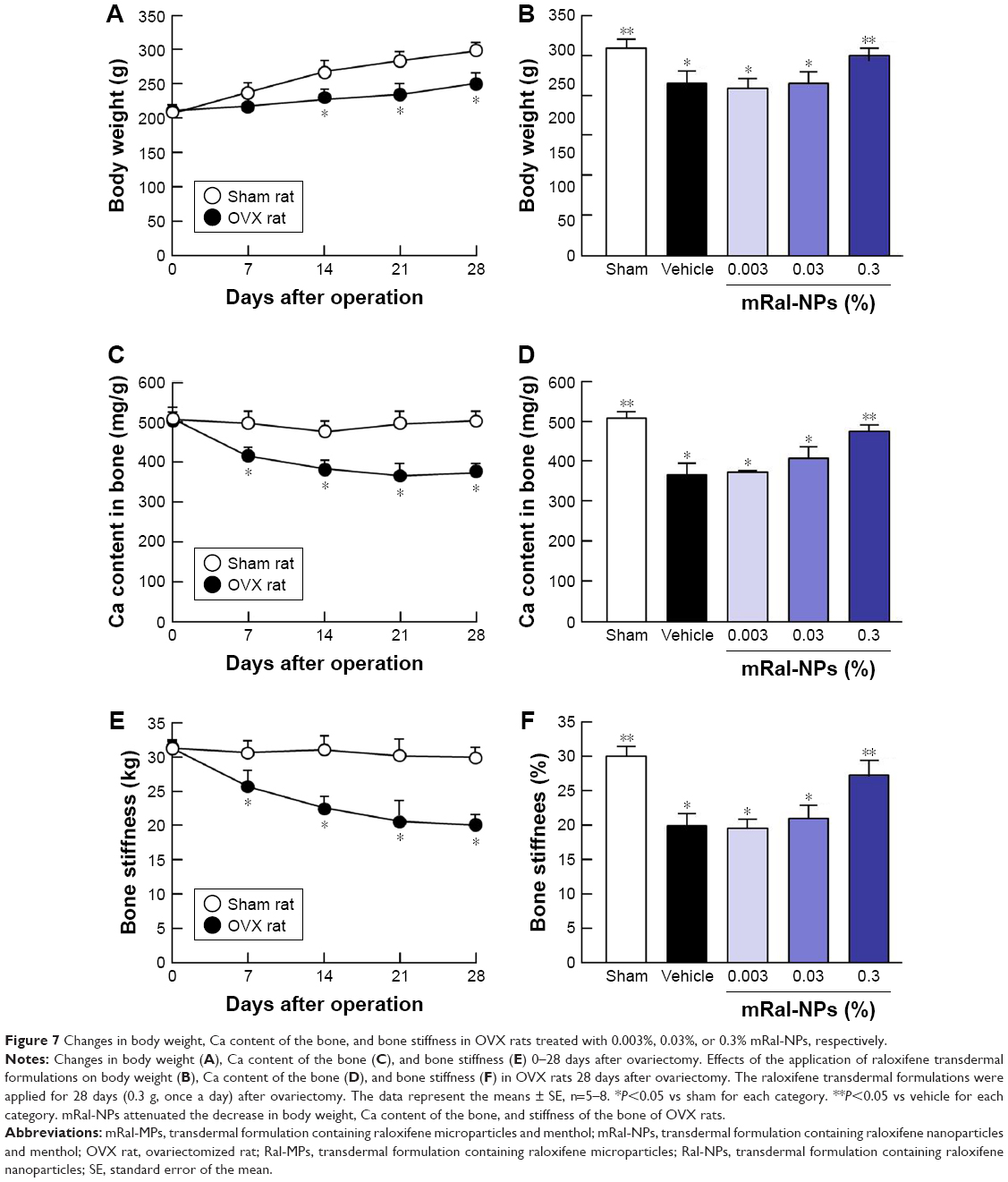 | Figure 7 Changes in body weight, Ca content of the bone, and bone stiffness in OVX rats treated with 0.003%, 0.03%, or 0.3% mRal-NPs, respectively. |
Discussion
Raloxifene is a second-generation SERM used as therapy for osteoporosis in postmenopausal women. In the clinical setting, raloxifene is administered orally; however, the absolute BA of raloxifene is only 2% because of its poor solubility in aqueous fluids and its extensive first-pass metabolism.29 Therefore, it is expected that the development of a transdermally delivered formulation may reduce the necessary dose without compromising therapeutic efficacy. Many techniques to enhance drug delivery through transdermal routes have been studied, such as micro-needles10,11 and iontophoresis.14,15 Recently, it was reported that the ability of a drug to penetrate across the skin can be improved by taking advantage of the nanoparticle size.19,24 In this study, we prepared raloxifene solid nanoparticles by bead mill methods and designed a novel transdermal formulation containing nanoparticles and permeation enhancers. These transdermal formulations overcome the barrier properties of the skin and show high drug penetration through the transdermal route (BA 8.5%). In addition, we found that raloxifene transdermal formulations are useful for the treatment of osteoporosis in OVX rat.
In previous studies, we showed that MC permits the preparation of nanoparticles by mill methods,24,25 and that carbopol, a water-soluble gel, is suitable for facilitating drug release from transdermal formulations containing nanoparticles.24 Therefore, MC and carbopol were selected as additives in the preparation of transdermal formulations containing raloxifene nanoparticles. The size of the raloxifene particles as measured by the NANOSIGHT LM10 decreased to 173.7±7.1 nm by bead mill treatment (Figure 1C, E, and G). On the other hand, the mean particle size as measured by the SALD-700 was 240 nm. This discrepancy may arise from the fact that SALD-7100 is a laser diffraction analyzer, whereas NANOSIGHT LM10 measures particle size by a dynamic light scattering method. The difference in the measurement methods may lead to the discrepancy in particle size. Raloxifene in the transdermal formulations is released as nanoparticles, and the release rate of raloxifene nanoparticles from Ral-NPs into the Franz diffusion cell was 88% (Figure 3D). In general, it is known that passage through the stratum corneum is the rate-limiting step in percutaneous absorption and presents the greatest resistance to penetration. The infiltration of raloxifene nanoparticles from the transdermal formulations is also impeded by the stratum corneum, with the skin penetration of rats treated with Ral-NPs similar to that of rats treated with Ral-MPs (Figure 4A and B). We previously reported that particles less than 100 nm in size (<100 nm) can penetrate skin tissue.24 However, the size of the raloxifene particles in Ral-NPs is greater than the drug particles in our previous studies using tranilast,24 indomethacin,30 and ketoprofe.31 These results show that it may be difficult for particles larger than 100 nm to penetrate through the stratum corneum.
Terpenes, terpenoids, essential oils, pyrrolidones, fatty acids, fatty acid ester sulfoxides, alcohols, and glycerides have been used as permeation enhancers for transdermal drug delivery and can help overcome the barrier properties of the stratum corneum.32 Menthol is a terpene alcohol that at concentrations of 1%–10% (w/v) have been used to enhance percutaneous absorption. Based on these findings, we prepared a transdermal formulation containing raloxifene nanoparticles and 2% menthol as an enhancer (mRal-NPs) to overcome the difficulties in penetration through the stratum corneum. In contrast to the results of Ral-NPs, mRal-NPs (with menthol) allowed the drug to penetrate through skin tissue containing stratum corneum (Figure 4B) and dissolved only raloxifene (not raloxifene particles) which was observed in the reservoir chamber of the Franz diffusion cell (not detectable by NANOSIGHT LM10). It is known that menthol enhances skin permeation by two mechanisms. In a transdermal delivery study using ibuprofen and menthol, Stott et al33 reported that a hydrogen bonding interaction is the primary mechanism by which some terpenes form binary eutectic mixtures with a drug (ibuprofen). The resultant melting point depression of the delivery system is correlated with a significant increase in transdermal permeation. As a second possible mechanism, menthol preferentially distributes into the intercellular spaces of the stratum corneum, thus altering the barrier properties of the stratum corneum and causing a reversible disruption of the lipid domains, resulting in an increase in drug skin absorption.34,35 Although there were no differences between the penetration of Ral-MPs through skin pretreated with or without menthol, menthol enhanced the penetration of raloxifene nanoparticles in the transdermal formulation (Figure 4). In addition, the solubility, particle number, and shape of raloxifene were similar among the transdermal formulations with or without menthol (Figure 1). These results indicate that menthol attenuates the barrier function of the stratum corneum, resulting in enhanced penetration of raloxifene nanoparticles into the skin tissue.
It is important to elucidate the mechanism of the transdermal absorption of raloxifene nanoparticles. We show that raloxifene nanoparticles penetrate the barrier of the stratum corneum, dissolve in the skin tissue, and are released from the skin as soluble raloxifene (Figure 4). Endocytosis is the major route by which nanomedicines are transported across the membrane, and pathways of endocytosis can be classified as phagocytosis or pinocytosis.36–38 Phagocytosis is a special endocytic pathway that occurs predominantly in phagocytes, such as macrophages, neutrophils, and monocytes.36 On the other hand, pinocytosis is the major route by which cells take up suspensions containing nanoparticles and can be divided into clathrin-dependent endocytosis, caveolae-dependent endocytosis, and macropinocytosis.37,38 Based on these reports, we investigated which endocytosis pathways are related to the skin penetration of raloxifene nanoparticles by using inhibitors specific for the individual pathways (nystatin [caveolae-dependent endocytosis inhibitor], dynasore [clathrin-dependent endocytosis inhibitor], rottlerin [macropinocytosis inhibitor], and cytochalasin D [phagocytosis inhibitor])26–28 and found that macropinocytosis accounts for the skin penetration of raloxifene nanoparticles (Figure 5). Zhang et al39 reported that the size of the vesicles varies with the specific pathway of pinocytosis in the endocytic process, and the sizes corresponding to clathrin-dependent endocytosis, caveolae-dependent endocytosis, and macropinocytosis are <120 nm, <80 nm, and 100 nm to 5 μm, respectively. The size frequency of raloxifene particles in mRal-NPs was in the range of 100–450 nm (mean particle size 167.2 nm). Taken together, it is possible that raloxifene nanoparticles reach the underlying epidermis via the stratum corneum and are absorbed into cells by macropinocytosis. After that, the raloxifene may dissolve and be diffused into the epidermis and dermis, resulting in transition into the blood circulation (Figure 8).
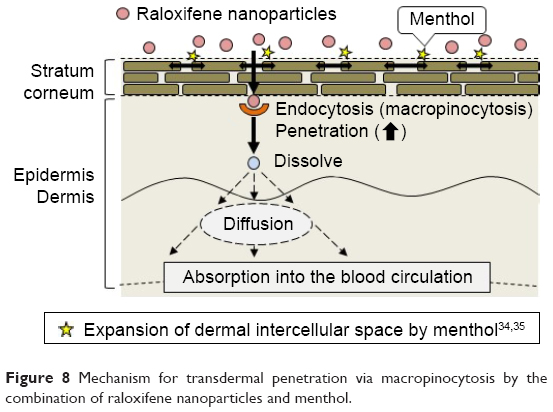 | Figure 8 Mechanism for transdermal penetration via macropinocytosis by the combination of raloxifene nanoparticles and menthol. |
We found that rottlerin reduced the skin penetration of mRal-NPs, but there was still about 25% penetration. Therefore, dissolved raloxifene on the stratum corneum may also reflect the skin penetration of raloxifene from the mRal-NPs.
In this study, we also demonstrated the therapeutic effect of mRal-NPs on osteoporosis using OVX rats as a model (Figure 7). The Ca contents and stiffness of the bones of OVX rats decreased following ovariectomy, and the application of 0.3% mRal-NPs attenuated the decreases in Ca content and stiffness of the bones of OVX rats (Figure 7D and F). These results support the data for the plasma concentration of raloxifene in rats treated with the raloxifene transdermal formulations (Figure 6) and show that mRal-NPs provide useful therapy for osteoporosis. On the other hand, the OVX rat was used as a model for post-menopausal osteoporosis, and the measurements of drug skin penetration and pharmacokinetics in rats have been used in many studies to evaluate transdermal formulations. However, the data obtained in rats are not consistent with the data in humans. Therefore, a study using human tissue needs to be carried out. Further studies are needed to elucidate the relationships of lysosomes via macropinocytosis and raloxifene dissolution in the percutaneous absorption process of raloxifene nanoparticles. Moreover, it is important to clarify the promoting effect of permeation enhancers on transdermal penetration. Therefore, we are now planning to investigate the correlation between drug particle size and endocytosis and demonstrate the activation of endocytosis (macropinocytosis) after the application of mRal-NPs using immunohistochemistry methods.
Conclusion
We designed a novel nanomedicine (transdermal formulation containing raloxifene nanoparticles) for which the BA of raloxifene in the transdermal formulation is 8.5% (BA in rats administered raloxifene orally is 1.7%). Moreover, we investigated the mechanism of percutaneous absorption and the therapeutic effect on osteoporosis. It is possible that both raloxifene dissolved on the stratum corneum and nanoparticles passed through the stratum corneum reflect the skin penetration of raloxifene in the mRal-NPs. In addition, it is hypothesized that menthol attenuates the barrier function of the stratum corneum, allowing raloxifene particles that are ~100–450 nm in size to transit into the underlying epidermis. After that, the raloxifene may dissolve and diffuse into the epidermis, dermis, and ultimately be absorbed into the blood circulation. The enhanced levels of plasma raloxifene provide efficient and effective therapy for osteoporosis. In addition, these results suggest that macropinocytosis is related to the skin penetration of raloxifene nanoparticles into the epidermis or dermis layers of the skin. These findings provide significant information that can be used to design further studies aimed at developing transdermal delivery systems using nanoparticles.
Author contributions
NN conceived and designed the study and wrote the manuscript. HO performed the experiments for the preparation of nanoparticles and analyzed the data. YN performed the transdermal penetration experiments. FO and NK performed the experiments to test the therapeutic effect using OVX rats. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Das S, Crockett JC. Osteoporosis – a current view of pharmacological prevention and treatment. Drug Des Devel Ther. 2013;7:435–448. | ||
Sultan N, Rao J. Association between periodontal disease and bone mineral density in postmenopausal women: a cross sectional study. Med Oral Patol Oral Cir Bucal. 2011;16(3):e440–e447. | ||
Jagadish B, Yelchuri R, K B, et al. Enhanced dissolution and bioavailability of raloxifene hydrochloride by co-grinding with different superdisintegrants. Chem Pharm Bull. 2010;58(3):293–300. | ||
Bikiaris D, Karavelidis V, Karavas E. Novel biodegradable polyesters. Synthesis and application as drug carriers for the preparation of raloxifene HCl loaded nanoparticles. Molecules. 2009;14(7):2410–2430. | ||
Cummings SR, Eckert S, Krueger KA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA. 1999;281(23):2189–2197. | ||
Delmas PD, Bjarnason NH, Mitlak BH, et al. Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N Engl J Med. 1997;337(23):1641–1647. | ||
Elsheikh MA, Elnaggar YS, Gohar EY, Abdallah OY. Nanoemulsion liquid preconcentrates for raloxifene hydrochloride: optimization and in vivo appraisal. Int J Nanomedicine. 2012;7:3787–3802. | ||
Garg A, Singh S, Rao VU, Bindu K, Balasubramaniam J. Solid state interaction of raloxifene HCl with different hydrophilic carriers during co-grinding and its effect on dissolution rate. Drug Dev Ind Pharm. 2009;35(4):455–470. | ||
Yousaf AM, Mustapha O, Kim DW, et al. Novel electrosprayed nanospherules for enhanced aqueous solubility and oral bioavailability of poorly water-soluble fenofibrate. Int J Nanomedicine. 2016;11:213–221. | ||
So JW, Park HH, Lee SS, Kim DC, Shin SC, Cho CW. Effect of microneedle on the pharmacokinetics of ketoprofen from its transdermal formulations. Drug Deliv. 2009;16(1):52–56. | ||
Katsumi H, Tanaka Y, Hitomi K, et al. Efficient transdermal delivery of alendronate, a nitrogen-containing bisphosphonate, using tip-loaded self-dissolving microneedle arrays for the treatment of osteoporosis. Pharmaceutics. 2017;9(3):pii: E29. | ||
Cagnie B, Vinck E, Rimbaut S, Vanderstraeten G. Phonophoresis versus topical application of ketoprofen: comparison between tissue and plasma levels. Phys Ther. 2003;83(8):707–712. | ||
Sharma DN, Hanesh M, Ali A. Phonophoresis with diclofenac versus ketoprofen for knee joint injuries Middle. East J Int Med. 1999;2:9–12. | ||
Tashiro Y, Kato Y, Hayakawa E, Ito K. Iontophoretic transdermal delivery of ketoprofen: effect of iontophoresis on drug transfer from skin to cutaneous blood. Biol Pharm Bull. 2000;23(12):1486–1490. | ||
Panus PC, Campbell J, Kulkarni SB, Herrick RT, Ravis WR, Banga AK. Transdermal iontophoretic delivery of ketoprofen through human cadaver skin and in humans. J Control Release. 1997;44(2–3):113–121. | ||
Shinkai N, Korenaga K, Okumura Y, Mizu H, Yamauchi H. Microdialysis assessment of percutaneous penetration of ketoprofen after transdermal administration to hairless rats and domestic pigs. Eur J Pharm Biopharm. 2011;78(3):415–421. | ||
Shinkai N, Korenaga K, Mizu H, Yamauchi H. Intra-articular penetration of ketoprofen and analgesic effects after topical patch application in rats. J Control Release. 2008;131(2):107–112. | ||
Maestrelli F, González-Rodríguez ML, Rabasco AM, Mura P. Preparation and characterisation of liposomes encapsulating ketoprofen-cyclodextrin complexes for transdermal drug delivery. Int J Pharm. 2005;298(1):55–67. | ||
Dragicevic N, Maibach H. Combined use of nanocarriers and physical methods for percutaneous penetration enhancement. Adv Drug Deliv Rev. 2018;127(18):pii:S0169. | ||
Kaur A, Katiyar SS, Kushwah V, Jain S. Nanoemulsion loaded gel for topical co-delivery of clobitasol propionate and calcipotriol in psoriasis. Nanomedicine. 2017;13(4):1473–1482. | ||
Huang HC, Barua S, Sharma G, Dey SK, Rege K. Inorganic nanoparticles for cancer imaging and therapy. J Control Release. 2011;155(3):344–357. | ||
Boisselier E, Astruc D. Gold nanoparticles in nanomedicine: preparations, imaging, diagnostics, therapies and toxicity. Chem Soc Rev. 2009;38(6):1759–1782. | ||
Delouise LA. Applications of nanotechnology in dermatology. J Invest Dermatol. 2012;132(3 Pt 2):964–975. | ||
Nagai N, Ito Y. Therapeutic effects of gel ointments containing tranilast nanoparticles on paw edema in adjuvant-induced arthritis rats. Biol Pharm Bull. 2014;37(1):96–104. | ||
Nagai N, Ito Y, Okamoto N, Shimomura Y. A nanoparticle formulation reduces the corneal toxicity of indomethacin eye drops and enhances its corneal permeability. Toxicology. 2014;319:53–62. | ||
Mäger I, Langel K, Lehto T, Eiríksdóttir E, Langel U. The role of endocytosis on the uptake kinetics of luciferin-conjugated cell-penetrating peptides. Biochim Biophys Acta. 2012;1818(3):502–511. | ||
Malomouzh AI, Mukhitov AR, Proskurina SE. Non-quantal release of acetylcholine in murine neuromuscular junctions. Dokl Biol Sci. 2014;459:330–333. | ||
Hufnagel H, Hakim P, Lima A, Hollfelder F. Fluid phase endocytosis contributes to transfection of DNA by PEI-25. Mol Ther. 2009;17(8):1411–1417. | ||
Heringa M. Review on raloxifene: profile of a selective estrogen receptor modulator. Int J Clin Pharmacol Ther. 2003;41(8):331–345. | ||
Nagai N, Yoshioka C, Ito Y. Topical therapies for rheumatoid arthritis by gel ointments containing indomethacin nanoparticles in adjuvant-induced arthritis rat. J Oleo Sci. 2015;64(3):337–346. | ||
Nagai N, Iwamae A, Tanimoto S, Yoshioka C, Ito Y. Pharmacokinetics and antiinflammatory effect of a novel gel system containing ketoprofen solid nanoparticles. Biol Pharm Bull. 2015;38(12):1918–1924. | ||
Sinha VR, Kaur MP. Permeation enhancers for transdermal drug delivery. Drug Dev Ind Pharm. 2000;26(11):1131–1140. | ||
Stott PW, Williams AC, Barry BW. Transdermal delivery from eutectic systems: enhanced permeation of a model drug, ibuprofen. J Control Release. 1998;50(1–3):297–308. | ||
Kunta JR, Goskonda VR, Brotherton HO, Khan MA, Reddy IK. Effect of menthol and related terpenes on the percutaneous absorption of propranolol across excised hairless mouse skin. J Pharm Sci. 1997;86(12):1369–1373. | ||
Kaplun-Frischoff Y, Touitou E. Testosterone skin permeation enhancement by menthol through formation of eutectic with drug and interaction with skin lipids. J Pharm Sci. 1997;86(12):1394–1399. | ||
Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. | ||
Rappoport JZ. Focusing on clathrin-mediated endocytosis. Biochem J. 2008;412(3):415–423. | ||
Wang J, Byrne JD, Napier ME, Desimone JM. More effective nanomedicines through particle design. Small. 2011;7(14):1919–1931. | ||
Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Size-dependent endocytosis of nanoparticles. Adv Mater. 2009;21(4):419–424. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.