Back to Journals » International Journal of Nanomedicine » Volume 14
D-α-tocopherol polyethylene glycol 1000 succinate-modified liposomes with an siRNA corona confer enhanced cellular uptake and targeted delivery of doxorubicin via tumor priming
Authors Tan X, Fang Y, Ren Y ![]() , Li Y, Wu P, Yang X, Liu W
, Li Y, Wu P, Yang X, Liu W
Received 23 October 2018
Accepted for publication 10 December 2018
Published 18 February 2019 Volume 2019:14 Pages 1255—1268
DOI https://doi.org/10.2147/IJN.S191858
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Xi Tan,1,* Yan Fang,1,* Yuanyuan Ren,1 Yinghuan Li,2 Peicheng Wu,3 Xiangliang Yang,1,4 Wei Liu1,4
1College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan 430074, People’s Republic of China; 2School of Pharmaceutical Sciences, Beijing Area Major Laboratory of Peptide and Small Molecular Drugs, Engineering Research Center of Endogenous Prophylactic of Ministry of Education of China, Capital Medical University, Beijing 100069, People’s Republic of China; 3School of Biosciences and Biopharmaceuticals, Institute of Biopharmaceuticals, Guangdong Pharmaceutical University, Guangzhou 510006, People’s Republic of China; 4National Engineering Research Center for Nanomedicine, Huazhong University of Science and Technology, Wuhan 430074, People’s Republic of China
*These authors contributed equally to this work
Background: Combination therapy employing siRNAs and antitumor drugs is a promising method for the treatment of solid tumors. However, regarding combined treatments involving siRNAs and chemotherapeutic reagents, most prior research has focused on the enhanced cytotoxicity against tumor cells conferred by downregulation of the targeted protein.
Purpose: We developed D-α-tocopherol polyethylene glycol 1000 succinate (TPGS)-modified cationic liposomes (LPs) to simultaneously deliver doxorubicin (Dox) and the Bcl-2 siRNA (siBcl-2) for synergistic chemotherapy. The co-loading of siBcl-2 onto the Dox-loaded cationic LPs (siBcl-2/Dox-TPGS-LPs) could promote cellular uptake, cytotoxicity against 3D H22 tumor spheroids, circulation in the blood, drug accumulation at tumor sites, and synergistic chemotherapy in vivo.
Methods: The siBcl-2/Dox-TPGS-LPs were constructed by co-loading siBcl-2 onto the Dox-loaded TPGS-modified cationic LPs (Dox-TPGS-LPs), and Dox entrapment into the LPs was achieved using an ammonium sulfate gradient method. The antitumor effects of siBcl-2/Dox-TPGS-LPs were studied in murine hepatic carcinoma H22 cells, 3D H22 tumor spheroids, and H22 tumor-bearing mice.
Results: Dynamic light scattering technique and transmission electron microscopy images revealed that siBcl-2 loaded onto the Dox-TPGS-LPs formed a prominent corona at an nitrogen to phosphorus (N/P) ratio of 4:1, resulting in particle size increase from 155 to 210 nm and a weak positive zeta potential (+12.5 mV). The siBcl-2/Dox-TPGS-LPs enhanced the cellular uptake of Dox, promoted toxicity against 3D H22 tumor spheroids via tumor priming, prolonged Dox circulation in the blood, and increased accumulation of Dox at tumor sites, thereby enhancing the cytotoxicity of Dox in vitro and its chemotherapeutic efficacy in vivo.
Conclusion: The siBcl-2/Dox-TPGS-LPs demonstrated a strong potential for application in synergistic chemotherapy. The co-loading of siRNAs both sensitized cells toward antitumor drugs by downregulating the expression level of a specific protein and influenced the pharmacokinetic behavior of the co-delivery system in vitro and in vivo.
Keywords: siRNA corona, cationic liposomes, co-delivery, Bcl-2, tumor priming, synergistic chemotherapy
Introduction
RNA interference (RNAi) is a widely used technology employed for targeted gene silencing.1 As an effective RNAi tool, siRNAs can be applied to specifically downregulate the expression of targeted proteins in treated cells.2 Thus, siRNAs have the potential to augment antitumor therapies when overexpression of a specific protein is closely related to tumor occurrence and development.3 Indeed, combined delivery of an antitumor drug and siRNAs has been recognized as a potential novel treatment approach for enhancing the efficacy of cancer chemotherapy and overcoming multidrug resistance.4 However, considering combined treatments involving siRNAs and chemotherapeutic reagents, most research has focused on the enhanced cytotoxicity against tumor cells conferred by downregulation of the targeted protein.5–8 It is rarely mentioned that co-loading of siRNAs onto nanoparticles can have uncertain impacts on cellular uptake, circulation in the blood, biodistribution, and penetration of the drug into solid tumor sites of such drug delivery systems.
The anti-apoptotic protein Bcl-2 is often overexpressed in many kinds of tumor cells and plays an essential role in preventing cancer cells from undergoing apoptosis and autophagy.9 It has been reported that co-delivery of Bcl-2-targeted siRNA (siBcl-2) and antitumor drugs via cationic liposomes (LPs) or nanoparticles can significantly downregulate the expression level of the Bcl-2 protein, increasing the sensitivity of the cells to chemotherapy.10,11 Here, we used siBcl-2 as an apoptosis-inducing RNA to expand the interstitial space and thereby enhance intratumoral penetration of chemotherapeutic agents via tumor priming, thus overcoming the poor diffusion caused by the high cell density at solid tumor sites. Tumor priming, first reported by Kuh et al in 1999, can be interpreted as an enhanced intratumoral diffusion of drugs due to enlargement of the interstitial space using apoptosis-inducing drugs, such as paclitaxel.12 As such, tumor priming induced by delivering siBcl-2 in conjunction with the co-delivery of antitumor drugs could serve as a superior method for enhancing the cytotoxicity of chemotherapeutic agents.
Cationic LPs are widely used in gene transfection and for delivering antitumor drugs, and therefore, are an excellent choice for co-delivery of siBcl-2 and chemotherapeutic agents.13 D-α-tocopherol polyethylene glycol 1000 succinate (TPGS), a polyethylene glycol (PEG) derivative of natural vitamin E succinate, can improve the stability of siRNA-loaded cationic LPs.14 In this study, we developed TPGS-modified cationic LPs to deliver both doxorubicin (Dox) and siBcl-2 (siBcl-2/Dox-TPGS-LPs). To evaluate the effect of co-loading siRNAs onto Dox-loaded LPs, we characterized the siBcl-2/Dox-TPGS-LPs in regard to the cellular uptake of Dox as well as its cytotoxicity in vitro in a 3D H22 tumor spheroid model, and biodistribution and antitumor efficacy in vivo. According to our results, siBcl-2/Dox-TPGS-LPs demonstrated the presence of an siBcl-2 corona on the surface of the LPs, increased cellular uptake, cytotoxicity against H22 cells, and toxicity toward H22 tumor spheroids in vitro, as well as prolonged circulation of Dox in the blood and increased Dox accumulation at tumor sites, thereby enhancing the chemotherapeutic efficacy of Dox. As such, these results highlight the potential of co-loading siRNAs onto drug delivery systems to influence cellular uptake, pharmacokinetic behavior, and biodistribution in vivo, thus increasing the efficacy of chemotherapy treatments.
Materials and methods
Materials
Dox hydrochloride was supplied by Beijing Mesochem Technology Co., Ltd. (Beijing, People’s Republic of China). TPGS was obtained from Sigma-Aldrich (St Louis, MO, USA). 1,2-Dioleoyl-3-trimethylammonium-propane (DOTAP) and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) were supplied by Avanti Polar Lipids, Inc. (Alabaster, AL, USA). Cholesterol (CHOL) was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, People’s Republic of China). 1,1′-Dioctadecyltetramethyl indotricarbocyanine iodide (DiR) was purchased from AAT Bioquest Inc. (Sunnyvale, CA, USA). All reagents and chemicals were from commercial sources and of HPLC or analytical grades.
Targeting mouse Bcl-2 siRNA (siBcl-2, sense: 5′-GCA UGC GAC CUC UGU UUG AdTdT-3′, antisense: 5′-UCA AAC AGA GGU CGC AUG CdTdT-3′) and fluorescein amidite (FAM)-labeled Bcl-2 siRNA (FAM-siRNA) were synthesized by GenePharma (Shanghai, People’s Republic of China).
Preparation and characterization of siBcl-2 and Dox co-loaded LPs
The cationic LPs were prepared using the film dispersion method.15 DOTAP, DPPC, and CHOL were dissolved in chloroform and mixed at a molar ratio of 40:40:20 in a round-bottom flask. After evaporation of the organic solvent using a rotary evaporator, the formed lipid film was immediately hydrated with 120 mM ammonium sulfate solution and further extruded through a polycarbonate membrane (100 nm pore size) using a mini-extruder (Avanti Polar Lipids, Inc.) to obtain bare LPs. To prepare TPGS-modified cationic LPs (TPGS-LPs), DOTAP, DPPC, TPGS, and CHOL were mixed at a molar ratio of 40:40:4:16 and then treated using subsequent procedures similar to that of LPs. The external phase of the blank LPs was replaced with 0.9% NaCl by diafiltration. Dox-loaded cationic LPs (Dox-LPs or Dox-TPGS-LPs) were obtained by adding free Dox solution to blank LPs (LPs or TPGS-LPs) at a drug-to-lipid ratio of 1:10 (w/w), and the mixture was then incubated at 40°C for 30 minutes. Subsequently, unloaded Dox was removed using a Sephadex G50 column to purify the Dox-loaded LPs.
To achieve co-delivery of siBcl-2 and Dox by TPGS-modified LPs, cationic LPs (Dox-TPGS-LPs) diluted in diethyl pyrocarbonate water were slowly added to siBcl-2 solution at different nitrogen to phosphorus (N/P) ratios. Then, the mixture was incubated for 30 minutes at room temperature to obtain siBcl-2/Dox co-loaded LPs (siBcl-2/Dox-TPGS-LPs).
The particle sizes, polydispersity indexes, and zeta potentials of Dox-LPs, Dox-TPGS-LPs, and siBcl-2/Dox-TPGS-LPs were determined using a Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK) instrument. Diluted LPs were placed on a carbon-coated copper grid, stained with phosphotungstic acid, and then observed under a transmission electron microscope (H-7000FA; Hitachi, Tokyo, Japan).
The entrapped Dox in the LPs was extracted with dimethyl sulfoxide (DMSO) to evaluate the loading and encapsulation efficiency (EE) of the drug.16 Dox concentration was measured at 482 nm using a UV spectrophotometer (UV-1750; Shimadzu, Kyoto, Japan). To evaluate the EE of siBcl-2 absorbed on the liposomal surface, FAM-labeled siBcl-2 (FAM-siRNA) was used to prepare FAM-siRNA/Dox-TPGS-LPs as described previously. The EE of siBcl-2 was then determined by ultracentrifugation at 20,000× g at 4°C for 2 hours, according to the ultrafiltration method.8
In vitro release studies
The in vitro release of Dox from siBcl-2/Dox-TPGS-LPs was studied using dialysis method.17 Briefly, 1 mL of siBcl-2/Dox-TPGS-LPs, Dox-TPGS-LPs, Dox-LPs, or free Dox was added to dialysis bags (MWCO 3,500 Da) at a Dox concentration of 100 μg/mL. Each dialysis bag was incubated in 100 mL of release media, PBS (0.01 M, pH 7.4) containing 1% Tween 80 (w/v), under shaking at 100 rpm and 37°C for 48 hours. Samples (1 mL) were removed from the release medium at predetermined time points (0.083, 0.25, 0.5, 1, 2, 4, 8, 12, 24, and 48 hours) and replaced with an equal volume of fresh release medium. Dox concentration of each sample was measured by fluorescence spectrophotometry (F-4500; Hitachi) at a λEx/Em of 485/574 nm, and the cumulative release percentage of Dox was calculated.
Cell culture
Murine hepatic carcinoma H22 cells were supplied by the China Center for Type Culture Collection (Wuhan University, Wuhan, People’s Republic of China). The cells were cultured in RPMI 1640 medium, supplemented with 10% FBS and 1% penicillin–streptomycin solution, and kept at 37°C in a humidified atmosphere with 5% CO2.
Cellular uptake and intracellular distribution of Dox and siRNA
H22 cells were seeded at a density of 2×105 cells per well into a 24-well plate and then treated with free Dox, Dox-LPs, Dox-TPGS-LPs, or siBcl-2/Dox-TPGS-LPs at a Dox concentration of 5 μg/mL for 4 hours. The cells were then washed twice with PBS, collected by centrifugation, and then were resuspended in PBS and analyzed using a flow cytometer (FC500; Beckman, Brea, CA, USA) to evaluate the intracellular accumulation of Dox.
Confocal laser scanning microscopy (CLSM) analysis was carried out to observe the cellular uptake and intracellular distribution of Dox and siBcl-2 within the H22 cells. The green fluorescent FAM-labeled siRNA was introduced to replace siBcl-2 for the preparation of FAM-siRNA/Dox co-loaded TPGS-modified LPs (FAM-siRNA/Dox-TPGS-LPs). The H22 cells were seeded in glass-bottom microwell dishes at a density of 4×105 cells and incubated to allow them to reach 80% confluence. Then, the medium was replaced with a fresh medium containing free Dox, Dox-LPs, Dox-TPGS-LPs, or FAM-siRNA/Dox-TPGS-LPs at a Dox concentration of 5 μg/mL. After treatment for 4 hours at 37°C, the cells were fixed with 4% paraformaldehyde for 15 minutes, washed with ice-cold PBS, and stained with DAPI (Biosharp Biotech Co., Hefei, People’s Republic of China) for an additional 15 minutes. The cells were further washed with ice-cold PBS and imaged by CLSM (FV1000; Olympus, Tokyo, Japan) under excitation at 405 nm (DAPI for nuclei), 488 nm (FAM-siRNA), and 559 nm (Dox).18
MTT assays
The in vitro cytotoxicity of the various Dox formulations against H22 cells was evaluated via MTT assays. H22 cells were seeded into 96-well plates at a density of 2.0×104 cells per well and incubated overnight. The cells were then treated with free Dox, Dox-LPs, Dox-TPGS-LPs, or siBcl-2/Dox-TPGS-LPs at a Dox concentration ranging from 0.05 to 10 μg/mL in serum-free medium for 24 hours. The culture medium in each well was replaced with a fresh medium, followed by the addition of 20 μL MTT solution (5 mg/mL). After incubation for another 4 hours, the medium in each well was removed, and 150 μL DMSO was added to each well to dissolve the purple formazan crystals. Absorbance was measured at 570 nm using a Multi-Mode Microplate Reader (Thermo Fisher Scientific, Waltham, MA, USA), and untreated cells were used as a control. The half-maximal inhibitory concentration (IC50) was calculated using GraphPad Prism 6.0 software. The cytotoxicity of empty LPs (LPs and TPGS-LPs) against H22 cells was also evaluated via MTT assays using the same protocol.
Toxicity of siBcl-2/Dox-TPGS-LPs in the 3D H22 tumor spheroid model
To accurately screen the efficacy of these cancer therapeutics and to study the tumor biology in vitro, multicellular tumor spheroids were generated to simulate solid tumors with similar morphologies and biological microenvironments.19 The H22 tumor spheroid model was established as previously described.20 H22 cells were seeded into a 96-well plate and cultured in complete RPMI 1640 medium containing fibrinogen gel (1 μg/mL) and thrombin (0.1 U). After incubation for 5 days, the medium in each well was replaced with a fresh medium containing free Dox, Dox-LPs, Dox-TPGS-LPs, or siBcl-2/Dox-TPGS-LPs at a Dox concentration of 5 μg/mL; treatment with normal saline served as a control. Finally, these H22 tumor spheroids were observed by CLSM after treatment with the above formulations for 72 hours to evaluate the cytotoxicity of siBcl-2/Dox-TPGS-LPs.
Animal and tumor model
BALB/c female mice (4–6 weeks old, weight of 18–22 g) were purchased from Liaoning Changsheng Biotechnology Co., Ltd (Shenyang, People’s Republic of China). All animals used in this research were maintained in a specific pathogen-free environment at a temperature of 22°C with free access to water and food. All animal experiments involved in this research were performed in compliance with the Guide for the Care and Use of Laboratory Animals of Huazhong University of Science and Technology, as approved by the Institutional Animal Ethical Committee of Huazhong University of Science and Technology.
For the subcutaneous hepatocellular carcinoma-bearing mouse model, BALB/c female mice were subcutaneously injected with 100 μL of cell suspension containing 1.0×106 H22 cells into their right flank. After injection, all mice were checked every 2 days to monitor the tumor size using a digital caliper and determine their body weights. The tumor volume was calculated using the following equation: tumor volume =0.5×(tumor length)×(tumor width)2. When the tumor size reached a predetermined volume, the mice could be used for the subsequent in vivo biodistribution and therapeutic studies.
Pharmacokinetic studies
Free Dox, Dox-LPs, Dox-TPGS-LPs, or siBcl-2/Dox-TPGS-LPs was administered intravenously to BALB/c female mice at a dose of Dox 5 mg/kg (n=5). Blood samples were collected via the retro-orbital vein at predetermined time points (0.083, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours) after injection and then stored at 4°C for 12 hours. The obtained blood samples were centrifuged at 12,000× g for 15 minutes, and the plasma supernatant was diluted 1:100 with methanol on a vortex mixer for 90 seconds. Following another centrifugation at 15,000× g for 30 minutes to precipitate the proteins, Dox was extracted with methanol. Dox concentration was further determined using a fluorescence spectrophotometer. The area under the curve (AUC) representing the plasma Dox concentration–time postinjection as well as the plasma half-time of Dox (T1/2) for the four groups was calculated according to a trapezoidal calculation.21
In vivo biodistribution
To investigate the tumor-targeting efficacy of the different LP formulations by passive targeting in vivo, DiR was entrapped into LPs to monitor the real-time biodistribution changes of the cationic LPs. DiR was loaded into the LPs using the film dispersion method after being mixed with the lipids, as described in the “Preparation and characterization of siBcl-2 and Dox co-loaded LPs” section. Free DiR and DiR-loaded LPs (ie, DiR-LPs, DiR-TPGS-LPs, and siBcl-2/DiR-TPGS-LPs) were intravenously injected into H22 tumor-bearing BALB/c female mice via the tail vein at a dose of 0.5 mg/kg DiR. Then, the mice were imaged using an in vivo imaging system (IVIS Lumina XR; Caliper Life Sciences, Waltham, MA, USA) at 3, 6, 12, and 24 hours after injection. Finally, the mice were sacrificed, and the tissues were harvested at 24 hours. The heart, liver, spleen, lungs, and kidneys of these mice were further imaged using an in vivo imaging system at an excitation wavelength of 748 nm and an emission wavelength of 780 nm.
To study the in vivo biodistribution of Dox, the mice were intravenously injected with free Dox, Dox-LPs, Dox-TPGS-LPs, or siBcl-2/Dox-TPGS-LPs via the tail vein at a Dox dose of 5 mg/kg. At 24 hours after injection, the mice were sacrificed, and the tissues were harvested and cleaned with PBS. The tissues were further dried with filter paper and weighed and then added to ultrapure water at a ratio of 20% (w/v). The homogenate was then obtained by homogenizing the tissue mixture and further mixed with methanol at a volume ratio of 1:5. The mixture was stored at −20°C for 24 hours and centrifuged at 15,000 rpm for 30 minutes to precipitate the proteins. The concentration of Dox was finally measured using a fluorescence spectrophotometer, and the untreated tissue homogenate was used as a blank control.
Tumor suppression assays
The tumor suppression effect of the different treatments was evaluated by measuring the tumor volume in a double-blinded manner. After reaching a tumor volume of around 150 mm3, the tumor-bearing BALB/c female mice were divided into five groups (Day 0). The mice were then injected with normal saline, free Dox, Dox-LPs, Dox-TPGS-LPs, or siBcl-2/Dox-TPGS-LPs (5 mg Dox equiv./kg or 1.2 mg siBcl-2 equiv./kg) via the tail vein at Days 1, 3, 5, 8, and 11. Body weight and tumor volume were monitored every 2 days. To compare the antitumor capacity among the various formulations, the tumor inhibitory rate (TIR) was calculated using the following equation:
|
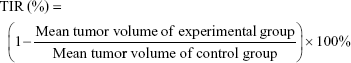
At Day 15, all mice were sacrificed, and the tumor and major organs were extracted and weighed. These samples were then stained with H&E and photographed via optical microscopy. The percentage of apoptotic cells in the frozen tumor sections fixed with 4% formalin solution was determined using TUNEL assays.
Statistical analyses
Results are presented as mean ± SD. Statistical significance was tested using a two-tailed Student’s t-test or one-way ANOVA, and was set at *P<0.05 or **P<0.01.
Results and discussion
Preparation and characterization of siBcl-2/Dox-TPGS-LPs
The cationic LPs were prepared using a film dispersion method, and Dox loading was established via an ammonium sulfate gradient to achieve a high rate of drug encapsulation. As shown in Table 1, the drug EE of Dox-LPs and Dox-TPGS-LPs was above 95%. TPGS-modified LPs (Dox-TPGS-LPs) displayed a slightly larger particle size of 155 nm and a significantly decreased zeta potential of +35.0 mV compared with non-coated LPs (Dox-LPs) (131 nm and +61.2 mV), indicating that the incorporation of TPGS changed these properties of the cationic LPs. TPGS, which is a PEGylated vitamin E, can form a PEG shell on cationic LPs that can enhance the stability of siRNA-loaded LPs in biological fluids.22 The positive effect of TPGS on the stability of siRNA delivery was further confirmed by promoted aggregation and formation of large-sized particles after mixing siRNA with Dox-LPs in contrast to Dox-TPGS-LPs.
 | Table 1 Characterization of Dox-loaded and siBcl-2/Dox co-loaded LPs |
After the successful encapsulation of Dox, siBcl-2 was further loaded onto the surface of the cationic LPs (Dox-TPGS-LPs) to prepare different siBcl-2/Dox co-loaded liposome complexes (siBcl-2/Dox-TPGS-LPs) with varied N/P ratios. As shown in Figure S1, the entrapment efficiency of siBcl-2 increased significantly from 27.5% to 96.8% as the N/P ratio value varied from 0.5 to 8. The optimized siBcl-2/Dox-TPGS-LPs were prepared at a ratio of 4:1 for further investigation, owing to a higher EE (~95%) of siBcl-2, a smaller particle size of 209.9 nm, and a suitable zeta potential of +12.5 mV. A higher N/P ratio (8:1) resulted in a slightly decreased particle size without substantially more siBcl-2 loading and a significantly increased positive zeta potential (+23.4 mV), which might promote the rapid clearance of siBcl-2/Dox-TPGS-LPs by the liver in vivo. Furthermore, the nanostructure of siBcl-2/Dox-TPGS-LPs was imaged via transmission electron microscopy (TEM) (Figure 1), indicating a significantly increased particle size (~200 nm). Importantly, a substantial outer corona on the siBcl-2/Dox-TPGS-LPs was apparent, indicating a highly efficient loading of siBcl-2 via electrostatic interactions. The TEM images (Figure 1) of Dox-loaded LPs, including Dox-LPs, Dox-TPGS-LPs, and siBcl-2/Dox-TPGS-LPs, all demonstrated suitable morphologies and distributions. It was also notable that loading of siBcl-2 had no apparent effect on the entrapment efficiency of Dox.
 | Figure 1 TEM images of Dox-LPs, Dox-TPGS-LPs, and siBcl-2/Dox-TPGS-LPs stained with phosphotungstic acid. |
Drug release from siBcl-2/Dox co-loaded LPs
As shown in Figure 2, the in vitro release profile of Dox from Dox-LPs, Dox-TPGS-LPs, and siBcl-2/Dox-TPGS-LPs showed a controlled and sustained release up to 48 hours, while the free Dox group presented a burst of release (~68%) within the first 4 hours. Dox-TPGS-LPs exhibited a higher accumulative release of Dox compared with Dox-LPs, and it was speculated that TPGS enhanced the solubility of the LPs, which would promote Dox release.23 Dox release from siBcl-2/Dox-TPGS-LPs was slightly slower than that from Dox-TPGS-LPs during the initial period, while the accumulative release from siBcl-2/Dox-TPGS-LPs exhibited no significant difference vs Dox-TPGS-LPs by the end of 48 hours, indicating that siBcl-2-loading might prolong the Dox release period, and this might serve to decrease systemic toxicity in vivo.
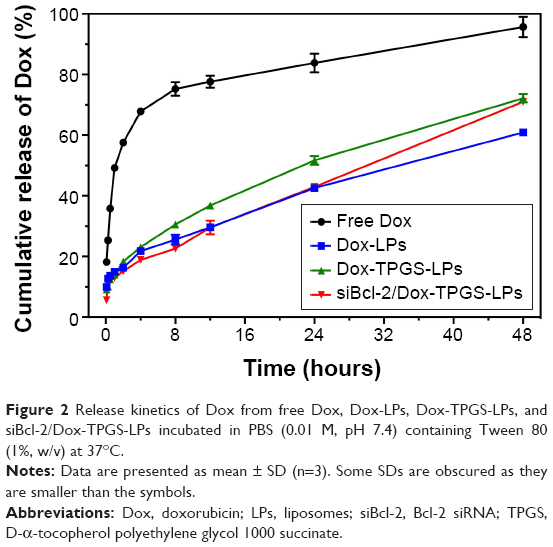 | Figure 2 Release kinetics of Dox from free Dox, Dox-LPs, Dox-TPGS-LPs, and siBcl-2/Dox-TPGS-LPs incubated in PBS (0.01 M, pH 7.4) containing Tween 80 (1%, w/v) at 37°C. |
Dox intracellular uptake by H22 cells
As shown in Figure 3, H22 cells treated with Dox-LPs demonstrated the highest intracellular uptake of Dox compared with other four experimental groups. This increased uptake was attributed to the higher positive surface charge of Dox-LPs, which likely promoted cellular uptake by the cells via a charge–charge interaction with the plasma membrane that would help mediate adsorptive endocytosis.24 However, siBcl-2/Dox-TPGS-LPs with an overall weaker positive zeta potential exhibited a higher intracellular uptake (~10%) by H22 cells compared with Dox-TPGS-LPs, as siBcl-2 binding (siRNA corona) increased the particle size and enhanced the cellular uptake by size-dependent endocytosis.25 Together, these results suggested that siBcl-2/Dox-TPGS-LPs were able to efficiently deliver Dox into the tumor cells as opposed to decreasing the cellular uptake of Dox due to the reduced positive surface charge following co-loading of siBcl-2.
 | Figure 3 The cellular uptake of Dox in H22 cells was measured by flow cytometer after treatment with free Dox, Dox-LPs, Dox-TPGS-LPs, or siBcl-2/Dox-TPGS-LPs at a Dox concentration of 5 μg/mL for 4 hours; untreated cells served as a control. |
Intracellular distribution of Dox and siRNAs co-delivered via siBcl-2/Dox-TPGS-LPs
To ensure siBcl-2 and Dox were delivered to subcellular sites, siBcl-2 was labeled with green fluorescent FAM (FAM-siRNA/Dox-TPGS-LPs) to observe their intracellular distribution using CLSM. As shown in Figure 4, the cationic LPs Dox-LPs showed significantly stronger red fluorescence in the nucleus of H22 cells than Dox-TPGS-LPs, indicating that Dox-LPs might deliver Dox to the nucleus at a higher efficiency. Free Dox exhibited weak fluorescence intensity in the nucleus of the tumor cells, owing to the internalization of Dox by passive diffusion, which would allow it to enter the nucleus easily. Notably, the FAM-siRNA/Dox-TPGS-LPs group exhibited significant green fluorescence in the cytoplasm rather than the nucleus of H22 cells, which highlighted both the targeted subcellular sites of Dox (nuclei) and siBcl-2 (cytoplasm). Furthermore, the Dox accumulation in the nucleus of the FAM-siRNA/Dox-TPGS-LPs group was slightly higher than that in the Dox-TPGS-LPs group. This result was attributed to enhanced cellular uptake conferred by size-dependent endocytosis and tumor priming.
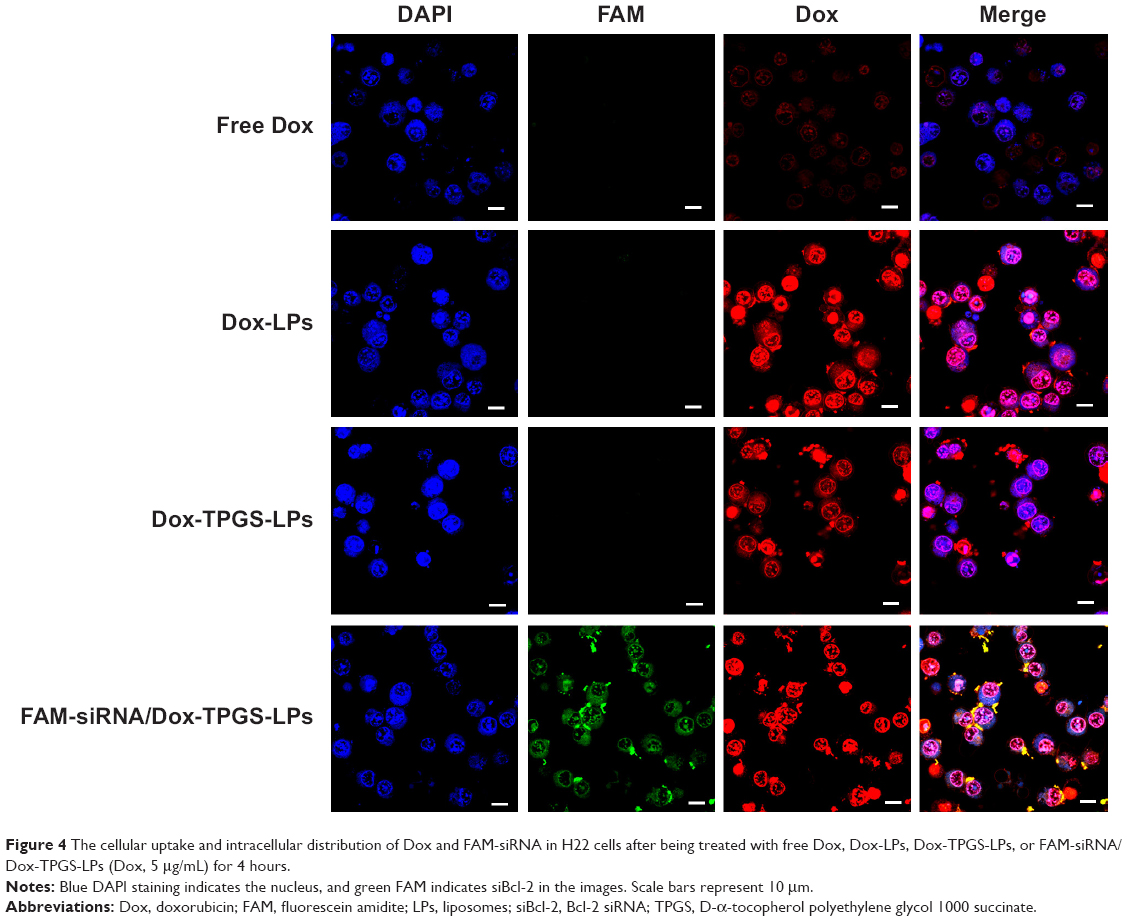 | Figure 4 The cellular uptake and intracellular distribution of Dox and FAM-siRNA in H22 cells after being treated with free Dox, Dox-LPs, Dox-TPGS-LPs, or FAM-siRNA/Dox-TPGS-LPs (Dox, 5 μg/mL) for 4 hours. |
Cytotoxicity assays
The effect of the chemotherapeutic drug alone and co-delivered LPs on growth inhibition of H22 cells was investigated via MTT assays, as shown in Figure 5. All of the formulations exhibited dose-dependent cytotoxicity toward the cancer cells. Dox-loaded LPs presented much stronger cytotoxicity than free Dox, owing to the increased cellular uptake of the cationic LPs in comparison to the cellular accumulation of Dox via passive diffusion. Notably, Dox-LPs did not show a higher cytotoxicity than Dox-TPGS-LPs, as both had almost identical IC50 values (0.83 vs 0.81 μg/mL) against H22 cells. Bcl-2-targeted siRNAs have been shown to downregulate the level of the anti-apoptotic protein Bcl-2 and sensitize different cancer cell lines toward chemotherapeutic agents.26 As expected, siBcl-2/Dox-TPGS-LPs showed a significant decrease in the IC50 (0.35 μg/mL) compared with all the other groups, indicating a clear synergistic antitumor effect of Dox and siBcl-2. As shown in Figure S2, empty TPGS-modified LPs (TPGS-LPs) exhibited a weaker cytotoxicity at therapeutic total lipid concentrations (1–200 μg/mL) compared to bare cationic LPs with a strong positive potential. It was apparent that TPGS-modified cationic LPs could be used as a potential platform for the co-delivery of siBcl-2 and Dox due to their weaker cytotoxicity.
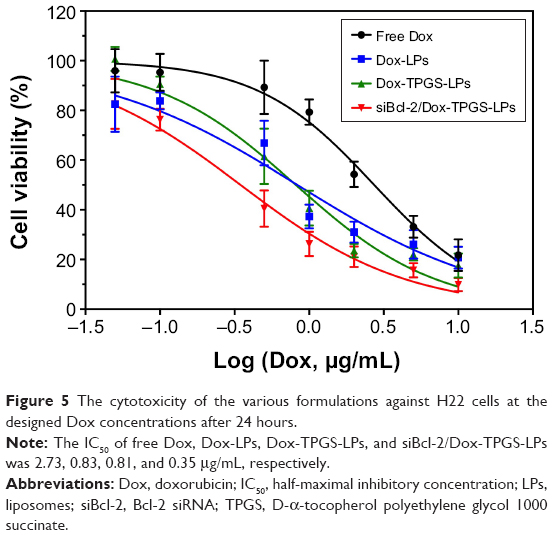 | Figure 5 The cytotoxicity of the various formulations against H22 cells at the designed Dox concentrations after 24 hours. |
Enhanced toxicity of siBcl-2/Dox-TPGS-LPs in H22 tumor spheroids
We established H22 tumor cell-derived multicellular tumor spheroids to evaluate the toxicity of siBcl-2/Dox-TPGS-LPs in vitro (Figure 6). Dox-LPs and Dox-TPGS-LPs showed a weaker toxicity, with the cells retaining a relatively regular spheroid morphology compared with free Dox, while cellular uptake of Dox-loaded cationic LPs was higher than that of the free Dox group. It was predictable that free Dox, as a small molecule, could enter tumor spheroids easily via passive diffusion, but the LPs (100–200 nm in size) were not able to penetrate deep within the tumor spheroids.24 However, siBcl-2/Dox-TPGS-LPs exhibited much better toxicity and more significant size shrinkage of the tumors compared to free Dox. This increased toxicity was a result of enhanced penetration due to tumor priming caused by partial cell apoptosis in the tumor spheroids, which was likely conferred by downregulation of Bcl-2 protein levels.
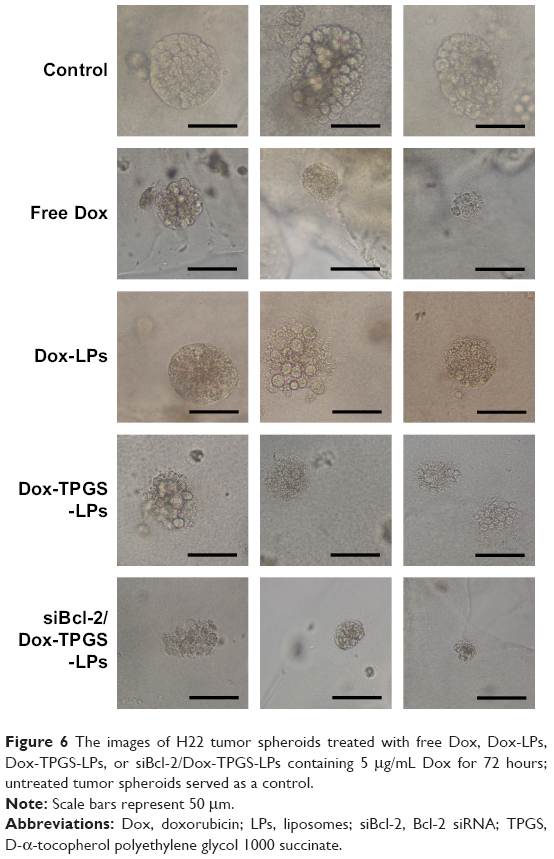 | Figure 6 The images of H22 tumor spheroids treated with free Dox, Dox-LPs, Dox-TPGS-LPs, or siBcl-2/Dox-TPGS-LPs containing 5 μg/mL Dox for 72 hours; untreated tumor spheroids served as a control. |
In vivo distribution and pharmacokinetics
Plasma concentration–time curves of the various Dox-loaded LPs were established to assess the retention time of Dox in the blood following LP delivery. As shown in Figure 7A, free Dox showed rapid clearance from the blood after intravenous injection. Dox-loaded LPs displayed a slower clearing rate than the free Dox group, indicating changes in pharmacokinetics conferred by Dox-loaded LPs. Importantly, siBcl-2/Dox-TPGS-LPs prolonged the elimination half-life time (T1/2) of Dox to 0.75 hour compared with free Dox (T1/2 of 0.20 hour), resulting in a chance for higher accumulation of Dox at the tumor sites. Moreover, siBcl-2/Dox-TPGS-LPs significantly increased the AUC in the blood by 16% compared with Dox-TPGS-LPs (17.77 vs 15.39 mg/L·h), which demonstrated that co-loading of siRNAs could prolong the circulation of Dox in vivo.
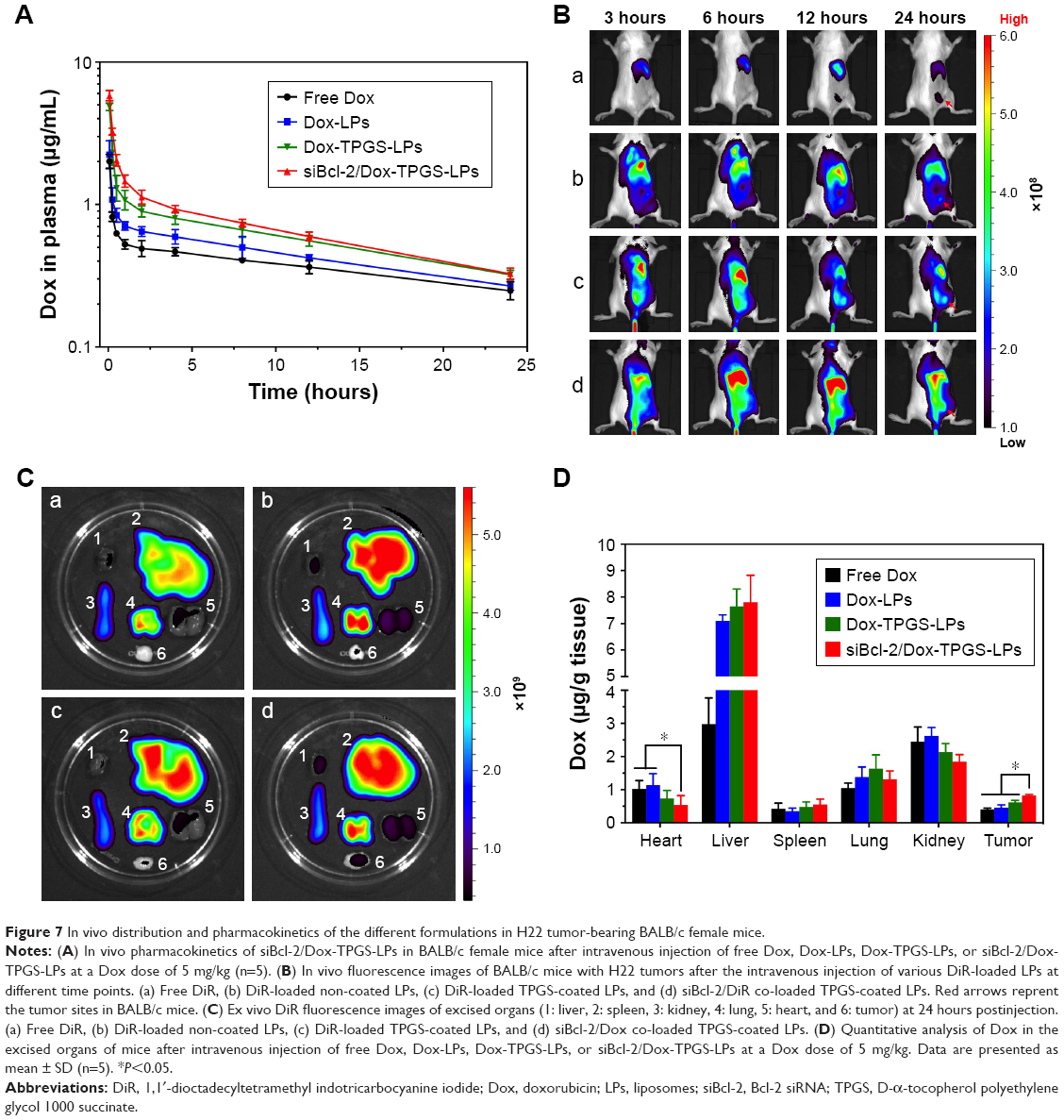 | Figure 7 In vivo distribution and pharmacokinetics of the different formulations in H22 tumor-bearing BALB/c female mice. |
Figure 7B depicts the real-time distribution of DiR-loaded LPs in vivo after intravenous injection, indicating enhanced accumulation of LPs at tumor sites conferred by TPGS and the siBcl-2 corona. DiR was used as a molecular marker due to its high transmission of infrared light through tissues in vivo. The fluorescence of non-coated DiR-loaded LPs (DiR-LPs) reached the maximum level in the liver within the first 3 hours, and then decreased rapidly over time. This result was attributed to the rapid clearance by the liver caused by the strong positive charge of the cationic LPs.27 DiR-loaded TPGS-modified LPs exhibited relatively slower clearance by the liver and achieved higher accumulation at the tumor sites compared with non-coated LPs, owing to their prolonged circulation time conferred by the PEG layer from TPGS.28 Moreover, siBcl-2 and DiR co-loaded LPs showed a lower fluorescence in the liver over the first 3 hours but a higher accumulation at tumor sites compared to the other DiR-loaded LPs at 24 hours after intravenous injection, which might be the result of the further decrease in their positive charge after siBcl-2 loading (Figure 7C).
Dox biodistribution in H22 tumor-bearing female mice was further quantified at 24 hours after intravenous injection, as shown in Figure 7D. The siBcl-2/Dox-TPGS-LPs presented a significantly decreased accumulation of Dox in the heart compared to free Dox and Dox-LPs, indicating the potential for a lower cardiotoxicity. Indeed, the cationic LPs Dox-LPs, Dox-TPGS-LPs, and siBcl-2/Dox-TPGS-LPs exhibited higher accumulation of Dox in the liver than the free Dox group, owing to the clearance of positively charged nanoparticles by the liver. It was noteworthy that siBcl-2/Dox-TPGS-LPs showed the highest accumulation of Dox at tumor sites compared with the other three Dox formulations, about twofold that of the free Dox group. Together, these findings demonstrated that siBcl-2/Dox-TPGS-LPs could prolong the retention time of Dox in the blood, significantly decrease the accumulation of Dox in the heart, and successfully deliver Dox and siBcl-2 to the tumor target sites.
Tumor growth inhibition
The antitumor growth effect of siBcl-2/Dox-TPGS-LPs was evaluated using an H22 tumor-bearing BALB/c female mouse model. siBcl-2/Dox-TPGS-LPs administered via the tail vein exhibited relatively higher tumor growth inhibition (75%) compared with Dox-TPGS-LPs (P<0.05). Free Dox and Dox-LPs only inhibited tumor growth by 46% (Figure 8A). As expected, siBcl-2/Dox-TPGS-LPs showed a significantly (P<0.05) smaller tumor weight and tumor size compared to the Dox-TPGS-LPs, Dox-LPs, and free Dox groups (Figure 8C and D). This effect could be attributed to a synergistic antitumor effect of siBcl-2 and Dox on tumor growth. Moreover, TUNEL staining results showed the largest number of apoptotic cells in the siBcl-2/Dox-TPGS-LPs group (Figure 8E), further clarifying the enhanced antitumor effect of siBcl-2/Dox-TPGS-LPs. The body weight of the mice injected with siBcl-2/Dox-TPGS-LPs, Dox-TPGS-LPs, and Dox-LPs did not appreciably decrease over the treatment period, while the free Dox group rapidly lost weight (>15%) (Figure 8B). These results were consistent with the potential cardiac toxicity caused by high Dox accumulation in the heart of the free Dox treatment group (Figure 7D), which was further confirmed by the higher levels of lactate dehydrogenase in the blood of mice treated with free Dox relative to the normal saline treatment group (Figure S3). As expected, these treatments did not cause significant cytotoxicity to other tissues, as shown in Figure S4. These results revealed that the TPGS-modified cationic LPs designed for co-delivery of siBcl-2 and Dox enhanced the chemotherapeutic efficacy of Dox via a combination of prolonged circulation in the blood (Figure 7A) and tumor priming due to the siBcl-2 corona and apoptosis-related gene silencing conferred by siBcl-2, respectively.
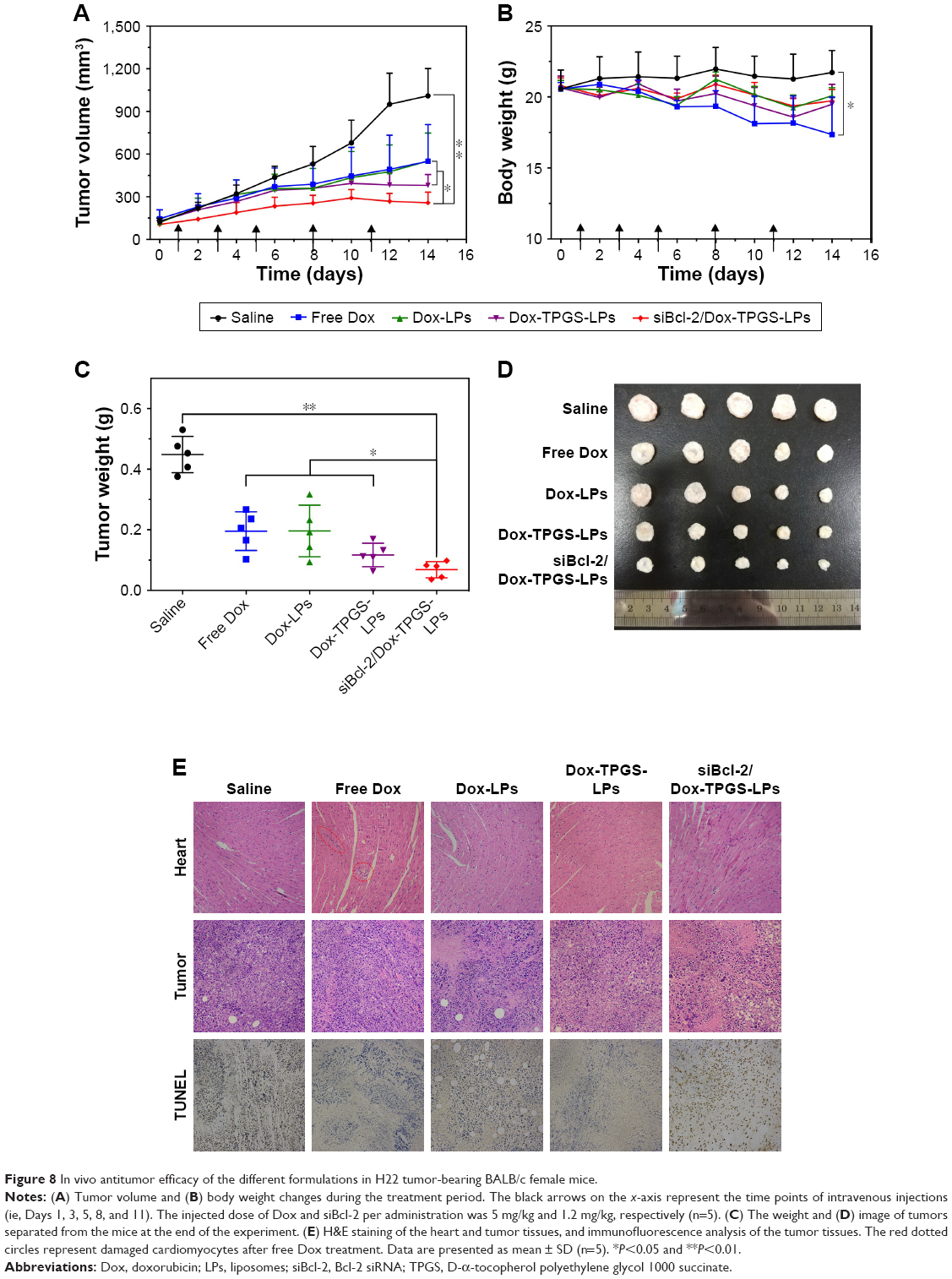 | Figure 8 In vivo antitumor efficacy of the different formulations in H22 tumor-bearing BALB/c female mice. |
Conclusion
In summary, we developed TPGS-modified LPs with an siBcl-2 corona to deliver Dox to tumor cells. This synergistic chemotherapy achieved enhanced cellular uptake, promoted diffusion of Dox, and increased accumulation of Dox at tumor sites. It was apparent that the siBcl-2 corona served to sensitize tumor cells toward the chemotherapeutic agent by downregulating the expression of the anti-apoptotic protein Bcl-2 and also increased the cellular uptake and intratumoral diffusion of the antitumor drug to enhance its chemotherapeutic efficacy. We believe that co-loading of siRNAs both sensitizes cells toward antitumor drugs by downregulating the expression level of a specific protein and influences the pharmacokinetic behavior of the co-delivery system in vitro and in vivo.
Acknowledgments
The authors acknowledge the support by National Basic Research Program of China (973 Program, 2012CB932500) and National Natural Science Foundation of China (31470968 and 81202486). The authors thank the Analytical and Testing Center of the College of Life Science and Technology at Huazhong University of Science and Technology for their help with TEM, confocal microscopy, and flow cytometry analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Xin Y, Huang M, Guo WW, Huang Q, Zhang LZ, Jiang G. Nano-based delivery of RNAi in cancer therapy. Mol Cancer. 2017;16(1):134. | ||
Chi X, Gatti P, Papoian T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov Today. 2017;22(5):823–833. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Creixell M, Peppas NA. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today. 2012;7(4):367–379. | ||
Zhou S, Chen W, Xiao ZL, Ye S, Ding CD, Fu JJ. Smart drug and gene co-delivery system for cancer therapy. Progr Chem. 2017;29(5):502–512. | ||
Sun W, Chen X, Xie C, et al. Co-delivery of doxorubicin and anti-Bcl-2 siRNA by pH-responsive polymeric vector to overcome drug resistance in in vitro and in vivo HepG2 hepatoma model. Biomacromolecules. 2018;19(6):2248–2256. | ||
Ye Z, Wu W-R, Qin Y-F, et al. An integrated therapeutic delivery system for enhanced treatment of hepatocellular carcinoma. Adv Funct Mater. 2018;28(18):1706600. | ||
Zhang J, Du Z, Pan S, et al. Overcoming multidrug resistance by Codelivery of MDR1-Targeting siRNA and doxorubicin using EphA10-Mediated pH-sensitive lipoplexes: in vitro and in vivo evaluation. ACS Appl Mater Interfaces. 2018;10(25):21590–21600. | ||
Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of Bcl-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16(2):99–109. | ||
Qu MH, Zeng RF, Fang S, Dai QS, Li HP, Long JT. Liposome-based co-delivery of siRNA and docetaxel for the synergistic treatment of lung cancer. Int J Pharm. 2014;474(1–2):112–122. | ||
Li JM, Zhang W, Su H, et al. Reversal of multidrug resistance in MCF-7/Adr cells by codelivery of doxorubicin and Bcl2 siRNA using a folic acid-conjugated polyethylenimine hydroxypropyl-β-cyclodextrin nanocarrier. Int J Nanomedicine. 2015;10:3147–3162. | ||
Kuh HJ, Jang SH, Wientjes MG, Weaver JR, Au JL. Determinants of paclitaxel penetration and accumulation in human solid tumor. J Pharmacol Exp Ther. 1999;290(2):871–880. | ||
Hu CM, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther Deliv. 2010;1(2):323–334. | ||
Li J, Cheng X, Chen Y, et al. Vitamin E TPGS modified liposomes enhance cellular uptake and targeted delivery of luteolin: an in vivo/in vitro evaluation. Int J Pharm. 2016;512(1):262–272. | ||
Li Y, Gao L, Tan X, Li F, Zhao M, Peng S. Lipid rafts-mediated endocytosis and physiology-based cell membrane traffic models of doxorubicin liposomes. Biochim Biophys Acta. 2016;1858(8):1801–1811. | ||
Jeong UH, Garripelli VK, Jo S, et al. Potential of pH-sensitive polymer-anchored cationic liposomes for combinatorial anticancer therapy with doxorubicin and siRNA. J Drug Deliv Sci Technol. 2014;24(1):27–32. | ||
Zhao X, Chen Q, Liu W, et al. Codelivery of doxorubicin and curcumin with lipid nanoparticles results in improved efficacy of chemotherapy in liver cancer. Int J Nanomedicine. 2015;10:257–270. | ||
Li X, Garamus VM, Li N, et al. Preparation and characterization of a pH-responsive mesoporous silica nanoparticle dual-modified with biopolymers. Colloids Surf A Physicochem Eng Asp. 2018;548:61–69. | ||
Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA. Spheroid-based drug screen: considerations and practical approach. Nat Protoc. 2009;4(3):309–324. | ||
Yong T, Hu J, Zhang X, et al. Domino-like intercellular delivery of Undecylenic acid-conjugated porous silicon nanoparticles for deep tumor penetration. ACS Appl Mater Interfaces. 2016;8(41):27611–27621. | ||
Cheng Q, Du L, Meng L, et al. The promising nanocarrier for doxorubicin and siRNA co-delivery by PDMAEMA-based amphiphilic nanomicelles. ACS Appl Mater Interfaces. 2016;8(7):4347–4356. | ||
Zhang M, He J, Zhang W, Liu J. Fabrication of TPGS-Stabilized Liposome-PLGA hybrid nanoparticle via a new modified Nanoprecipitation approach: in vitro and in vivo evaluation. Pharm Res. 2018;35(11):199. | ||
Muthu MS, Kulkarni SA, Xiong J, Feng SS. Vitamin E TPGS coated liposomes enhanced cellular uptake and cytotoxicity of docetaxel in brain cancer cells. Int J Pharm. 2011;421(2):332–340. | ||
Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172(3):782–794. | ||
Liang S, Yang XZ, Du XJ, et al. Optimizing the size of micellar nanoparticles for efficient siRNA delivery. Adv Funct Mater. 2015;25(30):4778–4787. | ||
Qian J, Xu M, Suo A, et al. Folate-decorated hydrophilic three-arm star-block terpolymer as a novel nanovehicle for targeted co-delivery of doxorubicin and Bcl-2 siRNA in breast cancer therapy. Acta Biomater. 2015;15:102–116. | ||
He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. | ||
Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. |
Supplementary materials
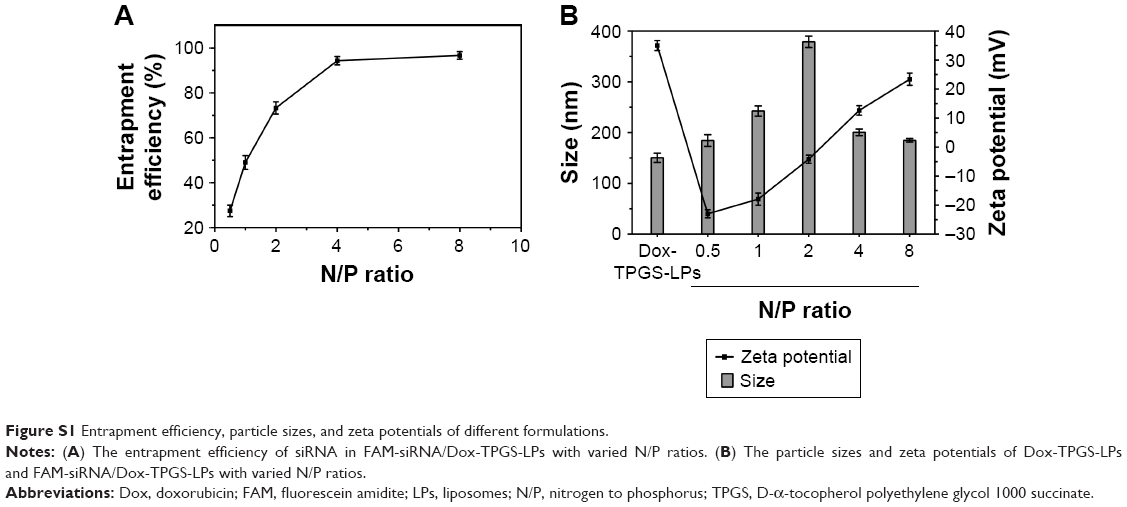 | Figure S1 Entrapment efficiency, particle sizes, and zeta potentials of different formulations. |
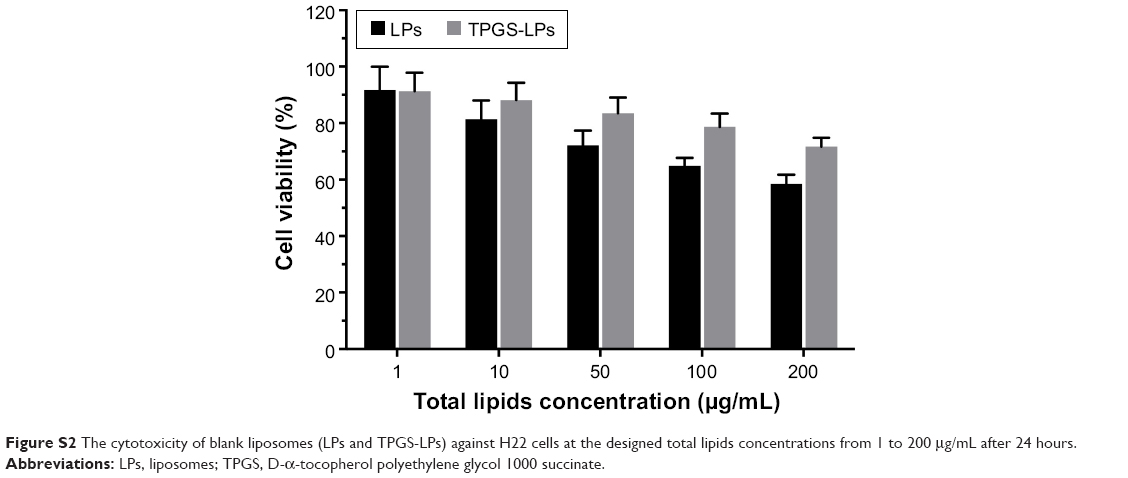 | Figure S2 The cytotoxicity of blank liposomes (LPs and TPGS-LPs) against H22 cells at the designed total lipids concentrations from 1 to 200 μg/mL after 24 hours. |
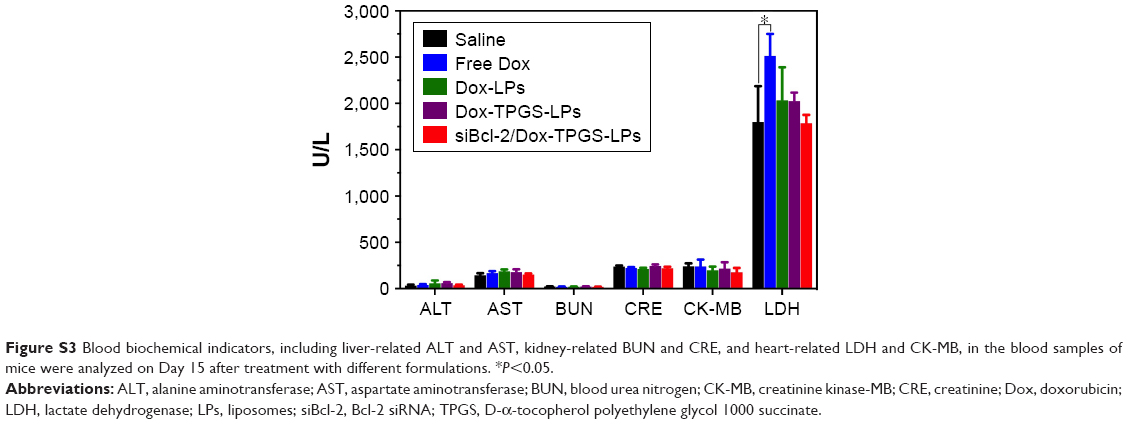 | Figure S3 Blood biochemical indicators, including liver-related ALT and AST, kidney-related BUN and CRE, and heart-related LDH and CK-MB, in the blood samples of mice were analyzed on Day 15 after treatment with different formulations. *P<0.05. |
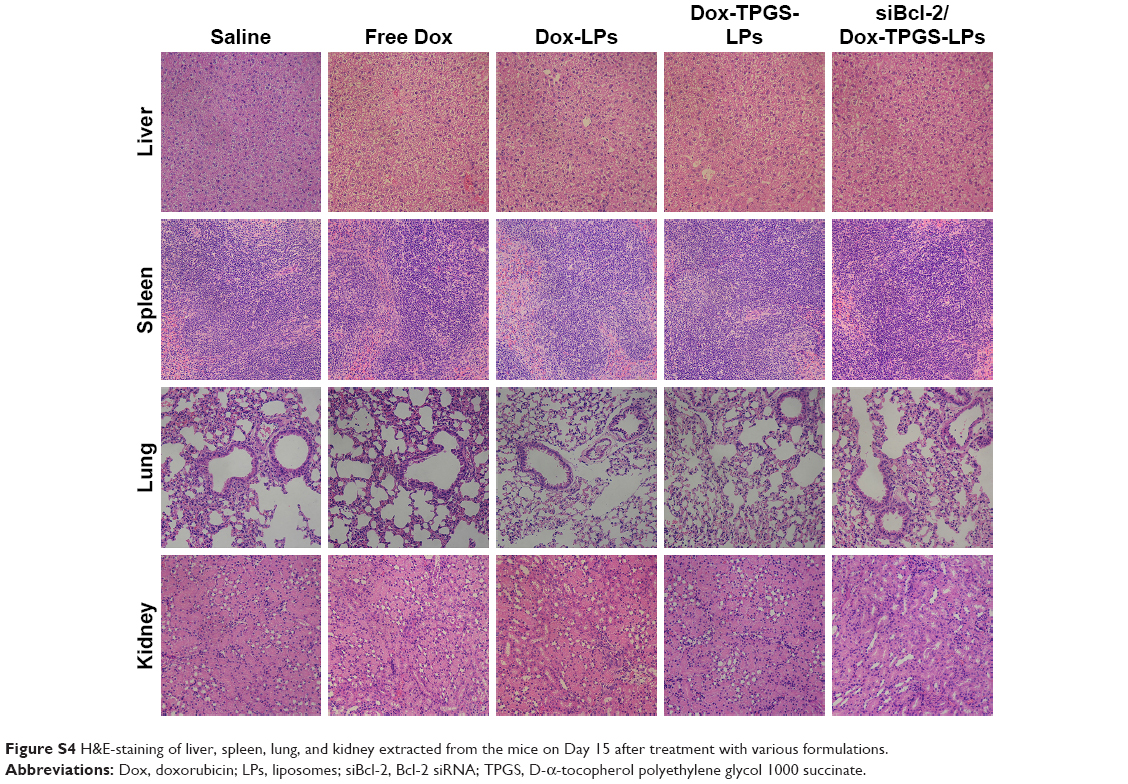 | Figure S4 H&E-staining of liver, spleen, lung, and kidney extracted from the mice on Day 15 after treatment with various formulations. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.