Back to Journals » Clinical Ophthalmology » Volume 10
DEBS – a unification theory for dry eye and blepharitis
Authors Rynerson JM, Perry HD ![]()
Received 9 June 2016
Accepted for publication 30 September 2016
Published 9 December 2016 Volume 2016:10 Pages 2455—2467
DOI https://doi.org/10.2147/OPTH.S114674
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
James M Rynerson,1 Henry D Perry2
1BlephEx, LLC, Alvaton, KY, 2Department of Ophthalmology, Nassau University Medical Center, Hofstra University School of Medicine, East Meadow, NY, USA
Abstract: For many years, blepharitis and dry eye disease have been thought to be two distinct diseases, and evaporative dry eye distinct from aqueous insufficiency. In this treatise, we propose a new way of looking at dry eye, both evaporative and insufficiency, as the natural sequelae of decades of chronic blepharitis. Dry eye is simply the late form and late manifestation of one disease, blepharitis. We suggest the use of a new term in describing this one chronic disease, namely dry eye blepharitis syndrome (DEBS). Bacteria colonize the lid margin within a structure known as a biofilm. The biofilm allows for population densities that initiate quorum-sensing gene activation. These newly activated gene products consist of inflammatory virulence factors, such as exotoxins, cytolytic toxins, and super-antigens, which are then present for the rest of the patient’s life. The biofilm never goes away; it only thickens with age, producing increasing quantities of bacterial virulence factors, and thus, increasing inflammation. These virulence factors are likely the culprits that first cause follicular inflammation, then meibomian gland dysfunction, aqueous insufficiency, and finally, after many decades, lid destruction. We suggest that there are four stages of DEBS which correlate with the clinical manifestations of folliculitis, meibomitis, lacrimalitis, and finally lid structure damage evidenced by entropion, ectropion, and floppy eyelid syndrome. When one fully understands the structure and location of the glands within the lid, it becomes easy to understand this staged disease process. The longer a gland can resist the relentless encroachment of the invading biofilm, the longer it can maintain normal function. The stages depend purely on anatomy and years of biofilm presence. Dry eye now becomes a very easy disease to understand. We feel that dry eye should be treated and prevented by early and routine biofilm removal through electromechanical lid margin debridement.
Keywords: biofilm, quorum-sensing gene activation, Demodex, MGD, meibomian gland disease, aqueous insufficiency
Introduction
In 1684, Antonie van Leeuwenhoek presented to the Royal Society of London and commented on the number of “animicules” noted within the scurf of a man’s teeth.1 This is the first known microscopic observation of a biofilm. For over 300 years, little was known about biofilms, and research was uncommon. Biofilm implications in all of human disease were vastly underappreciated.2 In the past 20 years, however, biofilm research has burgeoned, with complicated but fascinating interactions between bacteria, host, and their environment now being revealed.2,3 In a similar vein, the term “blepharitis” first appears in the literature in the 1800s, but like biofilm research, little progress was made over the subsequent 100+ years in terms of understanding or treating this disease.4 While we have made some strides since the days of “Great German Eye Water” to treat “weak or inflamed eyes”, blepharitis remains a poorly defined disease, with the use of confusing and inaccurate terminology and considerable misunderstanding on the part of practitioners.5,6 The fact that there are multiple overlapping symptoms and pathologies intertwined within dry eye syndrome makes the picture even murkier.7 However, if one examines the eyelid margins more closely in terms of biofilm formation, this natural bacterial phenomenon can serve as a bridge of understanding between two poorly understood diseases, namely dry eye and blepharitis. We can now link them together with one common source of pathology. What becomes clear is that dry eye and blepharitis become one entity, that is, dry eye blepharitis syndrome (DEBS), reflecting in fact one disease process, not two different ones. What may now be learned is that DEBS presents in different stages throughout a person’s life.
In 1946, Phillip Thygeson, MD, described blepharitis as “a chronic inflammation of the lid border”.8 Unfortunately, over time, this accurate definition of blepharitis has been somewhat lost, and replaced with the clinical finding of lash or lid “scurf” acting as one of our main diagnostic clues. This was due to a combined misunderstanding of the disease and the ease with which we can see the lash and lid debris at the slit lamp. Surprisingly, in spite of the very nature of the word blepharitis (blepha = lid, ritis = inflammation), inflammatory lid disease lacking lash and lid debris would often lead the practitioner away from the diagnosis of blepharitis, as if having “scurf” was a prerequisite for having the disease.9 Additionally, it is no coincidence that the word scurf, a nondescript, “catch-all” word, used in dentistry since 1684, but not in modern times, is still used as a vague, non-defining term in ophthalmology.9 This demonstrates a lack of precision and understanding of the lid margin disease process.
The first step to understanding DEBS is to understand and use the term blepharitis correctly. It should be used to describe lid inflammation. It does not mean “scurf” within the lash line. The second step is to realize that the normal lid margin flora bacteria, primarily Staphylococcus aureus and Staphylococcal epidermidis, become over-colonized and undergo a change in pathogenicity during a patient’s lifetime.10 Thygeson in 1946 recognized that “abnormal Staphylococcus colonization” was associated with blepharitis.11 The question then becomes, how do we go from a normal colonization of Staphylococcus with little pathology when we are young, to over-colonization of Staphylococcus with inflammation and significant pathology as we age? The answer lies in the biofilm, the most basic of bacterial survival strategies.12 This article takes the reader stepwise from bacterial survival through the six steps of lid margin disease (Figure 1), culminating in the four stages of DEBS which will tie together dry eye disease, blepharitis, and meibomian gland dysfunction (MGD).
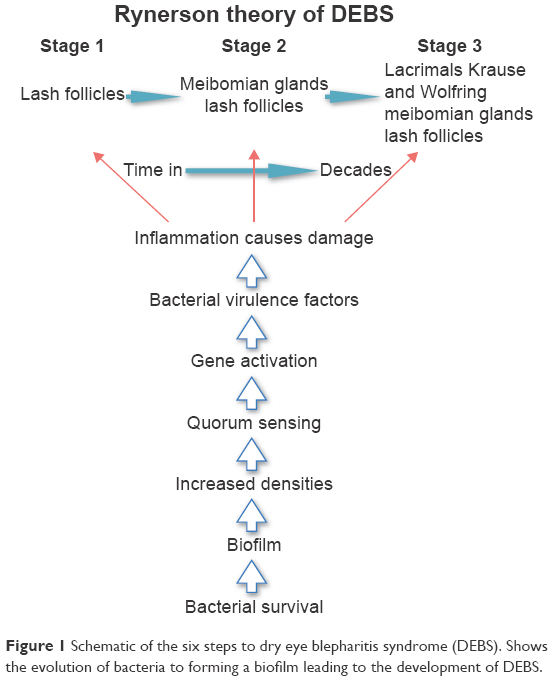 | Figure 1 Schematic of the six steps to dry eye blepharitis syndrome (DEBS). Shows the evolution of bacteria to forming a biofilm leading to the development of DEBS. |
To change the way we understand and treat dry eyes requires a reevaluation of existing evidence and intuitive reasoning. With the help of recent research regarding bacterial biofilms and virulence factors, we can now appreciate the change going from healthy lids when we are young, to inflammatory lid disease as we age in essentially six logical steps. However, none of these steps provide new information, new research, or is based on any new studies. Each step of this new theory, taken individually, is well proven, researched and available in the current literature. Understanding DEBS involves taking existing and proven concepts and putting them together with what we already know, to tell the story of blepharitis and dry eye disease in a new logical way that becomes intuitive based on the concept of the bacterial biofilm as basic to the pathophysiology of this syndrome.
Lid margin disease involves six steps, which are related to bacterial changes culminating in inflammation:
- Bacterial survival
- Biofilm formation
- Over-colonization
- Quorum-sensing gene activation
- Virulence factor production
- Inflammation.
Bacteria have survived on this planet for billions of years. They did not do this as free-floating planktonic bacteria. They were able to survive the eons by building a strong, rigorous, impenetrable defense structure known as a biofilm.13 Absalon et al14 and Pickering et al15 refer to the biofilm as “the prevailing microbial lifestyle” meaning that free-floating (planktonic) bacteria would be the exception in nature.
The biofilm proves to be an excellent defense structure by allowing bacteria to avoid desiccation, avoid host defense responses on or within other living systems, produce virulence factors, liberate and concentrate nutrients, and communicate with other bacterial species, thus enhancing survival strategies across species.16 Biofilms are easily encountered in nature as plaque on the teeth or “gunk” clogging industrial piping.17 They are what make rocks slippery in creek beds and your dog’s water bowl slimy within a few days of being outside on a summer day.17 They are manufactured and inhabited by a diverse mix of bacteria and are composed of a polysaccharide/protein matrix.18 These sticky strands of biofilm adhere easily to any surface and are not easily dislodged, allowing the bacteria to remain in a desirable environment. In fact, S. epidermidis and S. aureus produce a protein called adhesin, which essentially functions as a bacterial superglue, ensuring a tight adherence to the desired surface.19
“Biofilms grow virtually everywhere, in almost any environment where there is a combination of moisture, nutrients, and a surface.”20 Add in the inherent warmth of the lid margin and it makes an ideal surface for biofilm formation. When one considers that biofilms are found in deserts, on glaciers, and around deep-sea vents at the bottom of the ocean,21 it should come as no surprise that biofilms easily develop on the lid margin. It is well known that the lid margin is home to normal flora bacteria consisting of S. aureus, S. epidermidis, and to a lesser degree Corynebacterium spp. and Propionobacterium spp. It is also well known that both species of Staphylococcus are prolific biofilm formers, especially S. epidermidis.22 A recent study by Kivanç demonstrated that 32 out of 34 isolates cultured from eyes immediately after cataract surgery were indeed positive for being biofilm formers.23
The biofilm binds water and also provides protection from host defenses, two elements essential for bacterial survival. Biofilms act like an armor, impenetrable to white cells as well as antibiotics and povidone-iodine scrubs.24 This multi-laminar substrate also provides more surface area for bacterial replication, allowing for a vast over-colonization within the biofilm structure. Although the primary biofilm former is S. epidermidis, S. aureus also readily contributes to biofilm formation. It truly is, as Watnick and Kolter describe, a city of microbes, that is, diverse bacteria all living together in one complex community.25 Biofilms have been implicated in a wide array of different diseases, both chronic and acute. Otitis media, bacterial endocarditis, cystic fibrosis, and Legionnaires’ disease along with many nosocomial infections have all been related to biofilms.26
To avoid getting soap in our eyes when we wash our face, we instinctively keep our eyes tightly shut, lid margin against lid margin, effectively blocking access to an area that needs cleaning as much as or more than any other area of the body. In fact, there is only one place on the entire human body that never gets routinely cleaned during our entire life, the eyelid margin. Therefore, the biofilm accumulation builds up microscopically year after year, layer upon layer. It never willingly disperses, dissolves, or sloughs. No home scrub regimen can effectively reduce, and certainly eliminate, the adherent “superglued” biofilm layer upon the lid margin. Because of this, the population densities along the lid margin continue to increase within this biofilm as we age, eventually resulting in a vast over-colonization.27 We believe that as the biofilm thickens, the bacterial numbers continue to increase, and the inflammation continues to worsen, explaining why blepharitis and dry eye worsen with age. This process starts much earlier in contact lens wearers, since the contact lens is itself an inert foreign body allowing bacteria to avoid host defense and get a head start, producing a very early biofilm. Biofilm formation on contact lens and contact lens cases has been well documented.28 This also explains why dry eye disease is more common in contact lens wearers, 50% compared to 14% in controls.29
The over-colonization within the biofilm and increase in bacterial populations is what leads to quorum-sensing gene activation.30 Quorum-sensing gene activation (Figure 2), discovered by Hastings, was a groundbreaking study opening a brand new door to the understanding of bacterial virulence.31 Hastings demonstrated that populations of bacteria can sense when their densities achieve a certain quorum, and once that number or density is reached, dormant genes are activated.32 These newly activated genes produce a wide array of virulence factors, many of which are extremely inflammatory.33
 | Figure 2 Schematic showing quorum sensing and the development of two types of bacteria ie, planktonic and biofilm formers and how the latter can sense when there are sufficient numbers to liberate genes leading to increased inflammation. Reproduced with permission from Montana State University Center for Biofilm Engineering (http://www.biofilm.montana.edu/biofilm-basics/key_characteristics_of_biofilms.html).93 |
How do the bacteria communicate with one another once a gene-activation quorum has been reached? Bacteria communicate with one another using a chemical messenger called homoserine lactones (HSLs), and also, by using electric currents produced by potassium ions.34 While most bacteria produce HSLs, whether planktonic or within a biofilm, it is only the biofilm populations that are fixed within a close enough proximity to each other which are able to communicate, and only within a biofilm is the HSL and potassium ion concentration high enough for communication to take place.35 We believe that it is these virulence factors from the newly activated genes which are the real destructive force in inflammatory lid disease, causing low-grade, chronic inflammation on the lid surface and eventually within the structures of the lid margin such as lash follicles, meibomian glands and connective tissue, eventually affecting the accessory lacrimal glands as the inflammation inexorably marches forward.
Although S. epidermidis produces small amounts of a phenol-soluble modulin, which is a moderate cytolytic toxin, the primary virulence factor producer is S. aureus.36 S. aureus produces a vast array of highly toxic, highly destructive, and highly inflammatory proteins.37,38 These include two groups of exoproteins, namely exotoxins and enzymes. It is well known that exotoxins are extremely immunogenic, many of which are classified as super-antigens capable of activating large amounts of T cells resulting in immense release of cytokines, and thus, excessive inflammation. Important exotoxins include toxic shock syndrome toxin responsible for toxic shock syndrome, staphylococcal enterotoxins A, B, C, D, E, G, H, and I responsible for food poisoning and exfoliative toxins A and B responsible for scalded skin syndrome.39,40 The enzymes produced consist of nucleases, proteases, lipases, hyaluronidase, and collagenase, all capable of destroying host tissue.41 In addition to exoproteins, cytolytic toxins are also produced including at least three types of hemolysins and two types of leukocidins, all capable of destroying or damaging cells, further adding to the inflammatory cascade.42 One additional virulence factor worth special mention is polysaccharide intercellular adhesin (PIA), which is produced by a gene cluster known as the ica operon.43 PIA acts as the aforementioned superglue for biofilms, helping to hold them together as a structure, as well as anchoring them in place within the environment.43
This toxic virulence factor soup is within, on top of and under the biofilm, constantly exposing the lid margin tissue and related glands to a host of toxins that, when found in other areas of the body, can cause acute, severe debilitating disease as in scalded skin syndrome, food poisoning, and even death, as in the case of toxic shock syndrome.44 Wherever a thick biofilm exists, quorum-sensing gene activation eventually occurs followed by virulence factor production, whether on the lid margin or within the lash follicles, meibomian glands, or accessory lacrimal glands. We can see what this cauldron of inflammatory mediators does to not only these structures but eventually nerve endings and even the connective tissue of the eyelid, which can affect the structural integrity of the eyelids.45 When exposed to this toxicity on a continual, unrelenting basis for decades, the resulting inflammation is nonselective in what it damages.46 Nothing in the eyelid is immune.47 We believe that it is a testament to the body’s ability to defend itself that severe lid damage does not occur much more quickly than it does in this toxic environment. As opposed to toxic shock syndrome, food poisoning, scalded skin syndrome, etc., the disease of DEBS spans multiple decades of a patient’s life, and has multiple manifestations depending on whether it is an early or a late disease.48
This exceedingly slow but chronic disease process with its various presentations has led to much confusion over the years in identifying and understanding the etiology and progression of dry eye disease.49 We tend to focus on the immediate presenting problem and not what preceded it. For instance, when a patient presents with meibomian disease, we tend to ignore the involvement of the lash follicles, and tell the patient that he/she has evaporative dry eye.50 When the lacrimal glands are involved and there is little-to-no tear lake, we tell the patient that he/she has aqueous insufficiency, ignoring or not realizing that the meibomian glands were affected for many years prior to that visit.51,52
The four stages of DEBS
Inflammatory lid disease has essentially one etiology, namely the multiple virulence factors produced by the existing bacteria within a mature biofilm. Once we accept this domino effect of pathology origin, the clinical manifestations of inflammatory lid disease can then be examined, explained and easily learned. Every aspect of lid margin disease can be understood by looking at the lid anatomy, the duration of the presence of a biofilm and the associated virulence factors along the lid margin. We should avoid confusing terminology such as anterior or posterior blepharitis, staphylococcal blepharitis or seborrheic blepharitis. These distinctions are really distractions and do not serve to accurately describe the stage or duration of disease. We must learn that lid inflammation cannot be avoided once these highly inflammatory virulence factors begin to be produced. We must understand that inflammation does not discriminate. Every structure in the lid is eventually affected. It simply takes some structures longer to be affected than others. The biofilm does not self-destruct and rarely if ever gets effectively removed, and therefore, the inflammatory process is relentless.53,54 Once it begins, it exists for the remainder of the patient’s life.55
Biofilm on the lid margin (Figure 3) probably forms very early in life during the toddler stage of life, or even earlier. However, at this stage in life, pathology typically does not occur simply because the microscopic biofilm is not thick enough to support a large population for quorum sensing to occur. We certainly do see exceptions, however, with young children occasionally being afflicted with a severe form of blepharitis.56 For this to occur, a child probably requires two bacterial species simultaneously colonizing the eyelids, a prolific biofilm-forming S. epidermis in conjunction with a virulent strain of S. aureus whose quorum-sensing numbers are, for whatever reason, set too low.57 Further research into these children’s lid bacterial profiles would likely yield valuable information regarding pathogenesis.58
 | Figure 3 Scanning electron microscopic photograph showing biofilm on the lid margin with some accumulation at the base of the eyelash. Copyright © 2016 Photo Researchers, Inc.94 |
In the majority of the population, the biofilm must be present for decades before it can accommodate the numbers of bacteria necessary for quorum sensing to occur.59 As previously mentioned, biofilms form wherever there is a “combination of moisture, nutrients and a surface”.5 This certainly describes the lid margin, and therefore, it is safe to assume that the biofilm formation starts on the surface of the lid margin, including the lash line, extending back to just beyond the meibomian gland openings.60 The constant sweeping action of the lids across the cornea, conjunctiva, and lid margin (ie, blinking) and the flushing of tears play a mechanical role61 in preventing significant biofilm accumulation on palpebral or bulbar conjunctiva.62 Certainly, the antibacterial proteins of the tear film, lactoferrin, and lysozyme, play an important role.63 The goblet cells, however, probably play an even more significant part in mechanical prevention of biofilm accumulation as well as physically protecting the epithelium.64
Goblet cells of the bulbar and palpebral conjunctiva are similar to goblet cells elsewhere in the body.65 A common theme is that goblet cells are prominent in areas that are constantly exposed to bacteria of the outside environment, such as the gut, lungs, vagina, sinuses, and eyes.66 Goblet cell functions have been much better studied in the gut and lungs and less so in the conjunctiva.67,68 Inferences can be made, however, enabling us to draw some tentative conclusions as to the protective role of conjunctival goblet cell mucus.69 The function of the goblet cell is to produce mucus, the main component of which is mucin, in areas that, technically speaking, are “outside” of the human body. Mucin, a viscous fluid of highly glycosylated proteins, gives the mucus its gel-like characteristics. While the goblet cell has a baseline secretion at all times, the majority of mucus produced is done so through stimulated secretion due to irritants from the local environment.70,71
While goblet cell mucus appears to have at least some antibacterial properties, the abundance of research related to the mechanical characteristics leads one to suspect that this is the primary protective mechanism. In the large intestine for instance, there are two layers of mucus: the outer housing the commensural bacteria72 and the inner layer providing an impenetrable layer to bacteria, thus protecting the underlying epithelial cells from bacterial exposure.73 The small intestine lacks the inner, impermeable layer, but still protects by providing a diffusion gradient and a rapid turnover that bacteria must overcome to access the epithelial cells.74 Mucus also performs an immune-regulatory role by limiting antigen presentation to the epithelial cells, thus controlling excessive immune responses in the gut. It may well play the same role on the surface of the eye, an area constantly exposed to potential antigens, not only from the lids but also from the environment.75 This mechanical protection is certainly one of the roles, if not the main role, of conjunctival goblet cells as well. By providing either a resistant diffusion gradient or an impenetrable barrier, the result is the same: protecting the eye by preventing bacteria from accessing the epithelial cells. In addition, since biofilm creation requires a stable surface, moisture, and nutrients, the constant turnover of conjunctival mucus together with the sweeping action of blinking prevents a stable surface and thus prevents a successful buildup of biofilm. It is therefore important to maintain a healthy goblet cell population to help prevent biofilm buildup directly on the conjunctiva.
However, in spite of a protective mucus layer, certain ocular pathologies may be attributable to tear film virulence factors that, over time, have damaged goblet cell population much the same way that lash follicles and meibomian glands are damaged from long-term inflammation and cytolytic effects. While it is known that mucus can protect against the actual bacteria from contacting the epithelium, it is not known if it can prevent the abundance of inflammatory bacterial virulence factors from contacting the epithelium, and therefore affecting the goblet cells.76 In fact, it is doubtful that it can. If the mucus layer is permeable to twice-daily (BID), thrice-daily (TID), or once-a-day glaucoma drops, antibiotics, steroids, nonsteroidals, etc., it is quite possible, if not likely, that it is permeable to at least some of the abundant S. aureus exotoxins, cytolytic toxins, and enzymes that are not once daily, BID, or TID in contact with the eye, but are in constant contact with the ocular surface. If these virulence factors and cytokines were contacting the epithelium, they would eventually have a deleterious effect on the goblets cells. Like atrophy of meibomian glands, patients may be slowly losing their goblet cell population over time due to the same constant, chronic inflammatory process.77
Let us take it one step further. Many of these toxins may be able to do exactly what medicinal eye drops do: penetrate to the inside of the eye. Indeed, what would prevent them? If this were to occur, it could be responsible for a slow, long-term damage to the trabecular meshwork, either through a subclinical (little-to-no cell/flare) inflammatory process or by a slow but direct meshwork cellular damage from the cytolytic toxins and enzymes, and would therefore parallel lid margin disease. If this were true, it may in part explain why the incidence of glaucoma increases with age: a thicker biofilm, more toxins, and longer exposure on the surface and within the eye causing damage to the lid margin structures as well as structures within the eye, as we age. Would it not be an astounding revelation that glaucoma could be treated and possibly prevented by a regular and thorough lid hygiene and enhanced protection by regular electromechanical debridement (microblepharoexfoliation [MBE]) of the lid margin biofilm!
Stage 1 DEBS involves the lash follicles. Since there exists a potential space between the lash and surrounding follicle, the biofilm can fairly easily access this space by extending down along the inert eyelash and surrounding tissue. Once virulence factor production begins, the small lash bulb can become inflamed relatively quickly. This inflammation is evidenced clinically by noting the “volcano” sign that occurs as edematous follicular tissue swells up around the base of the lash. Additionally, the swollen area can take on a pale appearance (Figure 4). This may be due to compression of capillaries due to edema, or maybe the increased presence of transudate within the tissue, or both, causing pallor when compared to the surrounding inflamed tissue. As the lash grows, small pieces of biofilm adherent to the lash will be pulled off of the lid margin (Figure 5) resulting in what is known as collarettes. What has always been called scurf, debris or even lash dandruff along the lid margin should now be recognized as simply pieces of a sticky bacterial biofilm, pulled free from the primary layer on the lid margin. One can now understand why these collarettes appear at different levels along the eyelashes, since the lashes are all growing at different times and stages. In Figure 5, one can notice near the top of the picture a collarette that is just beginning to separate from the lid margin biofilm. Further down the lash line, the collarettes appear further from the lid margin since these are older eyelashes. Other manifestations can include “cylindrical dandruff”, hardly a term that implies any clinical accuracy.78 It is difficult to imagine how dandruff can form cylinders around an eyelash. “Cylindrical dandruff” most likely represents biofilm that accumulates around the lash while still deep within the follicle, in its dormant stage, or an accumulation of biofilm sticking to and surrounding the base, effectively sheathing the slow-growing lash. Since these lash follicles are usually damaged by this point, the rate of growth is slower allowing the biofilm accumulation to “keep up” with growth, effectively pipe-stemming the lash.78
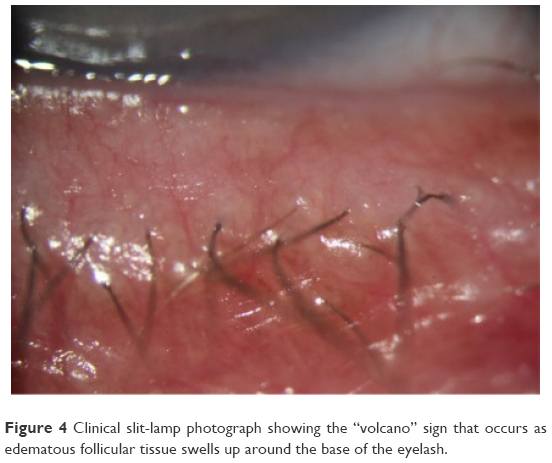 | Figure 4 Clinical slit-lamp photograph showing the “volcano” sign that occurs as edematous follicular tissue swells up around the base of the eyelash. |
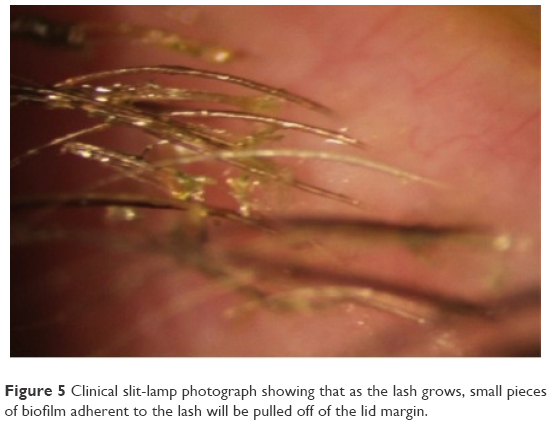 | Figure 5 Clinical slit-lamp photograph showing that as the lash grows, small pieces of biofilm adherent to the lash will be pulled off of the lid margin. |
It has been repeatedly suggested that cylindrical dandruff is pathognomonic for Demodex infestation.78 However, since the Demodex does not excrete waste products, it is equally difficult to imagine how this pipe-stemming can be attributed to Demodex. This notion of cylindrical dandruff being pathognomonic stems from a 2005 article by Gao et al, which found that all their patients with cylindrical dandruff had Demodex.78 They discount earlier studies without the same findings as cases of “miscounting”.78 Even if the 100% incidence in this one study is completely accurate, a cause and effect still cannot be drawn. In a later article by Tsubota et al, the authors state “Demodex was detected in the cilia of 8 out of 10 (80%), and 22 cilia out of 30 (73%) with cylindrical dandruff.”79 While these numbers certainly suggest an association, a high association should not imply causality. The fact that 73% of the lashes with cylindrical dandruff had Demodex should suggest just the opposite, no causality. The authors’ conclusion that “Demodex might be the pathogen causing blepharitis with cylindrical dandruff” is just simply overstated.79,80 It is much more likely that these eyelids have a longer history of an abundant biofilm, resulting in not only more diseased follicles with slower lash growth and a stacking of biofilm but also a higher likelihood that Demodex has, over the years, found its way to a copious and nutritious food supply in the form of a polysaccharide biofilm. In other words, it is probably a high association due to one common factor, namely an abundance of biofilm.
Collarettes, clumping, mattering, and eyelids stuck together upon awakening, are all manifestations and evidence of bacterial biofilm along the lid margin. Interestingly, in late-stage disease (40–50 years of chronic inflammation), we sometimes see significant lid inflammation without collarettes or “scurf” in the lash line, mistakenly leading the clinician to a diagnosis other than blepharitis. At this stage of disease, however, the lash bulb is likely so badly damaged that the lashes are barely growing, if at all, and thus are no longer pulling off small amounts of biofilm from the margin. What is seen at this stage is a paucity of lashes, which are falling out, and not growing back. By looking closely, one can notice extremely swollen lash follicles in Figure 4. Women, who complain of not being able to wear mascara any longer because their lashes are not growing back, have probably been suffering from 50 to 60 years of follicular inflammatory damage to the eyelashes.
Stage 2 DEBS involves the lash follicles and the meibomian glands. Meibomian involvement always occurs after follicular involvement due to the anatomical size and the relative difficulty of accessing the meibomian gland compared to the lash follicle. The meibomian gland has a narrow ductule. This characteristic, along with the constant flow of meibum out of the gland, effectively impedes the growth of biofilm into the gland. Once inflammation does start within the gland, the shear size of the gland (5–10 times larger than a lash follicle) means that it takes much longer for inflammation to negatively affect the working of the gland.81 How much longer will be determined by the biofilm-building and virulence factor characteristics of the particular bacteria, but we estimate that at least 10–15 years of follicular involvement would precede meibomian involvement, but this number may be much smaller given that early meibomian gland disease often goes unrecognized. Obvious vs nonobvious MGD is a recent topic of discussion and can also be explained with the biofilm theory. Nonobvious MGD (Figure 6) can be thought of as a layering of biofilm within meibomian gland.82 Inflammatory damage has begun, the ductule is probably blocked and decreased lipids or at least abnormal lipids now make up the meibomian secretions. Since lipid deficiency or abnormal lipids are characterized by an increased melting point, the secretions become thickened, mixing with biofilm, leading to a “toothpaste” quality, which eventually results in drastic decrease in functional lipids.83 Since the meibomian glands are just layered with biofilm, but not at capacity, expression may not elucidate the “toothpaste” secretions.84 Obvious MGD (Figure 7), on the other hand, is when inspissation and capping, or peaks over the gland openings, is noted on examination. While the composition of these peaks or caps has never been effectively studied, it is not difficult to imagine that it is a combination of biofilm mixed with diseased meibum, “meibofilm” (Figure 7). A meibomian gland will become full of this “meibofilm”, and as production of biofilm continues, these secretions have nowhere to go except out of the ductule, and will appear as little domes trapped under the original lid margin biofilm. When these glands are expressed, one is much likely to be “rewarded” with copious amounts of meibomian sludge or inspissated secretions.84 Nonobvious MGD is a layering of the gland with these altered secretions, while obvious MGD is a gland that is full and distended with the secretions trying to escape out of the ductule.84,85 Hence, the difference is simply one of degree. Lipid deficiency is noted in both states and is the main pathologic sequelae of both. An automated heat-and-express procedure, such as Lipiflow, is the best way to remove these altered secretions from within the glands, and therefore remove the source of virulence factors and inflammation within the meibomian gland.86
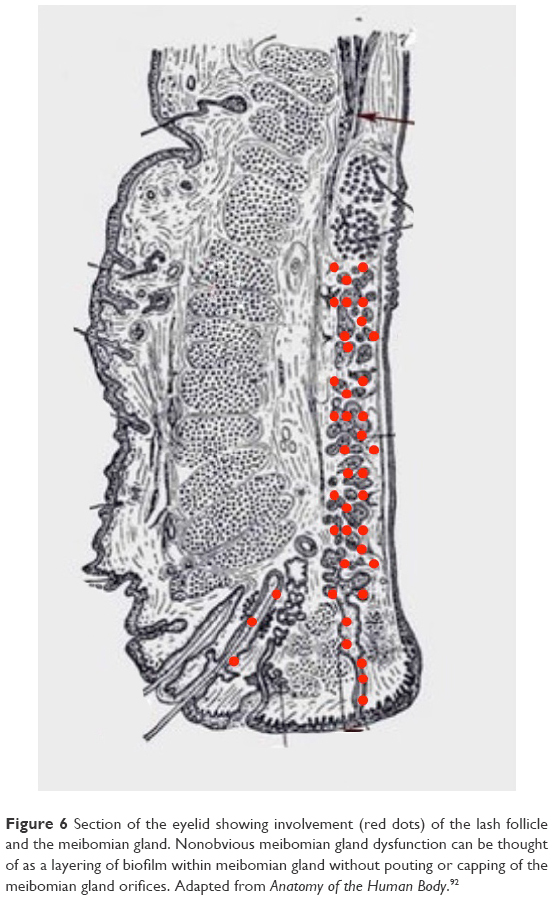 | Figure 6 Section of the eyelid showing involvement (red dots) of the lash follicle and the meibomian gland. Nonobvious meibomian gland dysfunction can be thought of as a layering of biofilm within meibomian gland without pouting or capping of the meibomian gland orifices. Adapted from Anatomy of the Human Body.92 |
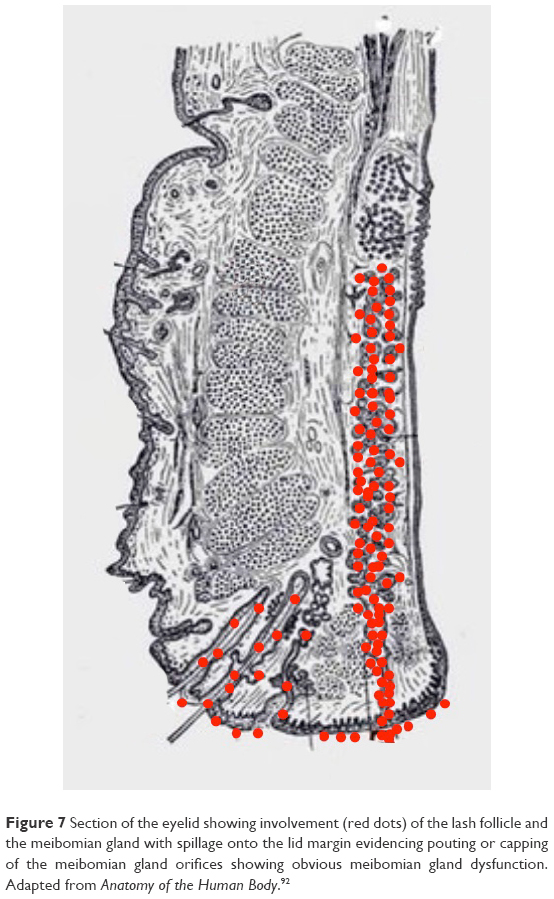 | Figure 7 Section of the eyelid showing involvement (red dots) of the lash follicle and the meibomian gland with spillage onto the lid margin evidencing pouting or capping of the meibomian gland orifices showing obvious meibomian gland dysfunction. Adapted from Anatomy of the Human Body.92 |
Stage 3 DEBS involves the follicles, meibomian glands, and now the accessory lacrimal glands of Wolfring and Krause. These numerous tear glands, responsible for baseline aqueous production, are well protected from the activity along the lid margin.87,88 The ducts of these glands empty along the inside lid, up near the fornices. The sheer distance from the lid margin biofilm, the narrow ducts, and the constant flushing activity of tear production all serve to protect these glands for many generations. However, the lid margin biofilm is constantly shedding small bits of biofilm into the tear film through dispersal (Figure 8). After 40–50 years of a daily dispersal of hundreds of bits of biofilm, eventually a bit of biofilm finds its way to the glands of Krause and Wolfring. We theorize that they are the last group of tear glands to become infiltrated by biofilm, and therefore, the last to succumb to the effects of inflammatory damage from biofilm virulence factors. We can prove to ourselves that these are simply different stages of the same disease, if we closely examine what we see clinically. We see many patients in our practices, probably every day, who present complaining of burning, irritation, difficulty seeing, and eyes that are tearing excessively. These are the patients who are difficult to refract because their vision changes with each blink. Examination reveals a deficient lipid layer and evaporative dry eye. Trying to explain to the patients that they have a form of dry eye is sometimes difficult, since watery eyes are a common complaint. Therefore, clinically, we see many patients with aqueous production who are deficient in lipid production. These patients have diseased meibomian glands but still healthy accessory glands of Krause and Wolfring. The question we should now ask is, do we ever see this clinical picture reversed, with lots of lipids, healthy meibomian glands but deficient aqueous production? Unless we are dealing with an autoimmune process, such as Sjogren’s syndrome, it is virtually impossible to see an aqueous-deficient patient without MGD. If aqueous deficiency were a separate disease process, we should, at least on occasion, expect to see diseased glands of Krause and Wolfring present together with healthy meibomian glands. We would expect to see aqueous deficiency sometimes occurring independently or before meibomian disease, but we rarely, if ever do. We always see aqueous deficiency follow MGD because these different states of dry eye are the same disease process, with one being the earlier manifestation, and one being the latter. Again, it is worth repeating that Stage 2 DEBS will always precede involvement of Krause and Wolfring due to the anatomical location and morphology of the glands.
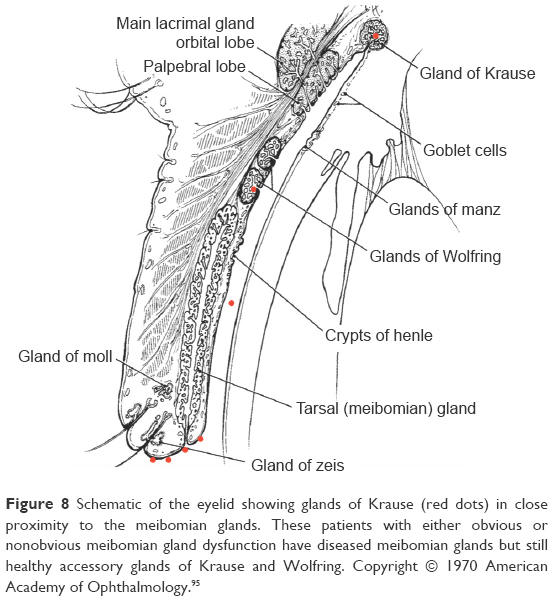 | Figure 8 Schematic of the eyelid showing glands of Krause (red dots) in close proximity to the meibomian glands. These patients with either obvious or nonobvious meibomian gland dysfunction have diseased meibomian glands but still healthy accessory glands of Krause and Wolfring. Copyright © 1970 American Academy of Ophthalmology.95 |
By the time we have Stage 3 DEBS (aqueous insufficiency), the follicles have been subjected to chronic inflammation for many decades and are sometimes so badly damaged in Stage 3 that lash growth is attenuated, and hence, there may be little-to-no biofilm noted among the eyelashes, since a growing lash is a requirement to having bits of biofilm pulled from the lid margin. Lashes fall out and may not regrow, or regrow very slowly. Looking closely, one may find significant swelling around the base of the lash along with pallor as in Figure 4.
Stage 4 DEBS is when the structural integrity of the eyelid begins to break down. Since inflammation eventually affects everything within the lid margin, connective tissue, muscle, and nerve endings all become damaged and lose their functionality.89 Lid laxity, entropion, ectropion, and floppy eyelid syndrome are all manifestations of end-stage chronic inflammatory lid disease.90 These are the patients who surprise us when we ask how bad their lids are bothering them, and they reply that they have no symptoms. After decades of inflammatory damage and lid destruction, the nerves endings are no longer capable of signaling discomfort. It is also possible to have very little biofilm noted on these eyelids when the lid has been dry for many years. Since bacteria need moisture to grow and produce biofilms, the eventual dry eye they cause may actually degrade their once-ideal environment to the point where they cannot sustain large colonies in the face of a desiccated lid margin and may actually have less biofilm. By this point, however, the damage to the lid and the tear glands is already done and may be irreversible. Once one understands that DEBS is a singular disease process that presents in stages, over decades, throughout a person’s lifetime, everything we see clinically makes sense. It explains the overlap of the so-called anterior blepharitis with posterior. DEBS explains why we do not see isolated cases of aqueous deficiency. It explains why the disease worsens with age. It explains the asymptomatic patient with severe lid disease findings. DEBS also explains the elderly lady who cannot wear her mascara any longer because she has lost so many lashes. It explains why we sometimes do not see biofilm within the lash line in advanced disease states. The lack of knowledge with regard to the presence of lid margin biofilm explains why everything we have thrown at this disease over the last 100 years has failed. Nothing we have done has had any impact whatsoever on the chronic, ever-present biofilm. It also suggests why a thorough mechanical biofilm removal of the lid margin may have such a profound impact on patient’s symptoms, quality of tears, and quality of life.91
A well-known fact is that the etiology of endophthalmitis is related most commonly to the patient’s own lid flora. These are biofilm-forming bacteria. The aforementioned Kivanç study demonstrated that these biofilm formers are present and even able to survive a surgical prep of Betadine.23 By performing a thorough electromechanical debridement (electric rotatary sponge cleaning) of the patient’s lids, we should be able to at least theoretically reduce the incidence of post-cataract infection. In addition, by removing the biofilm from the lid margin and meibomian glands, we should expect a better lipid layer and therefore better post-op vision. In a similar way, patients who have undergone refractive surgery, such as laser in situ keratomileusis, photorefractive keratectomy, and phototherapeutic keratectomy, should also benefit from an electromechanically debridement of their lid margin through improvement of the lipid layer. Contact lens wearers are known to develop early blepharitis and dry eye disease, and it is well known that contact lens develop adherent biofilms. All of these patients should benefit by eliminating or slowing down the progression of the lid biofilm with yearly electromechanical debridement.
We can do so much better in eye medicine by preventing damage to the critically important meibomian glands, rather than reacting to the damage once it is already done. Routine, electromechanical debridement of lids after age 50 (sooner in at-risk patients) should become as commonplace as routine dental cleaning, if we are to accept our new role as protectors of the meibomian glands and preventers of MGD.
One should imagine how much better pharmaceutical preparations intended for the lid margin would work without an impenetrable biofilm plugging the meibomian glands or blocking the lash follicles. How much better would topical cyclosporine work if it could better access the lid margin or meibomian glands? We have tried to make this disease so complicated for so many years when the simple answer has been right in front of our slit lamps the entire time. But as the most famous doctor in history once said, “Sometimes the questions are complicated and the answers are simple” (Dr Seuss). The answer here is also simple: The biofilm should be removed. A thorough electromechanical debridement and exfoliation of the lid margin should be performed. The meibomian glands and the rest of the lid structures should be given a chance to heal and a chance to return to normal function without the constant onslaught of inflammation from the biofilm’s virulence factors.
We should examine the amazing job that dentistry has done in educating society as to the importance of oral hygiene. Over 100 years ago, no one brushed their teeth or practiced any form of regular oral hygiene. After 20–30 years of chronic gingivitis, gum disease caused teeth to loosen and eventually fall out. Everyone had full sets of dentures in those days. If dentistry can effectively treat gingivitis and eventual tooth loss with patient education, and regular and routine oral hygiene, why cannot eye doctors effectively treat blepharitis and eventual meibomian gland loss? It is now possible, but it must start with a new understanding of this chronic disease, and an active role by the eye doctor in performing regular MBE procedures on all patients, particularly those at higher risk.
Disclosure
James M Rynerson is the President and CEO of BlephEx, LLC and Henry D Perry is the senior founding partner of Ophthalmic Consultants of Long Island.
The authors report no other conflicts of interest in this work.
References
Christersson LA, Zambon JJ, Genco RJ. Dental bacterial plaques. Nature and role in periodontal disease. J Clin Periodontol. 1991;18(6):441–446. | ||
Lappin-Scott H, Burton S, Stoodley P. Revealing a world of biofilms – the pioneering research of Bill Costerton. Nat Rev Microbiol. 2014;12(11):781–787. | ||
Bernhardt MJ, Myntti MF. Topical treatment with an agent disruptive to P. acnes biofilm provides positive therapeutic response: results of a randomized clinical trial. J Drugs Dermatol. 2016;15(6):677–683. | ||
Thygeson P. Etiology and treatment of blepharitis; a study in military personnel. Arch Ophthal. 1946;36(4):445–477. | ||
Dougherty JM, McCulley JP. Analysis of the free fatty acid component of meibomian secretions in chronic blepharitis. Invest Ophthalmol Vis Sci. 1986;27(1):52–56. | ||
Foulks GN. The correlation between the tear film lipid layer and dry eye disease. Surv Ophthalmol. 2007;52(4):369–374. | ||
Bzdrenga J, Daudé D, Rémy B, et al. Biotechnological applications of quorum quenching enzymes. Chem Biol Interact. Epub 2016 May 22. | ||
Wilson FM 2nd. Adverse external ocular effects of topical ophthalmic therapy: an epidemiologic, laboratory, and clinical study. Trans Am Ophthalmol Soc. 1983;81:854–965. | ||
Geerling G, Tauber J, Baudouin C, et al. The International Workshop on Meibomian Gland Dysfunction: report of the Subcommittee on Management and Treatment of Meibomian Gland Dysfunction. Invest Ophthalmol Vis Sci. 2011;52(4):2050–2064. | ||
Dougherty JM, McCulley JP. Comparative bacteriology of chronic blepharitis. Br J Ophthalmol. 1984;68(8):524–528. | ||
Thygeson P, Vaughan DG Jr. Seborrheic blepharitis. Trans Am Ophthalmol Soc. 1954;52:173–188. | ||
Brothers KM, Nau AC, Romanowski EG, Shanks RM. Dexamethasone diffusion across contact lenses is inhibited by Staphylococcus epidermidis biofilms in vitro. Cornea. 2014;33(10):1083–1087. | ||
Szczotka-Flynn L, Lass JH, Sethi A, et al. Risk factors for corneal infiltrative events during continuous wear of silicone hydrogel contact lenses. Invest Ophthalmol Vis Sci. 2010;51(11):5421–5430. | ||
Absalon C, Van Dellen K, Watnick P. A communal bacterial adhesin anchors biofilm and bystander cells to surfaces. PLoS Pathog. 2011;7(8):e1002210. | ||
Pickering BS, Smith DR, Watnick PJ. Glucose-specific enzyme IIA has unique binding partners in the Vibrio cholerae biofilm. MBio. 2012;3(6):e00228-12. | ||
Teles R, Teles F, Frias-Lopez F, Paster B, Haffajee A. Lessons learned and unlearned in periodontal microbiology. Periodontol 2000. 2013;62(1):95–162. | ||
McSwain BS, Irvine RL, Hausner M, Wilderer PA. Composition and distribution of extracellular polymeric substances in aerobic flocs and granular sludge. Appl Environ Microbiol. 2005;71(2):1051–1057. | ||
Baldi F, Marchetto D, Battistel D, et al. Iron-binding characterization and polysaccharide production by Klebsiella oxytoca strain isolated from mine acid drainage. J Appl Microbiol. 2009;107(4):1241–1250. | ||
Edwards AM, Bowden MG, Brown EL, Laabei M, Masey RC. Staphylococcus aureus extracellular adherence protein triggers TNFα release, promoting attachment to endothelial cells via protein A. PLoS One. 2012;7(8):e43046. | ||
Weiser J, Henke HA, Hector N, et al. Sub-inhibitory tigecycline concentrations induce extracellular matrix binding protein Embp dependent Staphylococcus epidermidis biofilm formation and immune evasion. Int J Med Microbiol. 2016;306(6):471–478. | ||
Jeffries TC, Ostrowski M, Williams RB, et al. Spatially extensive microbial biogeography of the Indian Ocean provides insights into the unique community structure of a pristine coral atoll. Sci Rep. 2015;5:15383. | ||
Paharik AE, Horswill AR. The Staphylococcal biofilm: adhesins, regulation, and host response. Microbiol Spectr. 2016;4(2):22–215. | ||
Kivanç SA, Kivanç M, Bayramlar H. Microbiology of corneal wounds after cataract surgery: biofilm formation and antibiotic resistance patterns. J Wound Care. 2016;25(1):12, 14–19. | ||
Monroe D. Looking for chinks in the armor of bacterial biofilms. PLoS Biol. 2007;5(11):e307. | ||
Watnick P, Kolter R. Biofilm, city of microbes. J Bacteriol. 2000;182(10):2675–2679. | ||
Doll K, Jongsthaphongpun KL, Stumpp NS, Winkel A, Stiesch M. Quantifying implant-associated biofilms: comparison of microscopic, microbiologic and biochemical methods. J Microbiol Methods. 2016;130:61–68. | ||
Saint-Ruf C, Garfa-Traoré M, Collin V, Cordier C, Franceschi C, Matic I. Massive diversification in aging colonies of Escherichia coli. J Bacteriol. 2014;196(17):3059–3073. | ||
Artini M, Cellini A, Scoarughi GL, et al. Evaluation of contact lens multipurpose solutions on bacterial biofilm development. Eye Contact Lens. 2015;41(3):177–182. | ||
OKhaireddin R, Hueber A. Lidrandhygiene bei Kontaktlinsenträgern mit Blepharitis Therapievergleich Babyshampoo versus Phospholipidlösung. [Eyelid hygiene for contact lens wearers with blepharitis. Comparative investigation of treatment with baby shampoo versus phospholipid solution]. Ophthalmologe. 2013;110(2):146–153. German. | ||
Wolf D, Rippa V, Mobarec JC, et al. The quorum-sensing regulator ComA from Bacillus subtilis activates transcription using topologically distinct DNA motifs. Nucleic Acids Res. 2016;44(5):2160–2172. | ||
Davis TH. Profile of J Woodland Hastings. Proc Natl Acad Sci U S A. 2007;104(3):693–695. | ||
Hastings JW, Greenberg EP. Quorum sensing: the explanation of a curious phenomenon reveals a common characteristic of bacteria. J Bacteriol. 1999;181(9):2667–2668. | ||
Knecht LD, O’Connor G, Mittal R, et al. Serotonin activates bacterial quorum sensing and enhances the virulence of Pseudomonas aeruginosa in the host. EBioMedicine. 2016;9:161–169. | ||
Mireille Ayé A, Bonnin-Jusserand M, Brian-Jaisson F, et al. Modulation of violace in production and phenotypes associated with biofilm by exogenous quorum sensing N-acylhomoserine lactones in the marine bacterium. Pseudoalteromonas ulvae TC14. Microbiology. 2015;161(10):2039–2051. | ||
Zhang C, Zhu S, Jatt AN, Zeng M. Characterization of N-acyl homoserine lactones (AHLs) producing bacteria isolated from vacuum-packaged refrigerated turbot (Scophthalmus maximus) and possible influence of exogenous AHLs on bacterial phenotype. J Gen Appl Microbiol. 2016;62(2):60–67. | ||
Cheung GY, Joo HS, Chatterjee SS, Otto M. Phenol-soluble modulins – critical determinants of staphylococcal virulence. FEMS Microbiol Rev. 2014;38(4):698–719. | ||
Prince LR, Graham KJ, Connolly J, et al. Staphylococcus aureus induces eosinophil cell death mediated by α-hemolysin. PLoS One. 2012;7(2):e31506. | ||
Stroh P, Günther F, Meyle E, Prior B, Wagner C, Hänsch GM. Host defence against Staphylococcus aureus biofilms by polymorphonuclear neutrophils: oxygen radical production but not phagocytosis depends on opsonisation with immunoglobulin G. Immunobiology. 2011;216(3):351–357. | ||
Liew YK, Awang Hamat R, van Belkum A, Chong PP, Neela V. Comparative exoproteomics and host inflammatory response in Staphylococcus aureus skin and soft tissue infections, bacteremia, and subclinical colonization. Clin Vaccine Immunol. 2015;22(5):593–603. | ||
Holmes A, Ganner M, McGuane S, Pitt TL, Cookson BD, Kearns AM. Staphylococcus aureus isolates carrying Panton-Valentine leucocidin genes in England and Wales: frequency, characterization, and association with clinical disease. J Clin Microbiol. 2005;43(5):2384–2390. | ||
Maloney WJ, Smith RL, Castro F, Schurman DJ. Fibroblast response to metallic debris in vitro. Enzyme induction cell proliferation, and toxicity. J Bone Joint Surg Am. 1993;75(6):835–844. | ||
Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13(1):16–34. | ||
Argudín MA, Vanderhaeghen W, Vandendriessche S, et al. Biofilm formation of ica operon-positive Staphylococcus epidermidis from different sources. APMIS. 2015;123(12):1081–1089. | ||
Krakauer T. Update on staphylococcal superantigen-induced signaling pathways and therapeutic interventions. Toxins (Basel). 2013;5(9):1629–1654. | ||
Baudouin CJ. Un nouveau schéma pour mieux comprendre les maladies de la surface oculaire. [A new approach for better comprehension of diseases of the ocular surface]. J Fr Ophtalmol. 2007;30(3):239–246. French. | ||
Brüning T, Bartsch R, Bolt HM, et al. Sensory irritation as a basis for setting occupational exposure limits. Arch Toxicol. 2014;88(10):1855–1879. | ||
Pflugfelder SC, Stern ME; Symposium Participants. Immunoregulation on the ocular surface: 2nd Cullen Symposium. Ocul Surf. 2009;7(2):67–77. | ||
Watanabe A, Imai K, Kinoshita S. Impact of high myopia and duration of hard contact lens wear on the progression of ptosis. Jpn J Ophthalmol. 2013;57(2):206–210. | ||
Ramadhani AM, Derick T, Holland MJ, Burton MJ. Blinding trachoma: systematic review of rates and risk factors for progressive disease. PLoS Negl Trop Dis. 2016;10(8):e0004859. | ||
Uchiyama E, Aronowicz JD, Butovich IA, McCulley JP. Pattern of vital staining and its correlation with aqueous tear deficiency and meibomian gland dropout. Eye Contact Lens. 2007;33(4):177–179. | ||
Messmer EM. The pathophysiology, diagnosis, and treatment of dry eye disease. Dtsch Arztebl Int. 2015;112(5):71–81; quiz 82. | ||
Yeotikar NS, Zhu H, Markoulli M, Nichols KK, Naduvilath T, Papas EB. Functional and morphologic changes of meibomian glands in an asymptomatic adult population. Invest Ophthalmol Vis Sci. 2016;57(10):3996–4007. | ||
Suzuki T, Kawamura Y, Uno T, Ohashi Y, Ezaki T. Prevalence of Staphylococcus epidermidis strains with biofilm-forming ability in isolates from conjunctiva and facial skin. Am J Ophthalmol. 2005;140(5):844–850. | ||
Miyanaga Y. A new perspective in ocular infection and the role of antibiotics. Ophthalmologica. 1997;211(Suppl 1):9–14. | ||
Earl CS, Keong TW, An SQ, et al. Haemophilus influenzae responds to glucocorticoids used in asthma therapy by modulation of biofilm formation and antibiotic resistance. EMBO Mol Med. 2015;7(8):1018–1033. | ||
Cetinkaya A, Akova YA. Pediatric ocular acne rosacea: long-term treatment with systemic antibiotics. Am J Ophthalmol. 2006;142(5):816–821. | ||
Nazir SA, Murphy S, Siatkowski RM, Chodosh J, Siatkowski RL. Ocular rosacea in childhood. Am J Ophthalmol. 2004;137(1):138–144. | ||
Suzuki T. Meibomitis-related keratoconjunctivitis: implications and clinical significance of meibomian gland inflammation. Cornea. 2012;31(Suppl 1):S41–S44. | ||
Huse HK, Kwon T, Zlosnik JE, Speert DP, Marcotte EM, Whiteley M. Pseudomonas aeruginosa enhances production of a non-alginate exopolysaccharide during long-term colonization of the cystic fibrosis lung. PLoS One. 2013;8(12):e82621. | ||
Pflugfelder SC, Karpecki PM, Perez VL. Treatment of blepharitis: recent clinical trials. Ocul Surf. 2014;12(4):273–284. | ||
Pult H, Riede-Pult BH, Murphy PJ. A new perspective on spontaneous blinks. Ophthalmology. 2013;120(5):1086–1091. | ||
Albar AH, Almehdar HA, Uversky VN, Redwan EM. Structural heterogeneity and multifunctionality of lactoferrin. Curr Protein Pept Sci. 2014;15(8):778–797. | ||
Tiffany JM. The normal tear film. Dev Ophthalmol. 2008;41:1–20. | ||
Dartt DA, Masli S. Conjunctival epithelial and goblet cell function in chronic inflammation and ocular allergic inflammation. Curr Opin Allergy Clin Immunol. 2014;14(5):464–470. | ||
McCauley HA, Guasch G. Three cheers for the goblet cell: maintaining homeostasis in mucosal epithelia. Trends Mol Med. 2015;21(8):492–503. | ||
McCauley HA, Liu CY, Attia AC, et al. TGFβ signaling inhibits goblet cell differentiation via SPDEF in conjunctival epithelium. Development. 2014;141(23):4628–4639. | ||
Specian RD, Oliver MG. Functional biology of intestinal goblet cells. Am J Physiol. 1991;260(2 Pt 1):C183–C193. | ||
Seibel J, Pergola C, Werz O, et al. Bronchipret® syrup containing thyme and ivy extracts suppresses bronchoalveolar inflammation and goblet cell hyperplasia in experimental bronchoalveolitis. Phytomedicine. 2015;22(13):1172–1177. | ||
Guzman-Aranguez A, Argüeso P. Structure and biological roles of mucin-type O-glycans at the ocular surface. Ocul Surf. 2010;8(1):8–17. | ||
Uchino Y, Uchino M, Yokoi N, et al. Impact of cigarette smoking on tear function and correlation between conjunctival goblet cells and tear MUC5AC concentration in office workers. Sci Rep. 2016;6:27699. | ||
Hayashi D, Li D, Hayashi C, Shatos M, Hodges RR, Dartt DA. Role of histamine and its receptor subtypes in stimulation of conjunctival goblet cell secretion. Invest Ophthalmol Vis Sci. 2012;53(6):2993–3003. | ||
Galley JD, Yu Z, Kumar P, Dowd SE, Lyte M, Bailey MT. The structures of the colonic mucosa-associated and luminal microbial communities are distinct and differentially affected by a prolonged murine stressor. Gut Microbes. 2014;5(6):748–760. | ||
Johansson ME. Mucus layers in inflammatory bowel disease. Inflamm Bowel Dis. 2014;20(11):2124–2131. | ||
Johansson ME, Jakobsson HE, Holmén-Larsson J, et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe. 2015;18(5):582–592. | ||
Nichols B, Dawson CR, Togni B. Surface features of the conjunctiva and cornea. Invest Ophthalmol Vis Sci. 1983;24(5):570–576. | ||
McGilligan VE, Gregory-Ksander MS, Li D, et al. Staphylococcus aureus activates the NLRP3 inflammasome in human and rat conjunctival goblet cells. PLoS One. 2013;8(9):e74010. | ||
Medel R, Alonso T, Vela JI, Calatayud M, Bisbe L, García-Arumí J. Conjunctival cytology in floppy eyelid syndrome: objective assessment of the outcome of surgery. Br J Ophthalmol. 2009;93(4):513–517. | ||
Gao YY, Di Pascuale MA, Li W, et al. High prevalence of Demodex in eyelashes with cylindrical dandruff. Invest Ophthalmol Vis Sci. 2005;46(9):3089–3094. | ||
Kawakita T, Kawashima M, Ibrahim O, Murato D, Tsubota K. [Demodex-related marginal blepharitis in Japan]. Nippon Ganka Gakkai Zasshi. 2010;114(12):1025–1029. Japanese. | ||
Kabataş N, Doğan AŞ, Kabataş EU, Acar M, Biçer T, Gürdal C. The effect of Demodex infestation on blepharitis and the ocular symptoms. Eye Contact Lens. Epub 2016 Jan 16. | ||
Elder MJ. Anatomy and physiology of eyelash follicles: relevance to lash ablation procedures. Ophthal Plast Reconstr Surg. 1997;13(1):21–25. | ||
Blackie CA, Korb DR, Knop E, Bedi R, Knop N, Holland EJ. Nonobvious obstructive meibomian gland dysfunction. Cornea. 2010;29(12):1333–1345. | ||
Gao Y, Zhuang M, Fan C, Ye K, Hu J, Hong Y. [Abnormal property of meibomian secretion and dry eye syndrome]. Yan Ke Xue Bao. 2007;23(2):121–125. Chinese. | ||
Uzunosmanoglu E, Mocan MC, Kocabeyoglu S, Karakaya J, Irkec M. Meibomian gland dysfunction in patients receiving long-term glaucoma medications. Cornea. 2016;35(8):1112–1116. | ||
Knop I, Korb DR, Blackie CA, Knop N. The lid margin is an underestimated structure for preservation of ocular surface health and development of dry eye disease. Dev Ophthalmol. 2010;45:108–122. | ||
Greiner JV. A single LipiFlow® Thermal Pulsation System treatment improves meibomian gland function and reduces dry eye symptoms for 9 months. Curr Eye Res. 2012;37(4):272–278. | ||
Conrady CD, Joos ZP, Patel BC. Review: the lacrimal gland and its role in dry eye. J Ophthalmol. 2016;2016:7542929. | ||
Stevenson W, Pugazhendhi S, Wang M. Is the main lacrimal gland indispensable? Contributions of the corneal and conjunctival epithelia. Surv Ophthalmol. 2016;61(5):616–627. | ||
Baudouin C. Ocular surface and external filtration surgery: mutual relationships. Dev Ophthalmol. 2012;50:64–78. | ||
Baudouin C, Messmer EM, Aragona P, et al. Revisiting the vicious circle of dry eye disease: a focus on the pathophysiology of meibomian gland dysfunction. Br J Ophthalmol. 2016;100(3):300–306. | ||
Romero JM, Biser SA, Perry HD, et al. Conservative treatment of meibomian gland dysfunction. Eye Contact Lens. 2004;30(1):14–19. | ||
Gray H. Anatomy of the human body. 20th ed. Lewis WH, ed. Philadelphia: Lea & Febiger; 1918. | ||
Montana State University Center for Biofilm Engineering. Quorum-sensing. Available from: https://www.biofilm.montana.edu/content/quorum-sensing. Accessed November 18, 2016. | ||
Science Source Images. Eyelash hairs and human skin, colored SEM. Available from: http://images.sciencesource.com/search?key=SB9970&lcn=rf&lcn=rm&ori=h&ori=v&ori=s&country=USA. Accessed October 21, 2016. | ||
Clinical neuro-opthalmology. Available from: http://content.lib.utah.edu/utils/getfile/collection/EHSL-NOVEL/id/1272/filename/image. Accessed November 23, 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.