Back to Journals » International Journal of Nanomedicine » Volume 13
Design and immunological evaluation of anti-CD205-tailored PLGA-based nanoparticulate cancer vaccine
Authors Jahan ST, Sadat SMA, Haddadi A ![]()
Received 18 June 2017
Accepted for publication 12 August 2017
Published 11 January 2018 Volume 2018:13 Pages 367—386
DOI https://doi.org/10.2147/IJN.S144266
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Sheikh Tasnim Jahan, Sams MA Sadat, Azita Haddadi
Division of Pharmacy, College of Pharmacy and Nutrition, University of Saskatchewan, Saskatoon, SK, Canada
Abstract: The aim of this research was to develop a targeted antigen–adjuvant assembled delivery system that will enable dendritic cells (DCs) to efficiently mature to recognize antigens released from tumor cells. It is important to target the DCs with greater efficiency to prime T cell immune responses. In brief, model antigen, ovalbumin (OV), and monophosphoryl lipid A adjuvant were encapsulated within the nanoparticle (NP) by double emulsification solvent evaporation method. Targeted NPs were obtained through ligand incorporation via physical adsorption or chemical conjugation process. Intracellular uptake of the NPs and the maturation of DCs were evaluated with flow cytometry. Remarkably, the developed delivery system had suitable physicochemical properties, such as particle size, surface charge, OV encapsulation efficiency, biphasic OV release pattern, and safety profile. The ligand modified formulations had higher targeting efficiency than the non-tailored NPs. This was also evident when the targeted formulations expressed comparatively higher fold increase in surface activation markers such as CD40, CD86, and major histocompatibility complex class II molecules. The maturation of DCs was further confirmed through secretion of extracellular cytokines compared to control cells in the DC microenvironment. Physicochemical characterization of NPs was performed based on the polymer end groups, their viscosities, and ligand-NP bonding type. In conclusion, the DC stimulatory response was integrated to develop a relationship between the NP structure and desired immune response. Therefore, the present study narrates a comparative evaluation of some selected parameters to choose a suitable formulation useful for in vivo cancer immunotherapy.
Keywords: nanoparticle, immunotherapy, ovalbumin, monophosphoryl lipid A, dendritic cells
Introduction
Cancer is the most detrimental disease in first world nations as well as developing countries. The goal of cancer therapy is to eradicate tumor cells without affecting normal healthy cells.1 Moreover, therapeutic approaches are smartly chosen based on their selective targeting of malignant cells. Nowadays, novel treatment options like immunotherapy have excelled with favorable outcomes and limited side effects. It is a well-accepted complementary intervention for patients with weakened immune system resulting from long-term cancer treatments. It has the potential to augment the therapeutic response with synergistic efficacy when combined with other treatment modalities.2 The immunotherapeutic approach appears as an ideal treatment option due to its unique mechanism of action.3
A cancer patient receives immunotherapy through therapeutic vaccine that induces antigen-specific T cells and tumor relevant chemokine receptors but avoids attraction of tumor-induced suppressor cells and their expansion.4 This type of vaccine aims in the clonal expansion of cytotoxic T cells that identifies cancer cells and kills them by sequential events of perforin and granzyme release, thereby activating the caspase cascade and finally leading to apoptosis.5,6 On the contrary, clinical trials were found to be ineffective initially. Failure in clinical trials is due to the lack of optimal immune stimulation (either for mature dendritic cell [DC] or suppressive DC), inadequate antigen loading, incorrect route of administration and dose, and prior analysis of DC transcriptosome.7,8 Therefore, researchers were inspired to manipulate DCs with target specific tools to fight the limitation of the immune system to detect cancer.
In vivo therapeutic vaccination involves the targeted delivery of antigen to DCs which will fuse tumor antigen to antibody (Ab) specific for a DC receptor. This strategy is cost-effective and less laborious compared to ex vivo DC vaccines.9 For example, when anti-CD205 Ab is conjugated with ovalbumin (OV), the receptor-mediated antigen presentation increases significantly for major histocompatibility complex (MHC) class I and MHC class II (MHCII). The target CD205 receptor could continually delete peripheral T cells at steady state to establish tolerance and solely provide immunity when DCs are matured.10 Whereas, in the case of ex vivo vaccination, when DCs are injected, problems such as failure to reach lymph node, insufficient amount of injected DCs, and disorder of immuno-function due to immunosuppressive environments occur. Besides, the molecules in that environment (CTLA-4, PD-1, CD200, interleukin [IL]-10, TGF-β) are capable of inhibiting T cell function by the formation of regulatory T cells.11
Nanoparticle (NP)-based delivery systems are popular carriers for delivery of antigenic material to antigen-presenting cells (APCs).12 To become an ideal delivery system, the combination of antigen and adjuvant is very important. This cargo system will be able to deliver specific antigen to the APCs to manipulate their response.13,14 The present study aimed to deliver surface-modified NP to obtain immunological responses as mentioned in Figure 1. Surface-modified NPs possess a hydrophilic layer that tends to escape the reticuloendothelial system to reach receptors on target APCs, specifically DCs.15
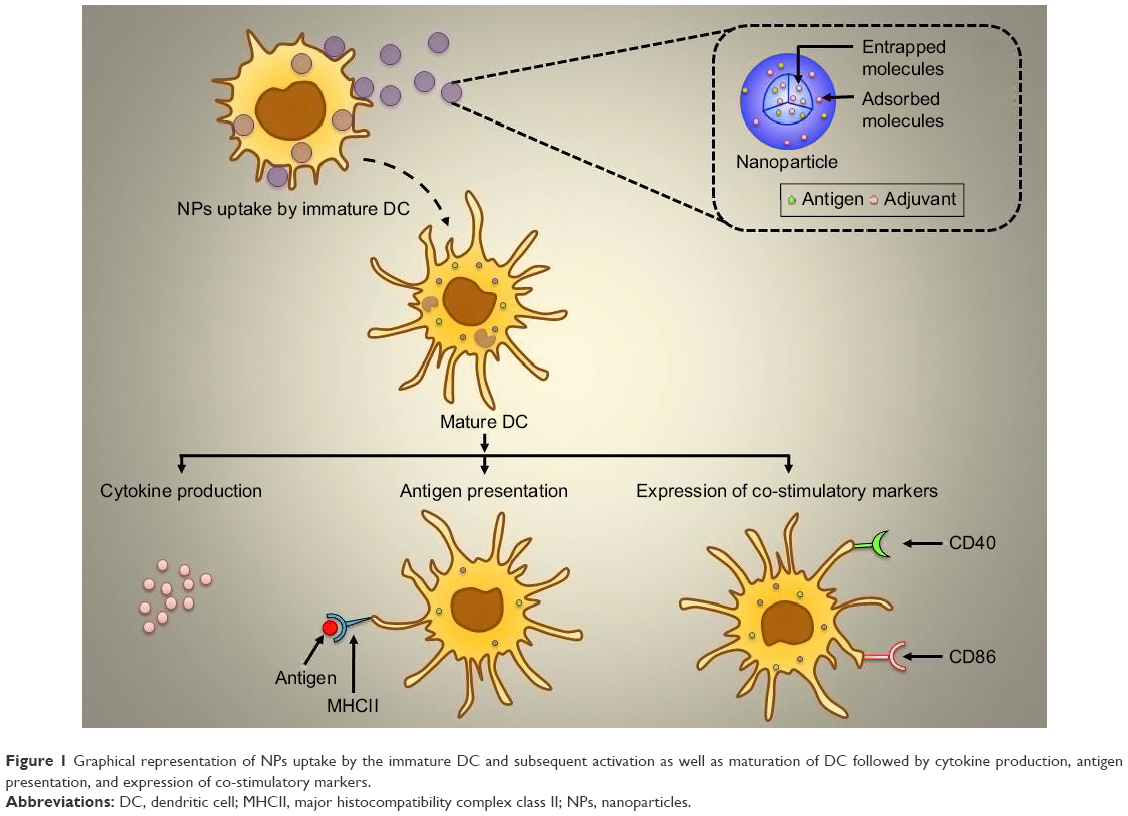 | Figure 1 Graphical representation of NPs uptake by the immature DC and subsequent activation as well as maturation of DC followed by cytokine production, antigen presentation, and expression of co-stimulatory markers. |
Immunostimulatory particulate delivery systems with biodegradable polymers have gained popularity due to their size, controlled release properties, and biocompatibility with tissues. Poly-lactic-co-glycolide (PLGA)-based delivery system has become an interesting strategy for antigen-based cancer immunotherapy.16 Biodegradable PLGA is approved by the US Food and Drug Administration and European Medicine Agency for use in humans.17 The encapsulation of protein antigen in the PLGA particle protects the protein from degradation in vivo. The antigen-loaded particles release antigen in a controlled manner to prolong the availability of antigen in vivo. However, the extended release of antigen as well as induction of innate and adaptive immune response are concerns.18,19
For this study, we have chosen anti-CD205 monoclonal Ab (MAb) to target CD205 receptor on DCs because it is an extensively studied receptor for antigen delivery.20–22 The formulations were designed using capped (ester) and uncapped (COOH) PLGA, both offering low and high viscosity grades. Results are discussed based on a relative evaluation of the physicochemical properties, in vitro studies, and immunological responses. The purpose of this study was to assess several formulations that would be safe to deliver their content in a controlled release manner to obtain necessary immunotherapeutic response.23
Materials and methods
Materials
Monophosphoryl lipid A (MP; molecular weight 1,763.469 Da) was obtained from Avanti Polar Lipids Inc (Alabaster, AL, USA). Ester-terminated PLGA 50:50 (inherent viscosity [iv] 0.15–0.25 dL/g and 0.55–0.75 dL/g) and COOH-terminated PLGA 50:50 (iv 0.18 dL/g and 0.55–0.75 dL/g) were purchased from LACTEL Absorbable Polymers (Birmingham, AL, USA). The ivs chosen for the respective end groups were designated as low- and high-viscosity ester/COOH-terminated PLGA. Polyvinyl alcohol (PVA), bis(sulfosuccinimidyl) suberate (BS), OV from chicken egg white (grade V), coumarin-6, alpha minimum essential medium (MEM), fetal bovine serum (FBS), and bicinchoninic acid (BCA) assay kit were purchased from Sigma-Aldrich Co (St Louis, MO, USA). Cell titer nonradioactive cell proliferation assay 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was purchased from Promega Corporation (Fitchburg, WI, USA). Biotin anti-mouse CD205 MAb was purchased from BioLegend (San Diego, CA, USA). JAWSII DC line was obtained from American Type Culture Collection (Manassas, VA, USA). Granulocyte-macrophage colony-stimulating factor (GM-CSF) and slow fade gold antifade mountant with 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Thermo Fischer Scientific (Waltham, MA, USA). Anti-mouse CD16/CD32 MAb (Fc blocker), CD40, CD86, MHCII Abs, and their respective isotype controls were purchased from BD Biosciences (San Jose, CA, USA). Murine interferon-γ (IFNγ), IL12p70, IL-6, and tumor necrosis factor (TNF)-α enzyme-linked immunosorbent assay (ELISA) kits were purchased from eBioscience (San Diego, CA, USA). All other reagents such as ethyl acetate, methanol, sodium hydroxide (NaOH), and sodium dodecyl sulfate (SDS) were of analytical grade.
Methods
Preparation, surface modification, and quantification of coumarin-6-loaded NPs
PLGA NPs were prepared using the oil/water emulsification solvent evaporation technique as previously mentioned.24 Briefly, 6.5% (w/v) PLGA was dissolved in ethyl acetate solvent. Subsequently, 100 μg of coumarin-6 was added to this organic phase. The organic phase was transferred in drops into the aqueous phase comprising 2.2% PVA. To obtain pre-activated NPs, BS was present in the aqueous PVA solution. The resulting primary emulsion was stirred, washed, and resuspended in water. Cryoprotectant (10% sucrose) was added to obtain aggregation-free lyophilized NPs.25 The freeze-dried NPs were surface modified with anti-CD205 Ab via adsorption (AD) and covalent (COV) attachment process as mentioned previously.26 Briefly, 20 mg of NPs were taken and dispersed in phosphate-buffered saline (PBS; pH 7.2), followed by stirring (4°C, 4 hours) and washing of unattached anti-CD205 Ab. The amount of Ab was quantified by BCA assay.27
The amount of coumarin-6 was quantified by fluorimetry using a microplate reader with an excitation wavelength at 430 nm and an emission wavelength at 485 nm. First, coumarin-6-loaded NPs were dispersed in dichloromethane for 24 hours.28 The supernatant was collected after washing the NPs twice by centrifugation. A standard curve of coumarin-6 was obtained in the concentration ranging from 0.78 to 100 μg/mL.25 The encapsulation efficiency (EE) and loading of coumarin-6 were calculated based on the following equations:29
|
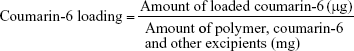
|

Preparation, surface modification, and quantification of OV-loaded NPs
OV-loaded NPs were prepared by water/oil/water double emulsification solvent evaporation method as previously described.30 In brief, 100% OV in PBS solution was transferred to PLGA-ethyl acetate solution (6.5%). MP (200 μg) dissolved in 1:4 methanol–chloroform mixture was added to the PLGA mixture for the formulations with MP. The sonicated primary emulsion was added to 2.2% of PVA to form a secondary emulsion. Once the organic solvent evaporated, the suspension was washed twice, freeze-dried, and stored at −20°C. All the formulations were cryopreserved with 10% sucrose solution.14
Surface modification of OV-loaded NPs was carried out using physical AD and COV attachment method as mentioned in the “Preparation, surface modification, and quantification of coumarin-6-loaded NPs” section. The amount of OV in NPs was quantified using BCA assay.31 Briefly, 5 mg of NPs were re-dispersed in 3 mL of 0.05 M NaOH containing 1% SDS. The suspension was incubated overnight in an orbital shaker. Next day, the supernatant was collected after centrifugation. Samples were placed in a 96-well plate (Corning Costar; Corning Inc, Corning, NY, USA), and absorbance was measured at 565 nm using a microplate reader. The result was compared with a standard curve obtained by OV in the concentration range of 15.6–1,000 μg/mL.30,32
Cell viability assay
DCs were seeded at a density of 10,000 cells per well in 96-well plates with complete media containing alpha MEM, 20% FBS, 5 ng/mL GM-CSF, and 1% penicillin–streptomycin–amphotericin B cocktail.33 For the assay, MTS reagent was used following the manufacturer’s directions. Briefly, NP suspension was diluted in complete medium and 20 μL of each dilution containing 1 mg/mL OV was added in triplicate wells. The samples were incubated for 72 hours. After that, 10 μL of MTS reagent was added to each well and the plate was incubated again for 2 hours at 37°C. The absorbance was measured at 490 nm with a microplate reader.34
OV release study
In vitro release studies of OV from PLGA NPs were carried out by incubating a specific amount of NPs in 2 mL of PBS at 37°C with agitation. At predetermined times (for a total of 20 days), the suspension was centrifuged. OV-containing supernatants were collected and stored at −20°C. Later, the protein present in samples was determined by BCA assay and read with a microplate reader at 565 nm.35
DC uptake study
DCs were treated with different formulations of coumarin-6-loaded NPs. DCs were harvested after 24 hours of incubation with the formulations. The cells were washed with ice-cold fluorescence activated cell sorting (FACS) buffer to remove non-internalized NPs. About 2×105 cells were incubated with anti-mouse Fc blocker for 15 minutes. The suspension was then washed with FACS buffer. Test groups were surface modified NPs and non-modified NPs. Untreated cells were used as negative control. Fold increase in DC uptake was calculated by dividing the mean fluorescence intensity (MFI) of coumarin-6-positive cells (test groups) by untreated cells. All samples were acquired on a BD FACS Calibur machine (BD Biosciences) and analyzed using Kaluza flow software (Beckman Coulter, Brea, CA, USA).30 For all flow cytometry analysis, cell debris and small particles were excluded by setting a gate on the dot plot of side scattering and forward scattering. A total of 10,000 gated events were used for the analysis. The detector was adjusted in a way so that the negative population appears in the first logarithmic decade.
Intracellular localization of NPs analyzed by confocal laser scanning microscopy (CLSM)
About 5×106 DCs were seeded in a six-well plate. After stabilization for 2 hours, the DCs were treated with coumarin-6-loaded NPs. After overnight incubation, the cells were washed with PBS and fixed with 4% paraformaldehyde solution for 20 minutes. The cell nuclei were then counterstained for 20 minutes with DAPI (1 mg/mL). Cells were examined under fluorescence microscopy (IX71-F22FL/PH; Olympus Corporation, Tokyo, Japan). Finally, the uptake was visualized in a glass bottom Petri dish with CLSM (LSM 510 Meta; Carl Zeiss Meditec AG, Jena, Germany).36
Characterization of surface phenotype by flow cytometry
Different NP formulations were incubated overnight with about 5×105 DCs in six-well plates. After overnight incubation, about 2×105 DCs were collected. Then, cells were washed thoroughly and incubated with Fc blocker to block any nonspecific binding. Next, the cells were stained with phycoerythrin conjugated specific Abs for CD40, anti-CD86, or MHCII for 30 minutes at 4°C. The isotype standard for each maturation marker was also used to measure background fluorescence intensity.37 Lipopolysaccharide (LPS) 1 μg/mL was used as positive control, and an unstimulated cell was used as negative control. NP-DC samples were acquired on a FACS Calibur machine and analyzed using CellQuest Pro software (BD Biosciences).
Detection of cytokine secretion
The supernatants from the DC culture were stored at −20°C for analysis of the level of IL-12, IL-6, IFNγ, and TNF-α using commercially available ELISA kits. The samples were then placed in a 96-well microplate using a microplate reader at 450 nm (background 570 nm) according to the manufacturer’s instructions.37 The minimum detection levels of the cytokines were 15, 4, 15, and 15 pg/mL for IL-12, IL-6, IFNγ, and TNF-α, respectively.
Statistical analysis
Data are given as mean ± standard deviation, and statistical significance was determined by Student’s t-test. Values of p<0.05 were considered statistically significant unless specifically mentioned. Data were analyzed using GraphPad Prism 5.03 software (GraphPad Software, Inc, La Jolla, CA, USA).
Results
Characterization of coumarin-6-loaded plain and Ab-modified NPs
Table 1 summarizes the NP size, polydispersity index (PDI), zeta potential (ZP), EE, and coumarin-6 loading (per mg of NPs). The particle size of plain NPs ranged from 121±5 to 162±5 nm. All the formulations were cryopreserved with 10% sucrose. Following Ab attachment (both AD and COV), the particle size ranged from 201±10 to 562±5 nm. The increase in polymer viscosity in turn increased both particle size and PDI although they were not significantly different. The EE of coumarin-6 ranged from 73.91%±2.87% to 88.13%±2.01% for plain NPs and 64.13%±2.98% to 73.75%±0.99% for Ab-modified formulations. The presence of BS did not show any effect on the formulations. Moreover, particle size demonstrated a significant relationship with PDI, ZP, EE, and loading of coumarin-6 (***p<0.05). The formulations (plain and modified) had ZP values of −8.32±0.19 to −1.93±0.18 mV. No significant relation within the groups was observed.
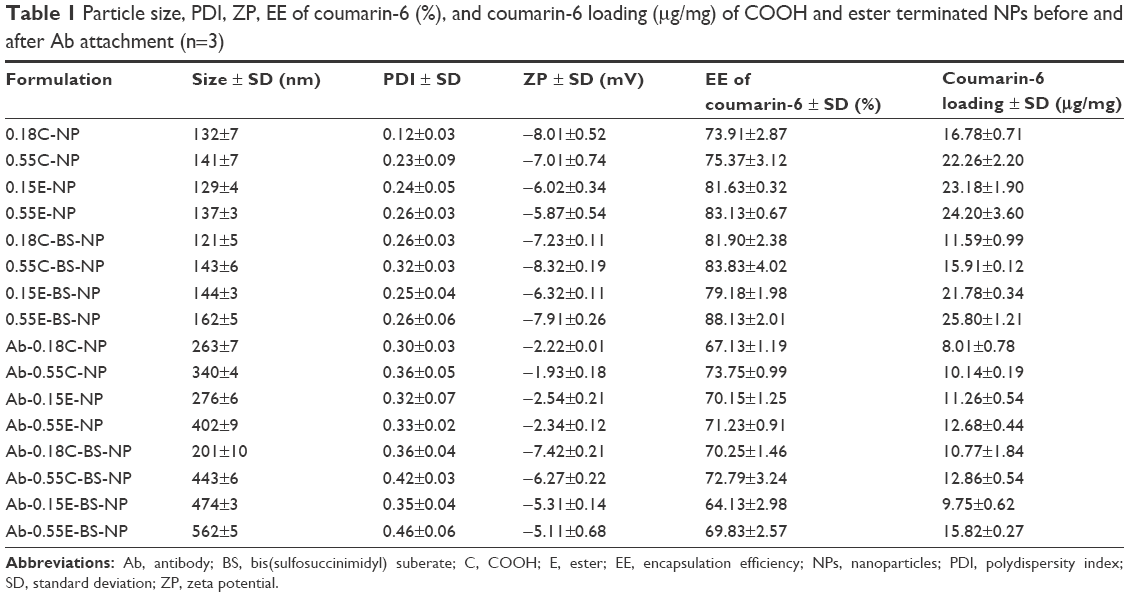 | Table 1 Particle size, PDI, ZP, EE of coumarin-6 (%), and coumarin-6 loading (μg/mg) of COOH and ester terminated NPs before and after Ab attachment (n=3) |
DC uptake study
A quantitative uptake of coumarin-6-loaded NPs by DCs is represented in Figure 2A and B. Treatment of DCs with Ab-modified NPs allowed higher uptake of particles compared to non-modified NPs as found in other studies.30 As shown in Figure 2A(a–h), compared to untreated cells (control), a clear significant shift for MFI toward the right for coumarin-6-loaded NPs was observed in all the histograms. Moreover, the highest shift was visible when Ab was present in the formulation.
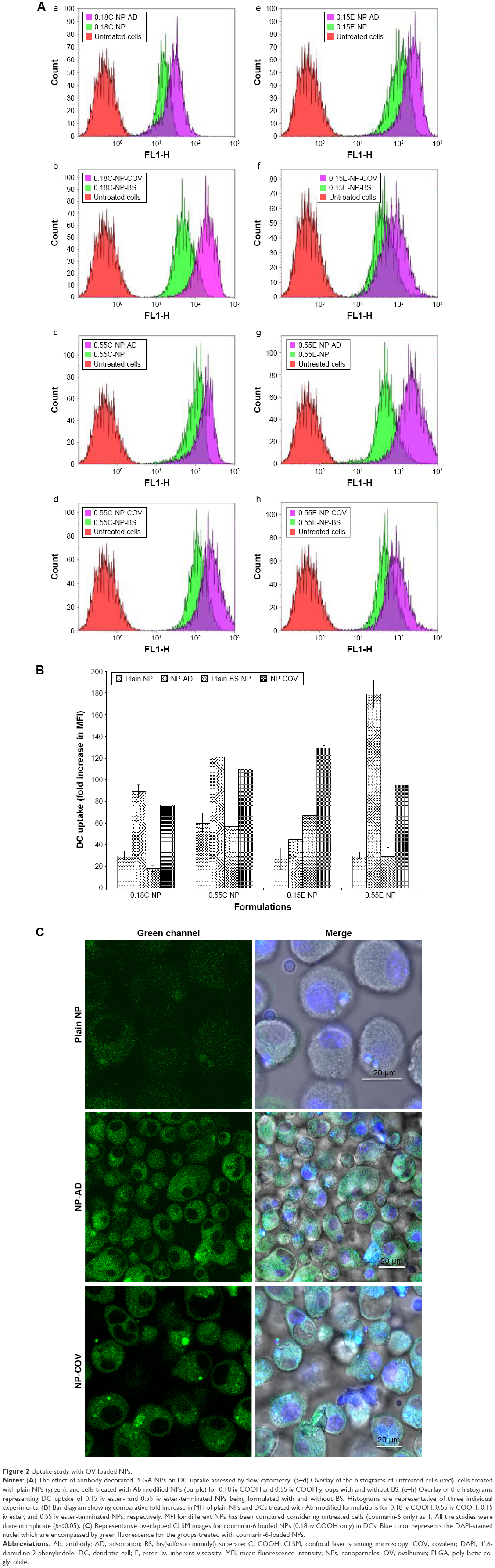 | Figure 2 Uptake study with OV-loaded NPs. |
The bar diagrams in Figure 2B represent the comparative fold increase in MFI for all the treatment groups. For example, there was a significant increase in MFI for the Ab adsorbed COOH (0.18 iv) terminated NPs compared to the plain unmodified formulations (***p<0.05). For these two groups, the fold increase in MFI was 30 [Plain NP] and 89 [NP-AD] compared to control (***p<0.05). Similarly, COV attached 0.18 iv COOH terminated NPs [NP-COV] showed a significantly higher fold increase in MFI (77) compared to that (MFI 18) of plain unmodified formulations [Plain-BS-NP] (***p<0.05). However, COV attached formulation of these PLGA NPs [NP-COV] had lower MFI than its Ab adsorbed [NP-AD] formulations (**p<0.05). A similar trend was observed for the high viscosity COOH terminated PLGA NPs and fold increase in MFI for [Plain NP], [NP-AD], [Plain-BS-NP], and [NP-COV] was 60, 121, 57, and 110, respectively. In addition, when comparing between two viscosity grades, the high viscosity formulations showed significantly higher fold increase in MFI compared to the low viscosity COOH terminated formulations (***p<0.05).
For ester-terminated NPs, there was a significant difference for fold increase in MFI for the plain and Ab-modified formulations (***p<0.05). The BS containing 0.15 iv ester formulations had comparatively higher fold increase in MFI when compared with the formulations without BS (***p<0.05). The fold increase in MFI for [Plain NP], [NP-AD], [Plain-BS-NP], and [NP-COV] of low viscosity ester terminated PLGA NPs was 27, 45, 67, and 129, respectively. When comparing between the [Plain NP] and [NP-AD] formulations of high viscosity ester PLGA, [NP-AD] formulations showed 179 times higher fold increase in MFI (***p<0.05). A similar result was observed for Ab-adsorbed ester-ended NPs of the two viscosity grades. When both viscosities of ester-terminated NPs were compared, the high-viscosity [NP-COV] formulations did not necessarily show a higher fold increase in MFI as observed for its low-viscosity grade. Among the low-viscosity PLGA NP formulations, 0.15E-COV had the highest targeting efficiency as represented in Figure 2B.
Figure 2C represents the intracellular localization of coumarin-6-loaded NPs (green color) that was further confirmed by CLSM. The images show that anti-CD205 Ab-modified NPs had comparatively higher uptake of NPs by DCs than unmodified NPs.
Characterization of OV/OV-MP-loaded plain and Ab-modified NPs
Table 2 represents the NP size, PDI, ZP, EE, and loading (per mg of NPs) of OV. The size of OV-, OV-MP-, Ab-OV-, and Ab-OV-MP-loaded PLGA NPs were within 171±3, 159±4, 501±9, and 486±4 nm, respectively. The ZP for all Ab free OV and OV-MP NPs ranged from −27.1±0.77 to −18.42±0.03 mV. The integration of Ab reduced the overall negative surface charge of NPs. For the Ab-modified formulations, the zeta values ranged from 1.31±0.17 to 4.54±0.21 mV. Good reproducibility in size and ZP was observed for different batches of NPs. The PDI, ZP, EE, and OV loading had significant relationship with particle size (***p<0.05). The OV EE for the OV/OV-MP NPs and Ab-modified OV/OV-MP NPs were within 59.15%±1.56% and 45.24%±0.09%, respectively. There was no significant difference within groups as analyzed by statistical software.
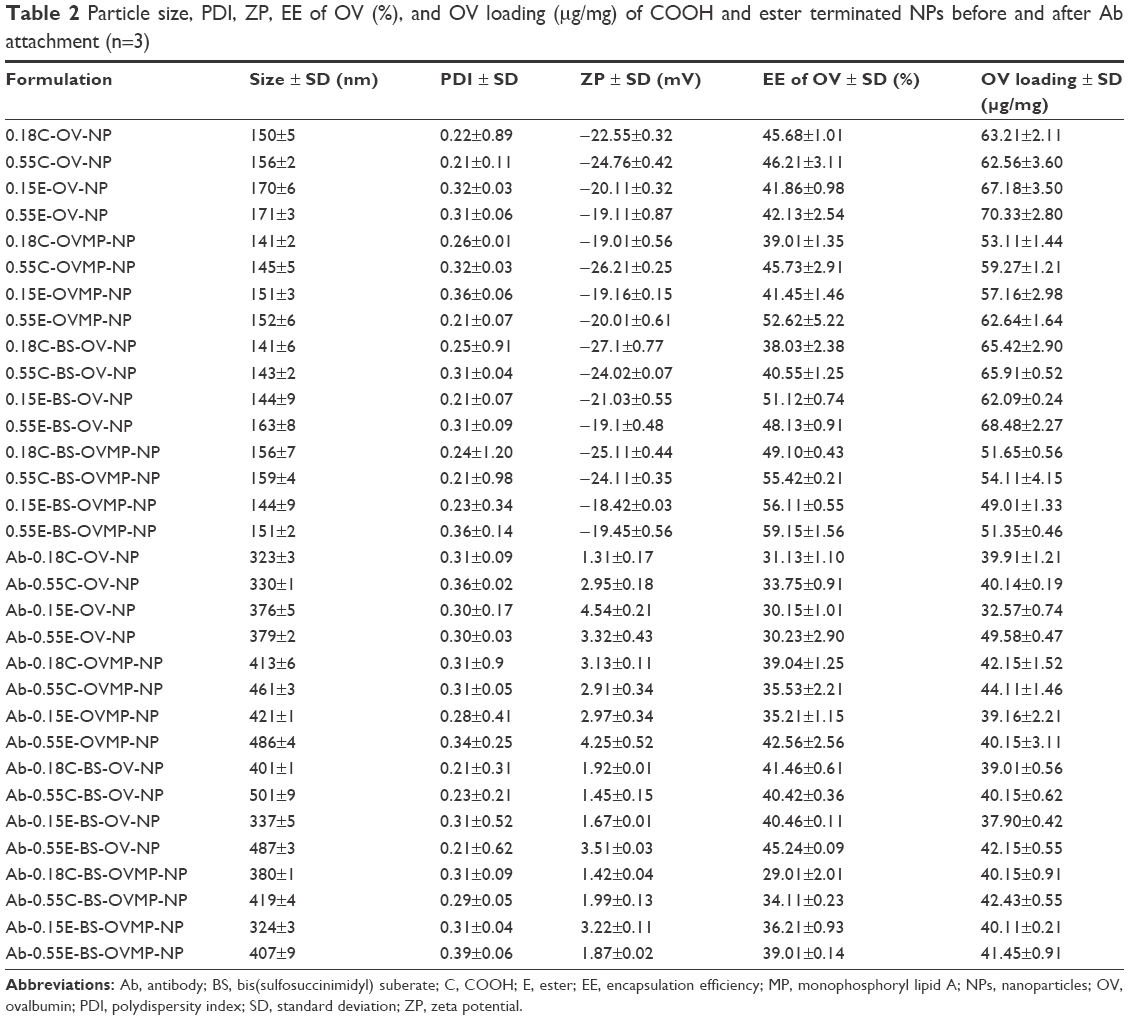 | Table 2 Particle size, PDI, ZP, EE of OV (%), and OV loading (μg/mg) of COOH and ester terminated NPs before and after Ab attachment (n=3) |
OV release study
The in vitro release study for OV-loaded NPs demonstrated a substantial effect on release percentage depending on the polymer’s molecular weight and type. After 12 hours, the cumulative release of OV from low- and high-viscosity COOH-terminated NPs was 39.88% and 30.87%, respectively (Figure 3A). Similarly (after 12 hours), OV release for the low- and high-viscosity ester-terminated NPs was 31.49% and 26%, respectively (Figure 3A). The release following 12 hours was significantly higher than the initial (first hour) release (*p<0.05). After day 7, the release of OV from both COOH- and ester-terminated NPs increased cumulatively (Figure 3B; *p<0.05). About 2 weeks were required to release about 50% of the OV from the high-viscosity PLGA NPs (COOH > ester; *p>0.05); whereas, low-viscosity PLGA NPs were found to release 50% of OV within 24 hours (COOH ended) and 7 days (ester ended). However, all OV-loaded PLGA NPs exhibited biphasic behavior over 20 days of release study as represented in Figure 3.23
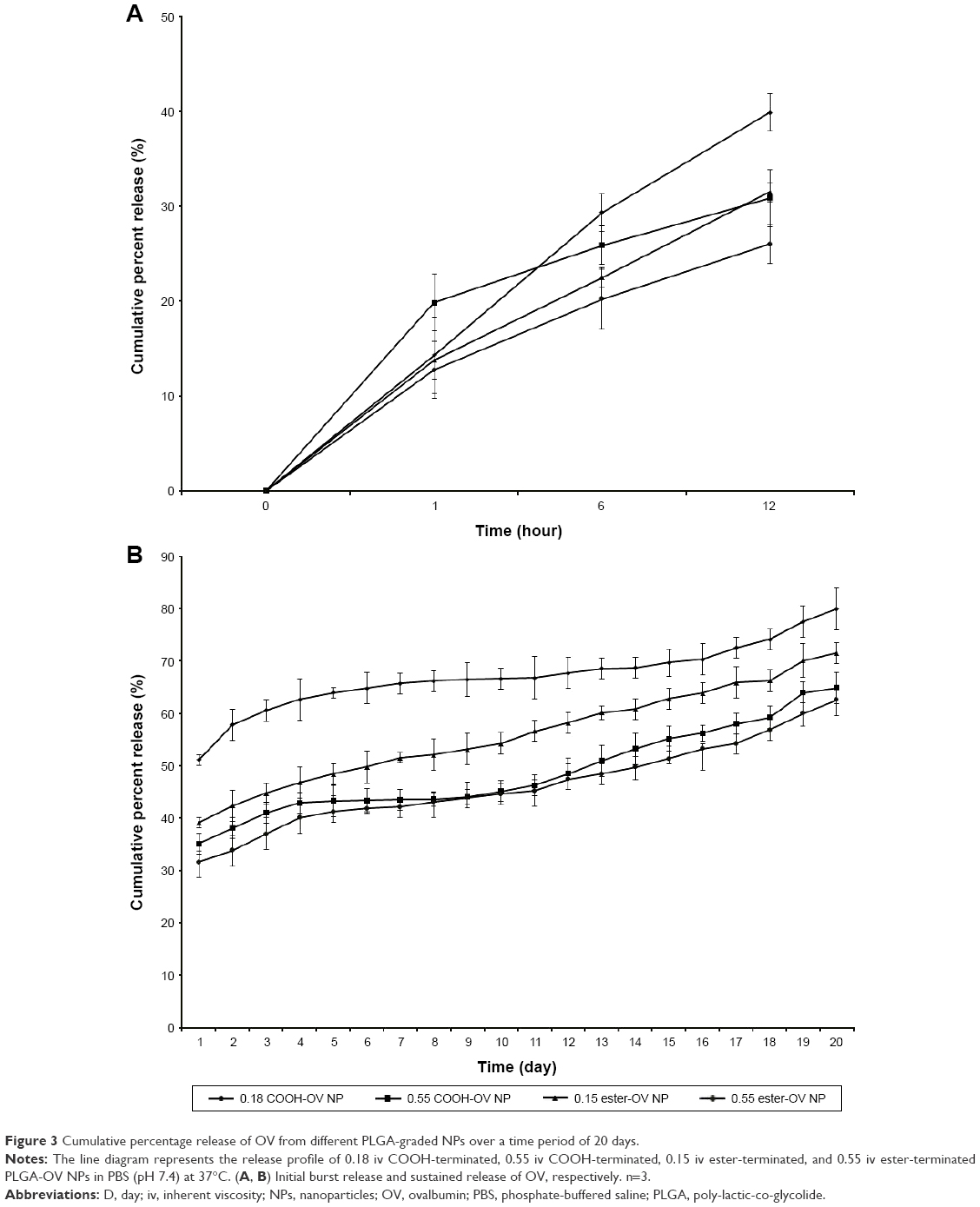 | Figure 3 Cumulative percentage release of OV from different PLGA-graded NPs over a time period of 20 days. |
Cell viability assay
Cells treated with DCs were about 79%–93% viable at an OV concentration of 0.5 mg/mL.38 Plain NPs without OV were less toxic with about 90%–100% viability as mentioned previously.24 Soluble OV showed reasonable toxicity to DCs (79.34%). No significant reduction of viability was observed due to the presence of MP in the formulations as represented in Figure 4.
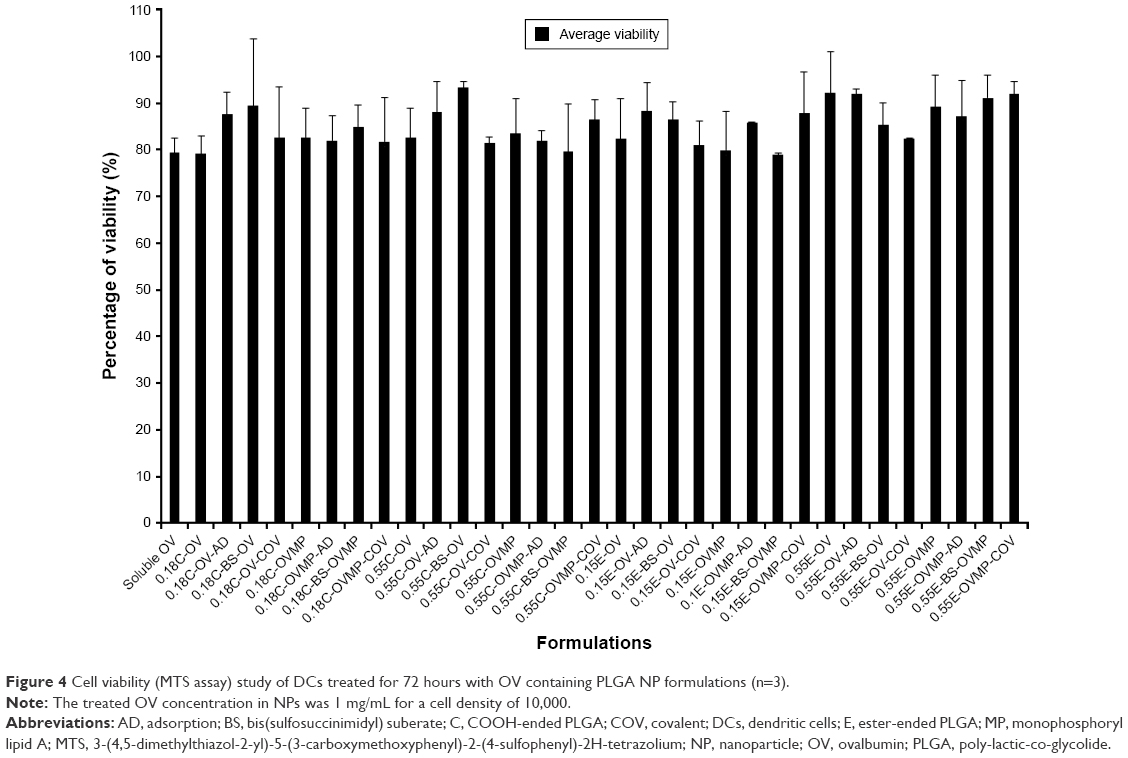 | Figure 4 Cell viability (MTS assay) study of DCs treated for 72 hours with OV containing PLGA NP formulations (n=3). |
Effect of surface modification with anti-CD205 Ab on DC maturation
The study groups were untreated DCs (negative control), DCs treated with LPS 1 μg/mL (positive control), and DCs treated with OV and OVMP-NPs (test groups). Figures 5 and 6 represent the comparative expressions of maturation markers (CD40, CD86, and MHCII). For all treatment groups, DCs were incubated with variable amounts of NPs containing a constant amount of loaded OV. It is evident that the presence of anti-CD205 Ab in formulations increases MFI and the percentage of positive cells compared to untreated DCs (**p<0.05).31 As represented in Figure 5A, Ab-adsorbed low-viscosity COOH-terminated NPs showed higher expression of all three markers (*p<0.05). As a result, the Ab-adsorbed OV-MP low-viscosity COOH-terminated NPs showed higher upregulation of all the markers (CD40 MFI: 68.80, CD86 MFI: 132.75, and MHCII MFI: 279.03) compared to their COV Ab-attached groups (CD40 MFI: 58.51, CD86 MFI: 78.64, and MHCII MFI: 131.48; Figure 5A). Considering the comparative fold increase in MFI for the same PLGA type, the Ab-modified high-viscosity COOH-terminated OV/MP-loaded NPs followed a similar trend as its low-viscosity grade. The highest maturation of DCs was obtained with 0.18C-OVMP-AD-NPs that showed 5-, 7-, and 9-fold increase in MFI for the expression of CD40, CD86, and MHCII molecules compared to the unstimulated cells, respectively (Figure 6A). Following similar trend as low viscosity, Ab-adsorbed formulations of high-viscosity COOH-terminated NPs showed higher expression of all three markers (Figures 5B and 6B).
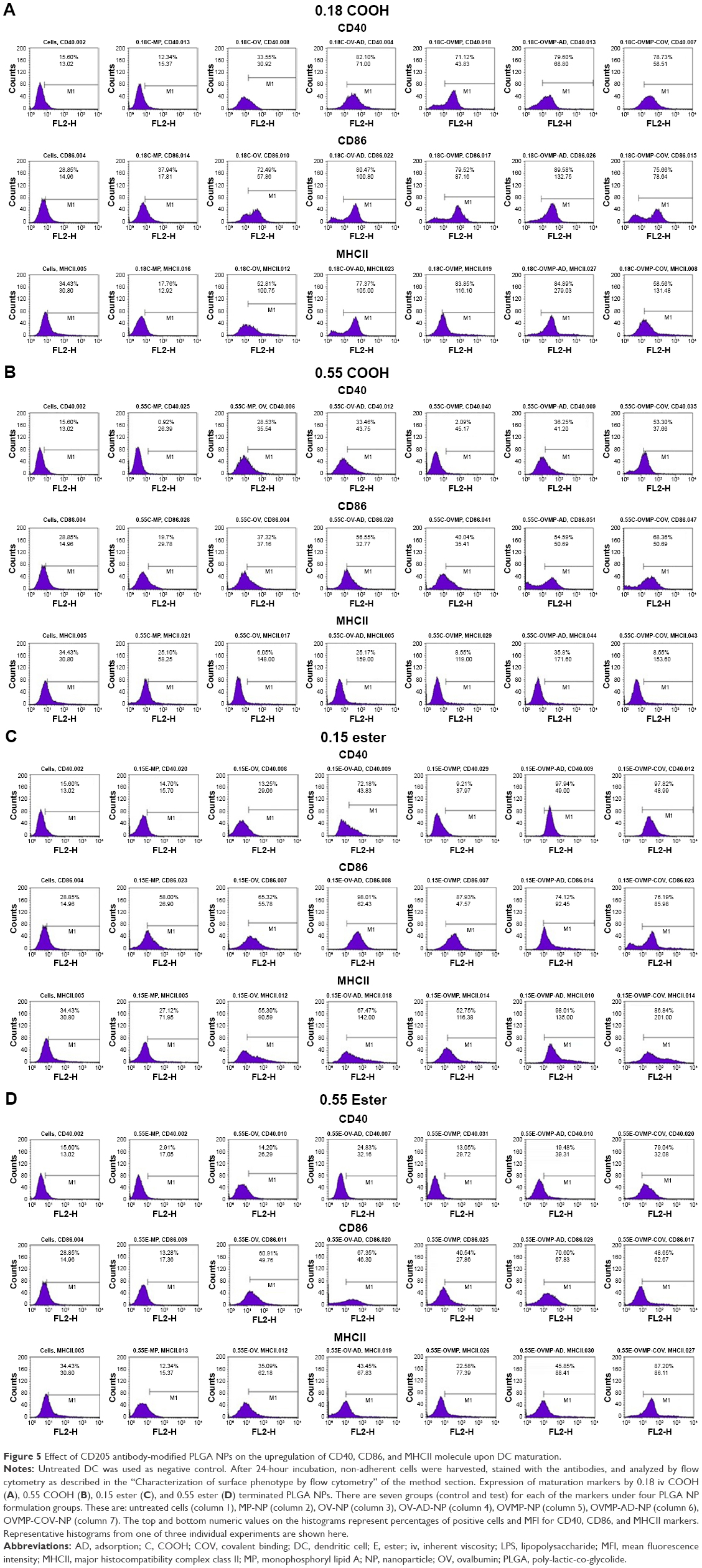 | Figure 5 Effect of CD205 antibody-modified PLGA NPs on the upregulation of CD40, CD86, and MHCII molecule upon DC maturation. |
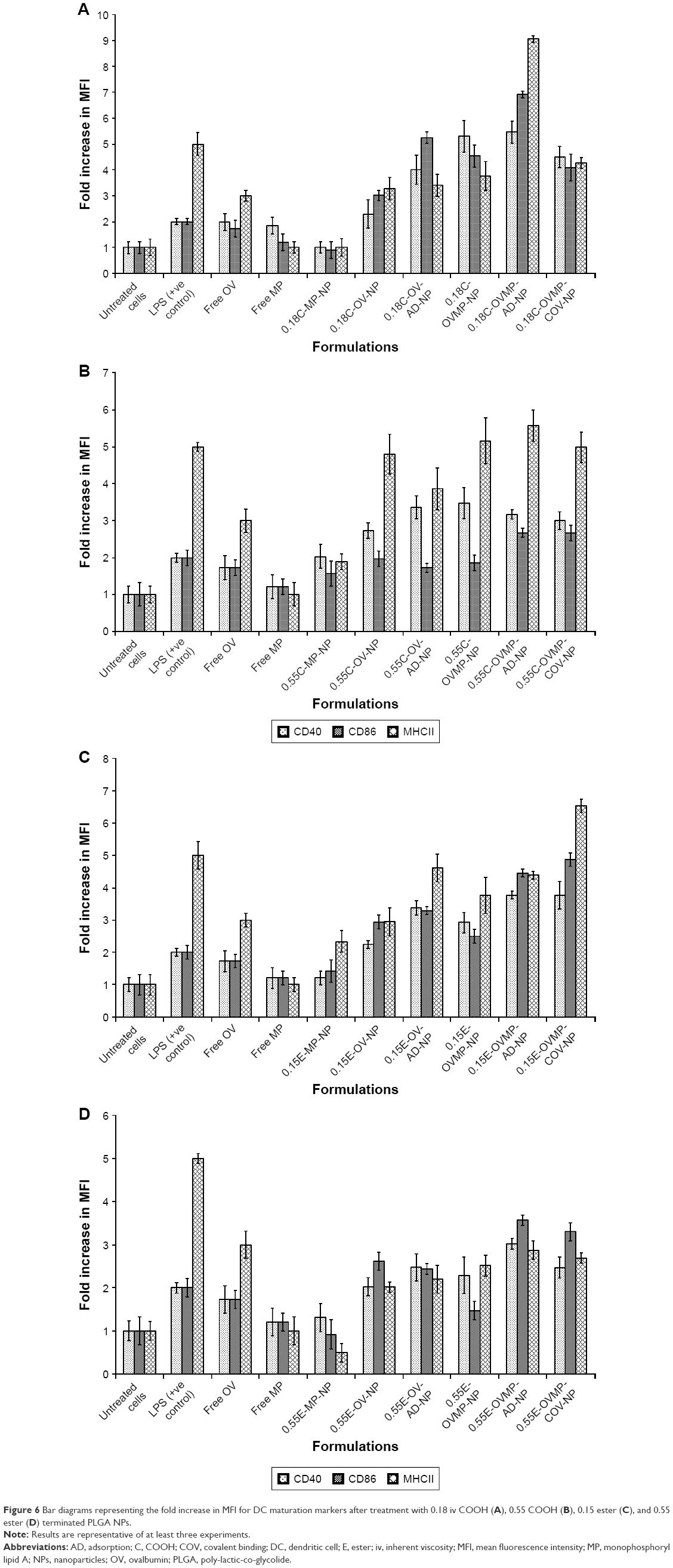 | Figure 6 Bar diagrams representing the fold increase in MFI for DC maturation markers after treatment with 0.18 iv COOH (A), 0.55 COOH (B), 0.15 ester (C), and 0.55 ester (D) terminated PLGA NPs. |
For ester-terminated NPs, 0.15E-OVMP-COV-NPs showed superior expression of activation markers than 0.15E-OVMP-AD-NPs. Simultaneously, the percentage of CD40+ cells and MFI was higher for the 0.15 iv ester-terminated OV/MP-modified NPs (Figure 5C). For example, 0.15E-OVMP-COV-NPs expressed 4-, 5-, and 7-fold increase in MFI than the 0.15E-OVMP-AD-NPs (Figure 6C). Between the two viscosities of ester-terminated PLGA NPs, a similar trend as the COOH-terminated OV/MP formulations was observed. Figures 5D and 6D represent the histogram and bar diagrams for the 0.55 iv ester-terminated PLGA NPs.
Presence of MP in all these formulations showed a significant change in MFI and the percentage of positive cells compared to untreated cells (*p<0.05). It is worth mentioning that both high-viscosity PLGAs NPs showed lower expression of three markers in comparison with their low-viscosity subtype. When comparing the effect of PLGA viscosity on maturation, the low-viscosity formulations showed comparatively higher shift in MFI.
Cytokine secretion profile
In parallel to the phenotypic upregulation of co-stimulatory molecules, mature DCs also secreted immunological playmakers such as IL-12, IL-6, IFNγ, and TNF-α upon exposure to free OV, OV-MP, Ab-AD, and Ab-COV NP groups.39 Figure 7 shows the secretion of cytokines in the DC supernatants upon stimulation with titrated amount of NPs. There was consistently high secretion of cytokines by the anti-CD205 Ab modified OVMP-NPs within similar groups (Figure 7A–H). The highest amount of IL-12 (2,165.22±215 pg/mL) was secreted after DC stimulation with 0.18C-OVMP-COV-NPs. Highest amount of IL-6 (1,343.23±87 pg/mL) and IFNγ (2,691.45±70 pg/mL) was detected in the supernatants of 0.18C-OVMP-AD-NPs. This was significantly high compared to the minimal secretion of the cytokines by free OV-treated DCs (***p<0.05). TNF-α concentration was significantly highest with the 0.55C-OVMP-COV-NPs (2,345.68±322 pg/mL) when compared with free OV (***p<0.05).
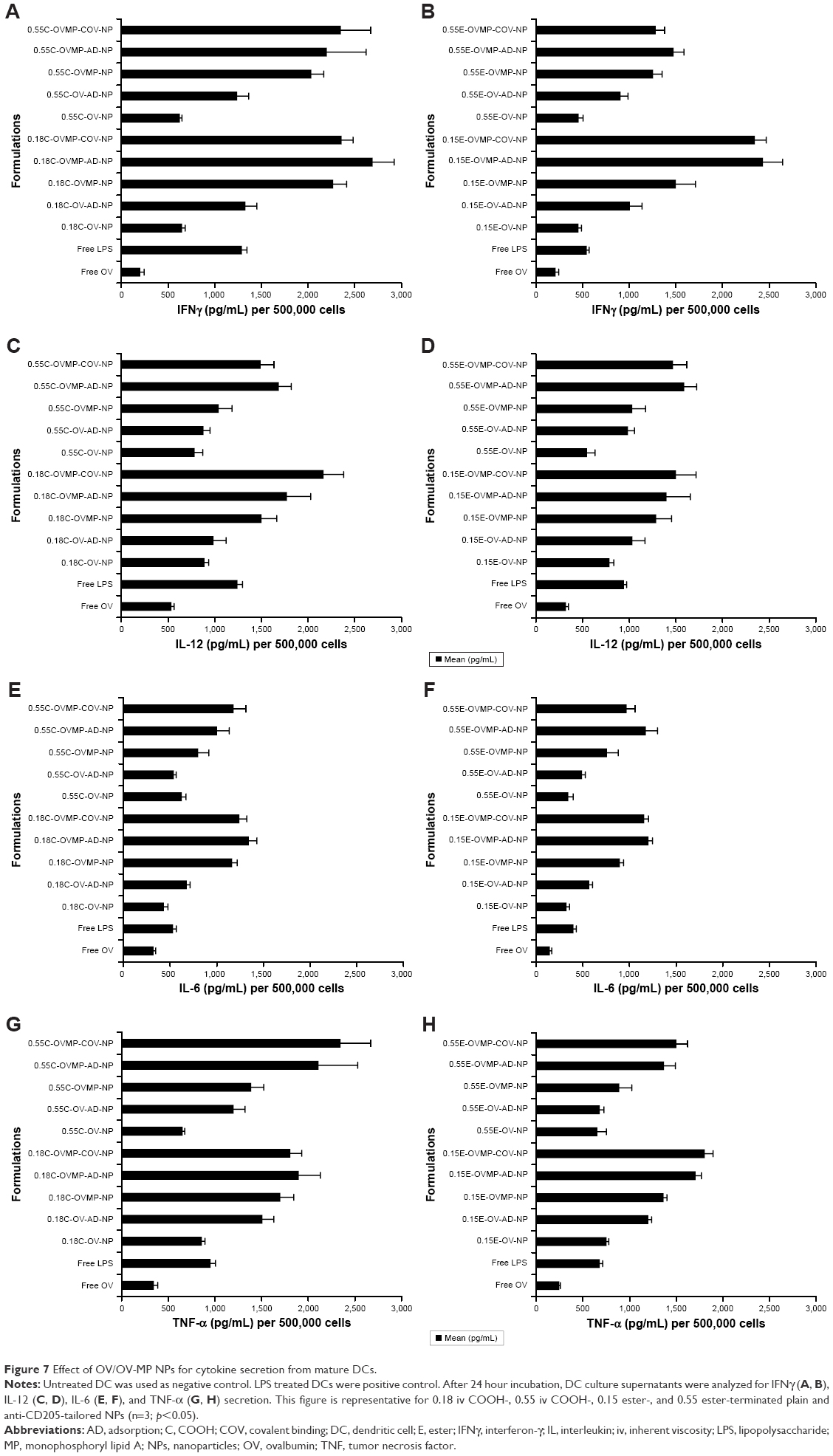 | Figure 7 Effect of OV/OV-MP NPs for cytokine secretion from mature DCs. |
Discussion
Coumarin-6 is widely used in uptake studies due to its biocompatibility, low leakage rate, and high fluorescence activity.40 In this study, coumarin-6-loaded NPs had suitable physicochemical properties to perform in vitro cellular uptake. All these formulations had an acceptable particle size range (121±5 to 562±5 nm; Table 1). Particle size has a proportional relationship with the NP uptake.36 Particles of 100–200 nm size are usually internalized by receptor mediated endocytosis. Larger particles (>500 nm) are taken up by phagocytosis process.41 This justifies the higher uptake of Ab-modified larger particles that occurred by combination of mechanisms. The large surface area of these particles is beneficial for higher loading of anti-CD205 Ab by AD mechanism.3 Consequently, anti-CD205 Ab-coated NPs had a greater affinity for DCs than other blood opsonins.42 This distinct surface modification escapes the body’s natural defense systems with improved circulation time and better chance to reach the target cells.43 Bar diagrams in Figure 2B represent the comparative change in MFI when coumarin-6 is present in the formulations. There was a substantial increase in MFI for the high-viscosity COOH-terminated PLGA NPs because these formulations encapsulated more coumarin-6 compared to low-viscosity PLGA formulations (Figure 2). On the contrary, BS-embedded low-viscosity ester-terminated NPs showed higher uptake compared to the high-viscosity ester formulations. This could be attributed to PLGA erosion and exposure of entrapped Abs in low-viscosity ester-terminated NPs.44 Relatively lower uptake in high-viscosity ester formulations (with BS) could be due to less embedment of BS available for linking with anti-CD205 Ab (Figure 2B). Moreover, masking of BS with the hydrophobic PLGA chain could also contribute to the lower uptake of high-viscosity ester formulations.26 Lower encapsulation of coumarin-6 was also observed in the BS-containing NPs, which justifies the lower MFI. Accordingly, the targeting efficiency of 0.55E-AD NPs was the highest (179 times vs 30) when compared with their unmodified NPs.
Physicochemical properties of NPs are also important to correlate with the desired immune response. Table 2 represents the cationic charged NPs after Ab conjugation with the desired size range (323±3 to 501±9 nm). For subcutaneous vaccine delivery, particle size ranging from 200 to 500 nm is immunogenic.45 Furthermore, positively charged particles are favorable for vaccination and ZP values close to zero were likely to result in aggregation.46 Formulations that possess a ZP value >+30 mV and <−30 mV are considered colloidally stable. Beyond this range, cryopreservation is required to maintain stability during shelf life of the vaccine.47 In the present study, 10% sucrose was suitable as that resulted in aggregation free slightly positive freeze-dried NPs. However, particle size and ZP are not the sole determinants of immune response in vivo.45 For the ester-terminated NPs, the highest EE of OV was with 0.55E-BS-OVMP-NPs (59.15%±1.56%), although there was no significant difference between the groups. In the case of COOH-terminated NPs, the highest amount of OV was loaded in 0.55C-BS-OVMP-NP (55.42±0.21 μg/mg of NPs). The ionic interactions between the amine groups of OV and the COOH groups of PLGA are responsible for the overall EE.35
PLGA is one of the most popular biodegradable polymers for drug delivery systems.15 The increase in viscosity will result in prolonged degradation rate of the polymer.43 PLGA follows a biphasic release behavior, where its content is released from the surface to the medium in the initial phase due to its solubility and water penetration into the polymer matrix (Figure 3A). In the second phase, progressive drug release occurred throughout the time frame by diffusion and erosion of the polymer (Figure 3B). The presence of the COOH-terminated group results in autocatalysis of the PLGA degradation process.44 A similar pattern of PLGA degradation might have occurred in this study. From the OV release study, it could be confirmed that the release of a protein from a polymeric matrix in aqueous media slows down due to polymer gelation. This change in polymer suspension is favorable for sustained release of protein from the NPs in vivo.48 In addition, the MTS assay shows that incorporating OV in the formulations did not show any significant effects on DC viability over 72 hours in the culture media (Figure 4).49
To confirm the maturation of DCs, the expression of maturation markers (CD40, CD86, and MHCII) and a cytokine cocktail (IL-12, IL-6, IFNγ, and TNF-α) was investigated to compare various formulations (Figures 5–7).50 The maturation of DCs was induced by the OV and OV-MP formulations rather than soluble OV. Within 24 hours period, about 30% of OV was released from all types of PLGA NPs. The expression of stimulatory and co-stimulatory molecules for 24 hours was due to the release of OV in the DC microenvironment. This is a good indication that the formulations would release sufficient amount of OV during the subcutaneous injection in the mice model over the 3 weeks period of vaccination. The DC maturation was further triggered by MP that increased the expression of all three markers, preferably in the Ab-tailored NPs.51 MP is able to induce antigen-specific immune response without affecting the systems in the body (cardiovascular, reproductive, and respiratory). It can boost both cell-mediated and humoral immunity when incorporated in vaccine formulations.35 The superiority of the Ab-AD groups over Ab-COV was also obvious in the formulations (with the exception of 0.15 iv ester-terminated NP formulations). This could be attributed to the rapid release of the loosely bound surface-linked OV from the OV-AD groups in the DC media. Although, Ab-adsorbed OV-MP formulations showed better maturation of DCs, both Ab-AD and Ab-COV formulations should be administered in the mouse model to conclude about their efficacy post-vaccination.
DCs are stimulated by secreting cytokines after recognition of certain patterns of pathogens. Secretion of the cytokines indicates the signal required for activation of T cells.52 The immunostimulatory effect of formulations was evaluated by the levels of various cytokines. The findings of this study reveal that Ab-modified NPs consistently showed higher cytokine secretion compared to unmodified NPs. Moreover, co-delivery of OV-MP resulted in significant secretion of T-helper 1 (Th1)-type cytokines including IL-12, IL-6, IFNγ, and TNF-α. It is known that IL-12 influences T cell differentiation, which is further regulated by IL-6. IL-6 is a potent regulator of DC maturation in vivo.53 In addition, IL-12 also increases the percentages of IFNγ-producing cells. Therefore, the release of these cytokines can efficiently convert immature DCs into T cell stimulatory mature DCs.54 The cytokine cocktail present in the DC supernatant shows a Th1-biased immune response. These results are consistent with earlier studies where MP was able to mature DCs with expression of markers as well as secretion of cytokines.37,39
Conclusion
In this study, we developed anti-CD205-targeted NPs for the subcutaneous delivery of therapeutic vaccines. PLGA NPs were produced by emulsification solvent evaporation method with suitable physicochemical properties. The low-viscosity ester-terminated NPs had the smallest size and suitable ZP among all the formulations. Compared with the Ab-free NPs, the functionalized PLGA NPs showed higher ability to recognize the CD205 receptors on DCs. The targeting efficiency of the Ab-adsorbed high-viscosity ester-terminated NPs was the highest. Among all the formulations, the ligand-attached low-viscosity COOH-terminated NPs could mature the DCs most with about 50% of OV released in 24 hours. In addition, ligand-attached low-viscosity COOH-terminated NPs were also able to stimulate DCs to secret rational amount of cytokines. This formulation can be chosen to conduct in vivo vaccination experiments due to its ability to maintain all the other desired physicochemical properties. Taken together, all results support our hypothesis that anti-CD205 Ab-modified NPs are effective in terms of in vitro DC stimulation with either type of polymers depending on the aim of the treatment. However, researchers could also design in vivo experiments by choosing any of the PLGA types mentioned in this study. In future, we aim to subcutaneously inject the formulations containing OV-MP (non-targeted and targeted) to develop the dose–response relationship in animal models. Therefore, this model vaccine delivery system could be adapted to design a therapeutic cancer vaccine to harness a patient’s immune responses.
Acknowledgments
This project was supported by a research discovery grant from Natural Sciences and Engineering Research Council of Canada (NSERC). The authors thank the College of Pharmacy and Nutrition, University of Saskatchewan for assisting with the flow cytometer. They also thank the Department of Biology, University of Saskatchewan for providing help with confocal microscopy.
Disclosure
The authors report no conflicts of interest in this work.
References
Han R, Zhu J, Yang X, Xu H. Surface modification of poly(D,L-lactic-co-glycolic acid) nanoparticles with protamine enhanced cross-presentation of encapsulated ovalbumin by bone marrow-derived dendritic cells. J Biomed Mater Res A. 2011;96(1):142–149. | ||
Roy A, Singh MS, Upadhyay P, Bhaskar S. Nanoparticle mediated co-delivery of paclitaxel and a TLR-4 agonist results in tumor regression and enhanced immune response in the tumor microenvironment of a mouse model. Int J Pharm. 2013;445(1–2):171–180. | ||
Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations. Adv Drug Deliv Rev. 2011;63(10–11):943–955. | ||
Kalinski P, Edington H, Zeh HJ, et al. Dendritic cells in cancer immunotherapy: vaccines or autologous transplants? Immunol Res. 2011;50(2–3):235–247. | ||
Sanchez-Ruiz Y, Valitutti S, Dupre L. Stepwise maturation of lytic granules during differentiation and activation of human CD8+ T lymphocytes. PLoS One. 2011;6(11):e27057. | ||
Whiteside TL. Immune responses to malignancies. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S272–S283. | ||
Mamedov IZ, Britanova OV, Zvyagin IV, et al. Preparing unbiased T-cell receptor and antibody cDNA libraries for the deep next generation sequencing profiling. Front Immunol. 2013;4:456. | ||
Butterfield LH. Dendritic cells in cancer immunotherapy clinical trials: are we making progress? Front Immunol. 2013;4:454. | ||
Xu H, Cao X. Dendritic cell vaccines in cancer immunotherapy: from biology to translational medicine. Front Med. 2011;5(4):323–332. | ||
Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med. 2002;196(12):1627–1638. | ||
Yanofsky VR, Mitsui H, Felsen D, Carucci JA. Understanding dendritic cells and their role in cutaneous carcinoma and cancer immunotherapy. Clin Dev Immunol. 2013;2013:624123. | ||
De Souza Reboucas J, Esparza I, Ferrer M, Sanz ML, Irache JM, Gamazo C. Nanoparticulate adjuvants and delivery systems for allergen immunotherapy. J Biomed Biotechnol. 2012;2012:474605. | ||
Demento S, Steenblock ER, Fahmy TM. Biomimetic approaches to modulating the T cell immune response with nano- and micro-particles. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:1161–1166. | ||
Hamdy S, Molavi O, Ma Z, et al. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine. 2008;26(39):5046–5057. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Silva AL, Rosalia RA, Sazak A, et al. Optimization of encapsulation of a synthetic long peptide in PLGA nanoparticles: low-burst release is crucial for efficient CD8(+) T cell activation. Eur J Pharm Biopharm. 2013;83(3):338–345. | ||
Han FY, Thurecht KJ, Whittaker AK, Smith MT. Bioerodable PLGA-based microparticles for producing sustained-release drug formulations and strategies for improving drug loading. Front Pharmacol. 2016;7:185. | ||
Rice-Ficht AC, Arenas-Gamboa AM, Kahl-McDonagh MM, Ficht TA. Polymeric particles in vaccine delivery. Curr Opin Microbiol. 2010;13(1):106–112. | ||
Diwan M, Elamanchili P, Lane H, Gainer A, Samuel J. Biodegradable nanoparticle mediated antigen delivery to human cord blood derived dendritic cells for induction of primary T cell responses. J Drug Target. 2003;11(8–10):495–507. | ||
Raghuwanshi D, Mishra V, Suresh MR, Kaur K. A simple approach for enhanced immune response using engineered dendritic cell targeted nanoparticles. Vaccine. 2012;30(50):7292–7299. | ||
Dhodapkar MV, Sznol M, Zhao B, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med. 2014;6(232):232ra251. | ||
Walters AA, Somavarapu S, Riitho V, et al. Assessment of the enhancement of PLGA nanoparticle uptake by dendritic cells through the addition of natural receptor ligands and monoclonal antibody. Vaccine. 2015;33(48):6588–6595. | ||
Solbrig CM, Saucier-Sawyer JK, Cody V, Saltzman WM, Hanlon DJ. Polymer nanoparticles for immunotherapy from encapsulated tumor-associated antigens and whole tumor cells. Mol Pharm. 2007;4(1):47–57. | ||
Jahan ST, Haddadi A. Investigation and optimization of formulation parameters on preparation of targeted anti-CD205 tailored PLGA nanoparticles. Int J Nanomedicine. 2015;10:7371–7384. | ||
Corrigan OI, Li X. Quantifying drug release from PLGA nanoparticulates. Eur J Pharm Sci. 2009;37(3–4):477–485. | ||
Thamake SI, Raut SL, Ranjan AP, Gryczynski Z, Vishwanatha JK. Surface functionalization of PLGA nanoparticles by non-covalent insertion of a homo-bifunctional spacer for active targeting in cancer therapy. Nanotechnology. 2011;22(3):035101. | ||
Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Control Release. 2007;120(1–2):18–26. | ||
Yu ZG, Wang Q, Li K, Li YQ, Gao XX. Determination and pharmacokinetics of 6,7-dimethoxycoumarin in rat plasma after intragastric administration of different decoctions of yinchenhao tang. J Chromatogr Sci. 2007;45(8):544–548. | ||
Haddadi A, Elamanchili P, Lavasanifar A, Das S, Shapiro J, Samuel J. Delivery of rapamycin by PLGA nanoparticles enhances its suppressive activity on dendritic cells. J Biomed Mater Res A. 2008;84(4):885–898. | ||
Hamdy S, Haddadi A, Shayeganpour A, Samuel J, Lavasanifar A. Activation of antigen-specific T cell-responses by mannan-decorated PLGA nanoparticles. Pharm Res. 2011;28(9):2288–2301. | ||
Bandyopadhyay A, Fine RL, Demento S, Bockenstedt LK, Fahmy TM. The impact of nanoparticle ligand density on dendritic-cell targeted vaccines. Biomaterials. 2011;32(11):3094–3105. | ||
Chen H, Gao J, Lu Y, et al. Preparation and characterization of PE38KDEL-loaded anti-HER2 nanoparticles for targeted cancer therapy. J Control Release. 2008;128(3):209–216. | ||
Ghotbi Z, Haddadi A, Hamdy S, Hung RW, Samuel J, Lavasanifar A. Active targeting of dendritic cells with mannan-decorated PLGA nanoparticles. J Drug Target. 2011;19(4):281–292. | ||
Heo MB, Lim YT. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials. 2014;35(1):590–600. | ||
Sarti F, Perera G, Hintzen F, et al. In vivo evidence of oral vaccination with PLGA nanoparticles containing the immunostimulant monophosphoryl lipid A. Biomaterials. 2011;32(16):4052–4057. | ||
Zhu L, Chen L, Cao QR, Chen D, Cui J. Preparation and evaluation of mannose receptor mediated macrophage targeting delivery system. J Control Release. 2011;152(Suppl 1):e190–e191. | ||
Haddadi A, Hamdy S, Ghotbi Z, Samuel J, Lavasanifar A. Immunoadjuvant activity of the nanoparticles’ surface modified with mannan. Nanotechnology. 2014;25(35):355101. | ||
Heo MB, Cho MY, Lim YT. Polymer nanoparticles for enhanced immune response: combined delivery of tumor antigen and small interference RNA for immunosuppressive gene to dendritic cells. Acta Biomater. 2014;10(5):2169–2176. | ||
Hamdy S, Haddadi A, Somayaji V, Ruan D, Samuel J. Pharmaceutical analysis of synthetic lipid A-based vaccine adjuvants in poly (D,L-lactic-co-glycolic acid) nanoparticle formulations. J Pharm Biomed Anal. 2007;44(4):914–923. | ||
Bao X, Gao M, Xu H, et al. A novel oleanolic acid-loaded PLGA-TPGS nanoparticle for liver cancer treatment. Drug Dev Ind Pharm. 2015;41(7):1193–1203. | ||
Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(15):2713–2722. | ||
Cruz LJ, Tacken PJ, Fokkink R, Figdor CG. The influence of PEG chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials. 2011;32(28):6791–6803. | ||
Dinarvand R, Sepehri N, Manoochehri S, Rouhani H, Atyabi F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int J Nanomedicine. 2011;6:877–895. | ||
Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. | ||
Slutter B, Bal S, Keijzer C, et al. Nasal vaccination with N-trimethyl chitosan and PLGA based nanoparticles: nanoparticle characteristics determine quality and strength of the antibody response in mice against the encapsulated antigen. Vaccine. 2010;28(38):6282–6291. | ||
Kumar A, Wonganan P, Sandoval MA, Li X, Zhu S, Cui Z. Microneedle-mediated transcutaneous immunization with plasmid DNA coated on cationic PLGA nanoparticles. J Control Release. 2012;163(2):230–239. | ||
Mukherjee B, Santra K, Pattnaik G, Ghosh S. Preparation, characterization and in vitro evaluation of sustained release protein-loaded nanoparticles based on biodegradable polymers. Int J Nanomedicine. 2008;3(4):487–496. | ||
Wu F, Jin T. Polymer-based sustained-release dosage forms for protein drugs, challenges, and recent advances. AAPS PharmSciTech. 2008;9(4):1218–1229. | ||
Alshamsan A, Haddadi A, Hamdy S, et al. STAT3 silencing in dendritic cells by siRNA polyplexes encapsulated in PLGA nanoparticles for the modulation of anticancer immune response. Mol Pharm. 2010;7(5):1643–1654. | ||
Kaka AS, Foster AE, Weiss HL, Rooney CM, Leen AM. Using dendritic cell maturation and IL-12 producing capacity as markers of function: a cautionary tale. J Immunother. 2008;31(4):359–369. | ||
Krishnamachari Y, Geary SM, Lemke CD, Salem AK. Nanoparticle delivery systems in cancer vaccines. Pharm Res. 2011;28(2):215–236. | ||
Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000;1(4):311–316. | ||
Demento SL, Bonafe N, Cui W, et al. TLR9-targeted biodegradable nanoparticles as immunization vectors protect against West Nile encephalitis. J Immunol. 2010;185(5):2989–2997. | ||
Mahnke K, Schmitt E, Bonifaz L, Enk AH, Jonuleit H. Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol Cell Biol. 2002;80(5):477–483. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.